Introduction
Rheumatoid arthritis (RA) is a complicated systemic
disease that manifests as chronic synovial inflammation and leads
to the gradual damage of the articular cartilage, loss of joint
functions and comorbidity with the extraarticular organs (1). A study in Italy reported that the
incidence rate of RA was 0.038% in women and 0.013% in men
(2,3). A study in South Korea reported that
the incidence of RA was higher for individuals >60 years old
(4). The pathogenesis of RA is not
yet fully understood.
RA is associated with a number of factors, such as
dysregulated gene expression, and environmental and stochastic
factors. Several studies have demonstrated that environmental
factors, including smoking (5),
exposure to inhaled particulate air pollution (6) and multiple dietary (7), are risk factors for rheumatoid-factor
positive RA. Early diagnosis of RA is important for effective
treatment, and a longer course of disease can lead to worse
outcomes (8). Thus, it is necessary
to develop efficient diagnostic and therapeutic methods.
Fibroblast-like synoviocytes (FLSs) are one of the most common and
dominant type of effector cells found in synovial joints that bear
several features of malignant cells, such as abundant cytoplasm,
large pale nuclei with several prominent nucleoli and a dense,
rough endoplasmic reticulum (9-12).
Results of our previous studies demonstrated that dual-specificity
tyrosine-regulated kinase 1A (Dyrk1A) can promote the
proliferation, migration and invasion of FLSs through the
suppression of protein sprouty homolog 2 and the activation of the
ERK/MAPK signaling pathway in patients with RA (13). In addition to the aggressive
proliferative and migratory patterns, FLSs also secrete matrix
metalloproteinases (MMPs), for example MMP-9 and MMP-3(14), which destroy collagen bundles within
the cartilages, thereby assisting the invasion of cartilage and
bone (15).
Researchers have been focusing on suppressing the
abnormal activation of FLSs as a novel method to treat RA (16). The repressor element-1 silencing
transcription factor (REST) is a transcription factor consisting of
a DNA-binding domain and two repressor domains (17,18).
REST corepressor (CoREST) is a functional corepressor that assumes
several roles in regulating the expression of neuron-specific
genes. CoREST contains two SANT (SW13/ADA2/NCoR/TFIIIB B) domains,
a structural feature of the nuclear receptor, and silencing
mediator for retinoid and thyroid human receptors (SMRT)-extended
corepressors that mediate inducible repression by steroid hormone
receptors (19). The role of
lysine-specific histone demethylase 1 (LSD1) in RA has been
investigated. In CD4+T cells obtained from active RA
synovial fluids, LSD1 knockdown can significantly promote cell
proliferation, while also significantly increasing the synthesis of
interferon-γ (IFN-γ), interleukin (IL)-17 and IL-10(20). An in vivo study indicated
that LSD1 knockdown significantly alleviated disease severity
(20). The repressed activity of
LSD1 has been shown to depend on its interaction with CoREST
(21-23).
It has been reported that LSD1 can act as a negative regulator of
the Notch signaling pathway through its interaction with the
deacetylase sirtuin 1 (SIRT1) in cell cultures (24). Previous studies have shown that
interfering with the CoREST/LSD1 complex can slow down the
development of the cerebral cortex by delaying cell differentiation
(25,26). However, the roles of CoREST in the
pathogenesis of RA are still unknown.
In the present study, the expression levels of
CoREST in the synovial tissues of patients with RA were
investigated. Furthermore, the effects of CoREST on the
proliferative, migratory and invasive patterns of RA-FLSs were
evaluated, and the fundamental processes involved in the
pathological process of RA was explored.
Materials and methods
Patient selection
A total of 14 patients with RA were involved in this
study, including 7 female and 7 male patients. Patients
hospitalized in The Second Affiliated Hospital of Nantong
University (Nantong, China) from October 2018 to April 2019 were
selected. They were selected according to the 2010 American College
of Rheumatology/European League Against Rheumatism Classification
Criteria for Rheumatoid Arthritis and other immune system related
diseases were excluded (27). The
inclusion criteria were as follows: i) DAS-28 score was >2.6;
ii) no previous diagnosis of other immune-related diseases; and
iii) had not taken any medication for RA within the year before the
operation. The synovial tissues were obtained from the patients
while they underwent total knee replacement or arthroscopic
surgeries. Normal synovial tissues were obtained from another 14
healthy volunteers through arthroscopic procedures. The average
ages of patients were 45±6 and 47±5 years old for patients with RA
and healthy controls, respectively. The procedures and processes of
the present study were reviewed and approved by the institutional
medical ethics committee of Affiliated Hospital 2 of Nantong
University, Nantong, China (approval no. 2019KY126). Patients
provided written informed consent to participate before they
received any treatment.
Cell culture and TNF-α treatment
The synovial tissues were sliced into 2-4 mm
sections and left to degrade for 4 h at 37˚C with collagenase I
(Gibco; Thermo Fisher Scientific, Inc.) in Hanks' balanced salt
solution (Beyotime Institute of Biotechnology). Following
centrifugation at 111.8 x g and room temperature for 5 min, cells
obtained from the synovial tissues were cultured in Dulbecco's
modified Eagle's medium (DMEM)/F12 medium (Gibco; Thermo Fisher
Scientific, Inc.) with 100 U/ml penicillin, 100 µg/ml streptomycin
(HyClone; Cytiva) and 10% fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc.). Cells were incubated in a humidified
incubator with 5% CO2 at 37˚C. Cells at passages 3-8
were utilized for the present study. A total of 10 ng/ml TNF-α
(PeproTech, Inc.) was used to stimulate FLSs for 24 h in a
humidified incubator with 5% CO2 at 37˚C. Following
treatment with TNF-α, FLSs were collected for subsequent
experiments.
Immunohistochemistry (IHC)
analysis
Harvested synovial tissues were fixed in 4%
paraformaldehyde at 4˚C for 24 h and subsequently embedded in
paraffin. The embedded tissue was cut into 5-µm slices. A total of
100 µl blocking buffer [10% FBS (cat. no. F8318; Sigma-Aldrich;
Merck KGaA) in 1X PBS (Sangon Biotech)] was added to the sections,
and these were incubated in a humidified chamber at room
temperature for 1 h. The blocking buffer was subsequently drained
from the slices. The IHC analysis was carried out using a primary
antibody against CoREST (1:100; cat. no. 07-455; Sigma-Aldrich;
Merck KGaA) at 4˚C overnight, and the slices were subsequently
washed with 1X PBS (Sangon Biotech) twice for 5 min each. The
slices incubated in a humidified chamber at room temperature for 1
h with a horseradish peroxidase (HRP)-conjugated secondary antibody
(1:4,000; cat. no. 12-348; Sigma-Aldrich; Merck KGaA). The slices
were subjected to immunoperoxidase staining performed using an
HRP/diaminobenzidine IHC detection kit (cat. no. ab64264; Abcam).
The slices were photographed using a Bx53 LED fluorescent
microscope (Olympus Corporation).
Knockdown of CoREST with small
interfering (si)RNA transfection
The CoREST expression in FLSs was knocked down with
human CoREST siRNA (siCoREST) and the synthetic siRNA was applied
as a negative control (siNC; Guangzhou RiboBio Co., Ltd.). The
target sequence of the CoREST siRNA was
5'-AAGAUUGUCCCGUUCUUGACU-3', and the sequence of the control siRNA
was 5'-UUGAUGUGUUUAGUCGCUA-3'. The control siRNA sequence was
purchased from Guangzhou RiboBio Co., Ltd. The treatment
concentration of all siRNAs was 40 nmol. Transient transfection of
siRNA was achieved using riboFECTTM CP Reagent (Guangzhou RiboBio
Co., Ltd.), according to the manufacturer's recommendations.
Following the transfection of siRNA, cells were incubated and
cultured in serum-free medium for 6 h, and then transferred to
total medium that contained 10% FBS for a further 48 h. After
verifying the knockdown of CoREST, the cells were applied for
further experiments.
Cell Counting Kit-8 (CCK-8) assay
In order to examine the proliferation of FLSs, a
CCK-8 assay was performed. Cells were seeded into 96-well plates at
a density of 1x103 cells/well and tested for viability
at 24 h. To determine cell proliferation, CCK-8 reagent was added
into each well (10 µl/well). The cells were incubated at 37˚C for
1.5 h with 5% CO2, and the absorbance was recorded at
450 nm with an ELISA plate reader for each well. The histogram of
cell viability was constructed using GraphPad Prism 8.0 software
(GraphPad Software, Inc.).
Wound healing assay
FLSs were seeded into 6-well plates at a density of
2x105 cells/well. Once the cell confluence reached 90%,
sterile pipette tips were used to create wounds. Detached cells
were washed with phosphate buffer saline and the medium was
replaced with DMEM/F12 containing 2% FBS. Images were captured at 0
and 24 h using a Bx53 LED fluorescent microscope (Olympus
Corporation) and subsequently examined using ImageJ software
(version 1.51j8; National Institutes of Health) to count the cells
beyond the reference line.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) was used mainly for total mRNA extraction
from fibroblast-like synoviocytes, following which, the products
were reverse transcribed into cDNA using a RevertAid First Strand
cDNA Synthesis Kit according to the manufacturer's protocol (Thermo
Fisher Scientific, Inc.). qPCR was performed using PowerUp™ SYBR™
Green Master Mix (Thermo Fisher Scientific, Inc.) on a StepOnePlus™
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycling conditions of qPCR were as follows:
Initial denaturation at 95˚C for 5 min, followed by 35 cycles of
denaturation at 95˚C for 30 sec, annealing at 60˚C for 30 sec and
extension at 72˚C for 30 sec. The relative expression of target
mRNA was normalized to β-actin as an endogenous control and
quantified using the 2-ΔΔCq method (28). The primers applied in this study
were as follows: MMP-3 forward (F), 5'-GACAAAGGATACAACAGGGACCAAT-3'
and reverse (R), 5'-TGAGTGAGTGATAGAGTGGGTACAT-3'; MMP-9 F,
5'-TGCCCGGACCAAGGATACAG-3' and R, 5'-CAGGGCGAGGACCATAGAG-3'; and
β-actin F, 5'-GTCGGTGTGAACGGATTTG-3' and R,
5'-TCCCATTCTCAGCCTTGAC-3'.
Western blotting
Total protein was extracted from cells obtained from
the synovial tissues of patients using 1X PBST [1X PBS (Sangon
Biotech) and 1% Triton X-100, (Sigma-Aldrich; Merck KGaA)]. Total
protein was quantified using a BCA assay kit (Beyotime Institute of
Biotechnology). A total of 10 µg protein per lane was loaded on a
10% gel, and proteins were separated by SDS-PAGE. Proteins were
transferred onto PVDF membranes, and these were incubated with 10%
milk at room temperature for 1 h. Subsequently, the PVDF membranes
were incubated with the following antibodies: Anti-CoREST (1:1,000;
cat. no. 07-455; Sigma-Aldrich), rabbit anti-human LSD1 (1:1,000;
cat. no. A8711; Abclonal), anti-β-actin (1:4,000; cat. no. ab8227;
Abcam), anti-proliferating cell nuclear antigen (PCNA; 1:1,000;
cat. no. ab18197; Abcam), anti-MMP-3 (1:1,000; cat. no. AF7482;
Beyotime Institute of Biotechnology) and anti-MMP-9 (1:1,000; cat.
no. AF5234; Beyotime Institute of Biotechnology) at 4˚C overnight.
Following primary incubation, membranes were washed using PBS with
0.05% Tween-20 five times, followed by incubation for 1 h at room
temperature with a HRP-conjugated secondary antibody (1:4,000; cat.
no. 12-348; Sigma-Aldrich; Merck KGaA). The membrane was developed
using an electrochemiluminescence kit (Pierce; Thermo Fisher
Scientific, Inc.). β-actin expression was used for normalization.
All the experiments were performed in triplicate. The results of
western blots were analyzed using the ImageJ software (version
1.38; National Institutes of Health).
Data analysis
Results are presented as the mean ± standard
deviation (SD). Data were analyzed using SPSS software version 19.0
(IBM Corp.). An unpaired Student's t-test was performed to compare
statistically significant differences between two groups, while
one-way ANOVA followed by Dunnett's post hoc test was applied for
>2 groups. P<0.05 was considered to indicate a statistically
significant difference. All experiments in our study were performed
independently at least three times.
Results
Upregulation of CoREST in RA synovial
tissues
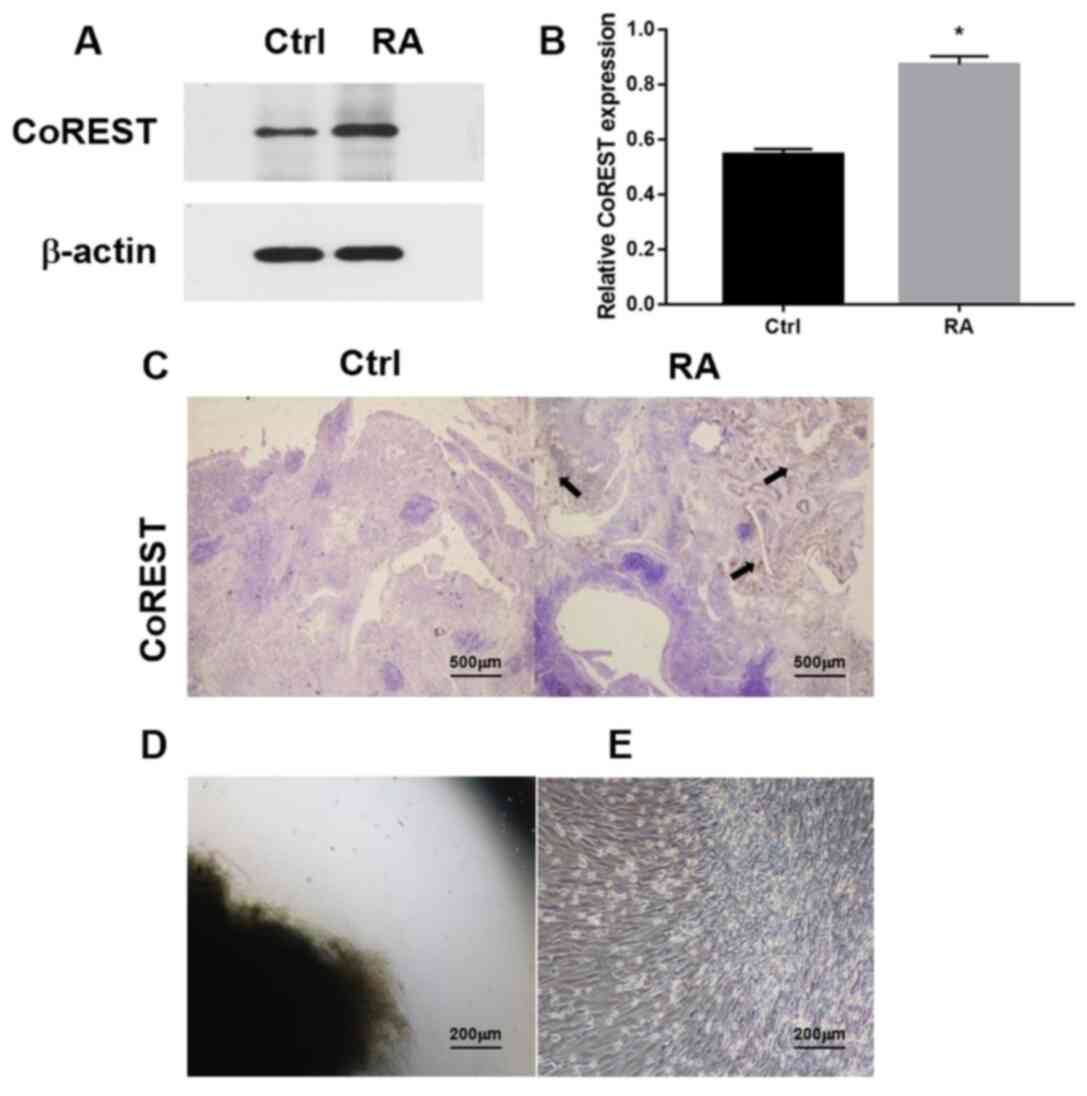
Western blotting was performed to detect the
expression of CoREST in the synovial tissues of patients with RA
and healthy controls (Fig. 1A).
There was a significant increase in the expression of CoREST in the
RA group compared with in the control group (Fig. 1B). The IHC analysis demonstrated
that CoREST expression was higher in the RA group compared with the
control group (Fig. 1C). FLSs,
which have important roles in the onset and disease progression of
RA (29), were found in both RA and
healthy synovial tissues. The shredded tissue appeared as a black
mass under the microscope (data not shown). After 3-5 days of
adhesion, the edge of the tissue appears as a gray image (Fig. 1D), indicating that cells had moved
from the border of the adherent tissue. By passage three, the
RA-FLSs showed spindle-shaped and whirlpool-like morphological
characteristics while growing under a light microscope (Fig. 1E).
CoREST knockdown alleviates
TNF-α-induced CoREST expression and the proliferation of FLSs
To further evaluate the function of CoREST in
RA-FLSs, a TNF-α-induced FLS activation model was included as
previously described (30-32).
Briefly, following treatment with TNF-α (10 ng/ml), CoREST
expression was significantly upregulated in FLSs in a
time-dependent manner (Fig. 2A). To
elucidate the potential influence of CoREST on the proliferation of
FLSs, CoREST expression was knocked down with siCoREST (Fig. S1). Following transfection with
siCoREST, the expression of CoREST in TNF-α-induced FLSs was
significantly inhibited as compared with that of the control group
(Fig. 2B). PCNA is a marker of cell
proliferation (26). After the
silencing of CoREST expression by siCoREST, the expression of PCNA
was also significantly reduced as compared with that in the control
group (Fig. 2B). The effects of
CoREST on the cell proliferation of FLSs stimulated with TNF-α were
determined via a CCK-8 assay. The cell viability of
TNF-α-stimulated FLSs was significantly suppressed after the
silencing of CoREST compared with the control group (Fig. 2C).
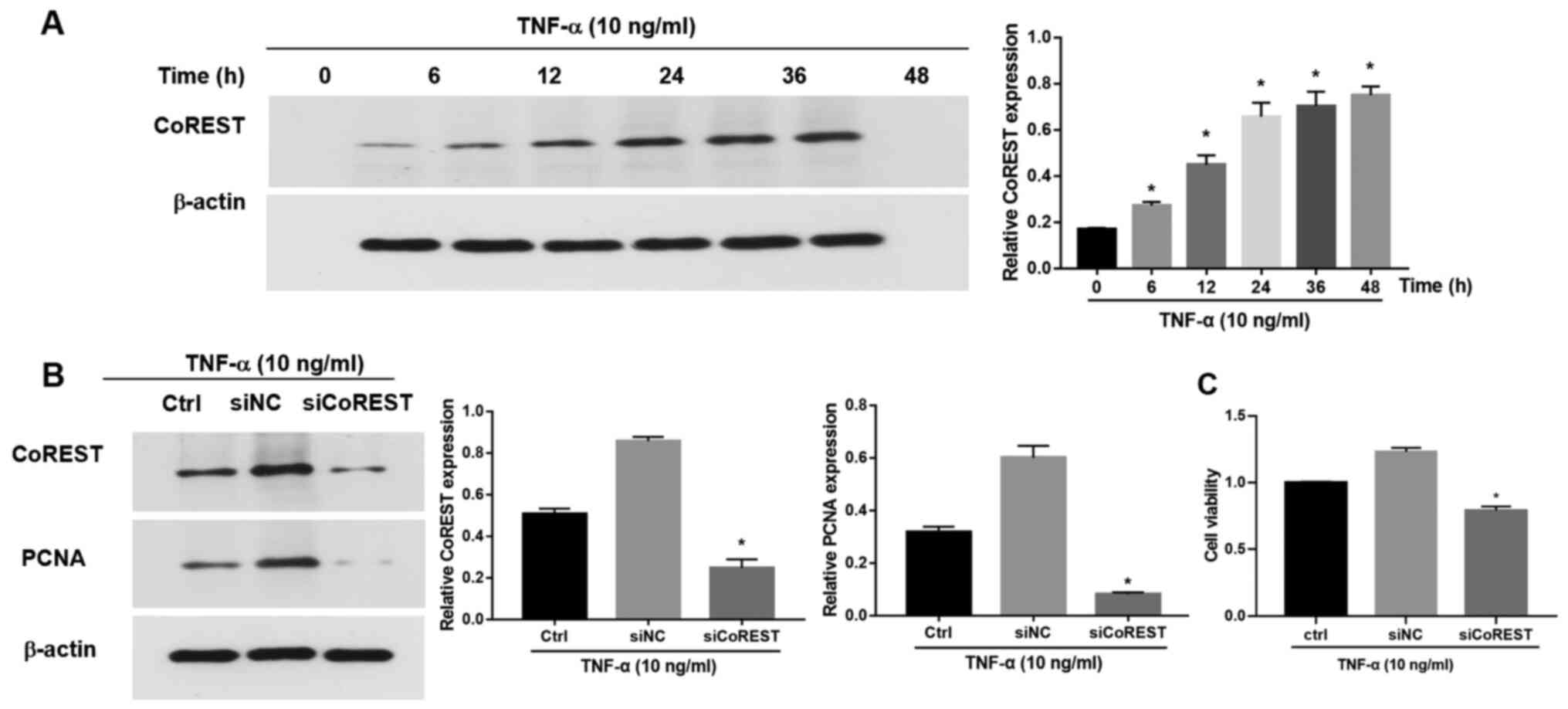 | Figure 2CoREST expression in FLSs stimulated
with TNF-α. (A) CoREST expression in FLSs stimulated with TNF-α (10
ng/ml) for 0, 6, 12, 24, 36 and 48 h, as determined via western
blotting. (B) The expression levels of CoREST and PCNA in
TNF-α-stimulated FLSs were determined by western blotting following
the knockdown of CoREST expression with siCoREST. The processed
siRNA was applied as the negative control (siNC). (C) Transfection
with siCoREST suppressed the proliferation of TNF-α-stimulated
FLSs. Data are presented as the mean ± SD. *P<0.05
vs. the siNC group. CoREST, corepressor element-1 silencing
transcription factor; FLSs, fibroblast-like synoviocytes; PCNA,
proliferating cell nuclear antigen; si, small interfering RNA; NC,
negative control. |
Knockdown of CoREST inhibits the
migratory and invasive abilities of TNF-α-stimulated FLSs
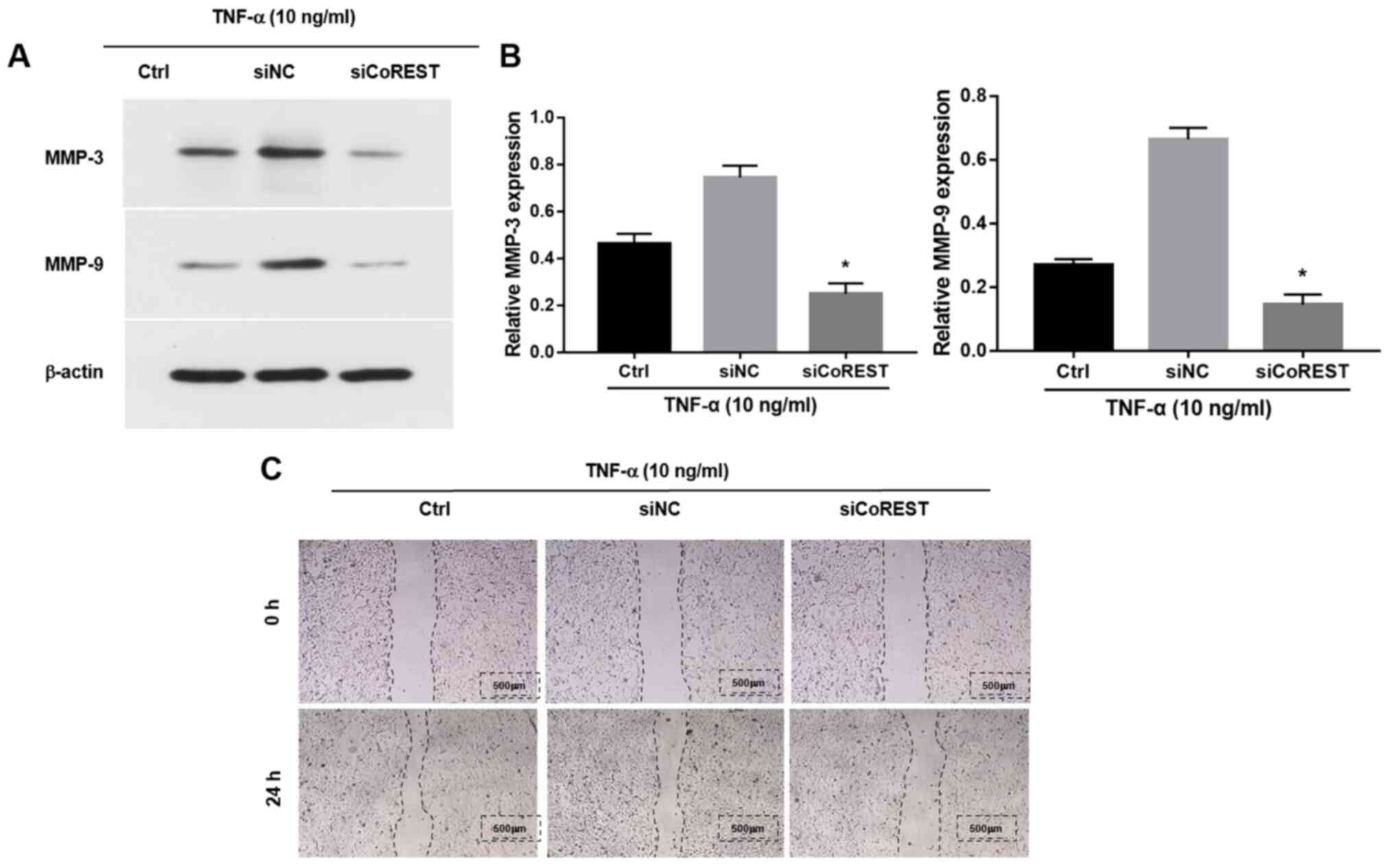
MMPs are a group of zinc-dependent homologous
proteases (33). A distinct and
typical clinical indicator observed in patients with RA is elevated
levels of MMPs including MMP-9 and MMP-3, which are associated with
the increased migration and invasion of FLSs (14). Previous studies have shown that
stimulation with TNF-α elevated the expression levels of MMP-9 and
MMP-3(34). However, following
transfection with siCoREST, the TNF-α-induced upregulation of MMPs
was significantly inhibited compared with the control group
(Fig. 3A and B). The effects of CoREST on the migration
of FLSs were further explored by performing a wound healing assay.
Transfection with siCoREST notably inhibited the migratory ability
of FLSs compared with the control group (Fig. 3C).
Knockdown of CoREST downregulates the
expression of LSD1
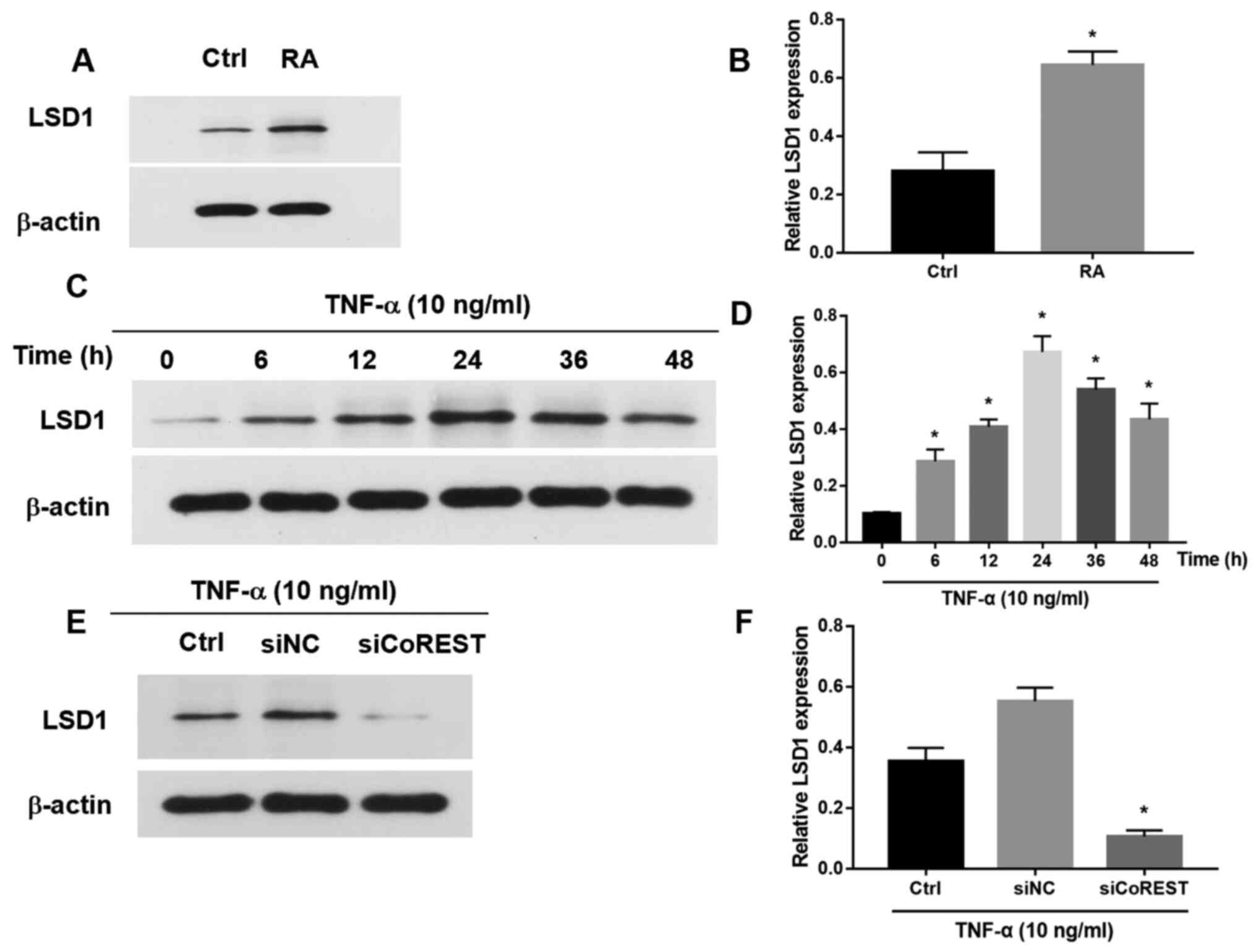
The expression of LSD1 was upregulated in patients
with RA (Fig. 4A and B). The effect of CoREST on the expression
of LSD1 was further explored. Following treatment with TNF-α, the
expression of LSD1 in FLSs was significantly increased at 12, 24
and 36 h, and then began to decrease at 48 h (Fig. 4C and D). Following transfection with siCoREST,
the expression of LSD1 was significantly downregulated compared
with the control group (Fig. 4E and
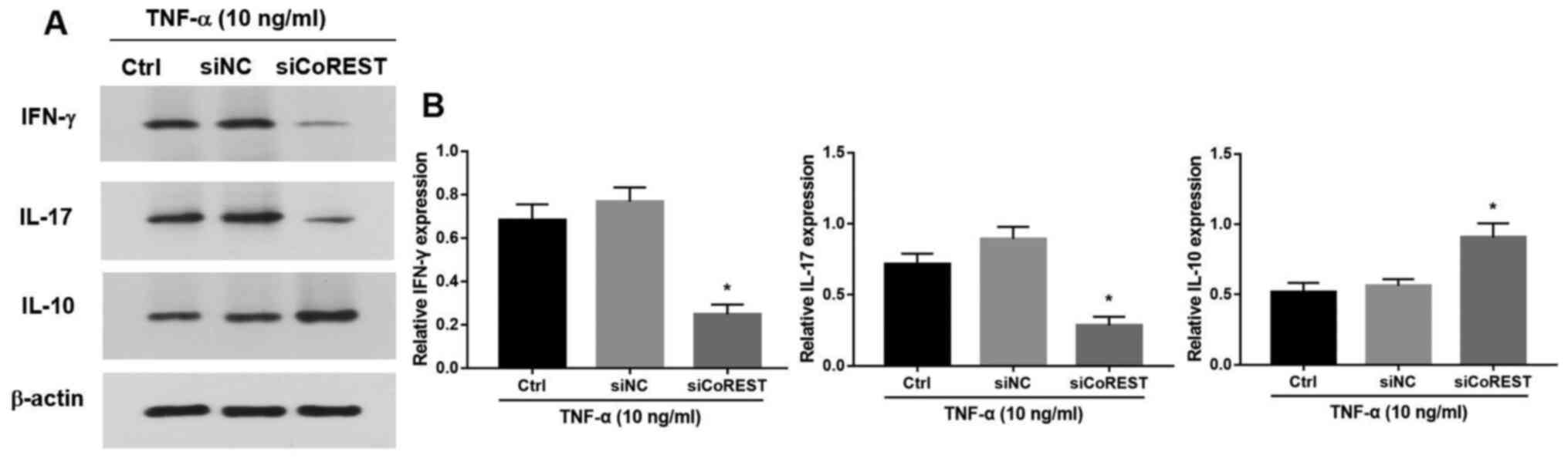
F). Furthermore, silencing of
CoREST significantly suppressed the production of IFN-γ and IL-17
and promoted the expression of IL-10 (P<0.05; Fig. 5A and B).
Discussion
RA is a complicated systemic disease (1), and a considerable number of studies
have demonstrated that increased proliferation, migration and
invasion of FLSs leads to the damage of arthrodial cartilage that
advances the pathogenesis of RA (12,35,36).
The present study revealed that CoREST and its corepressor protein
LSD1 were highly expressed in RA synovial tissues. A previous in
vivo study indicated that LSD1 knockdown significantly
alleviated disease severity (20).
The results of the current study gave support for the hypothesis
that there is a molecular relationship between the LSD1/CoRST
complex in RA-FLSs. However, to the best of our knowledge, there
have been few functional investigations into the LSD1/CoREST
complex outside of the neural system.
LSD1 is a well-characterized histone demethylase
that regulates gene transcription and chromatin configuration
through epigenetic modifications (37). The tower motif, which is crucial for
the catalytic activity of LSD1, acts as an adaptor to recruit other
proteins, such as CoREST (37).
LSD1 demethylates H3K4me1/me2 in a CoREST complex-dependent manner
and functions as a transcription repressor (38). In the present study, the elevated
expression of CoREST was observed in RA-FLSs, which confirmed that
the LSD1/CoREST complex may exert effects on RA-FLSs.
FLSs are transferred to an activated state by
stimulation with proinflammatory cytokines, such as TNF-α, which
induces increased proliferative, migratory and invasive abilities
(39). The aforementioned effects
resemble the features of active RA progression. The current study
demonstrated that proliferation, migration of FLSs treated with
TNF-α could be reduced by the inhibition of CoREST. With TNF-α
stimulation, the expression of CoREST in FLSs was increased as
compared with that in the controls. Meanwhile, the expression of
LSD1 was also upregulated.
In the TNF-α-stimulated RA-FLSs, CoREST expression
was upregulated in a time-dependent manner. LDS1 expression was
mildly elevated within the first 12 h, reaching a peak at 24 h and
gradually deceasing at 36 h. This variation may be the result of
effects in the early phase, in which TNF-α may function in the
activation process and also trigger the mechanisms responsible for
self-protection within FLSs. The expression of CoREST was
relatively low, with limited effects, thus, the expression of LSD1
was only slightly elevated. After 36 h, with the accumulation of
CoREST, the function and expression of LSD1 was mostly eliminated.
This hypothesis was further confirmed by the knockdown of CoREST
with siRNA, which caused the expression of LSD1 to decrease, thus
suggesting that CoREST had a positive regulatory effect on LSD1 in
RA-FLSs. These results indicated that the LSD1/CoREST complex may
worked together towards the TNF-α-stimulated RA-FLSs. The molecular
mechanism of the role of the LSD1/CoREST complex in RA is still
unclear. The Notch signaling pathway, a crucial pathway in RA
pathogenesis, has been demonstrated to be upregulated in FLSs after
stimulation with proinflammatory cytokines, such as TNF-α and IL-1β
(40). LSD1 has been reported to
act as a negative regulator of the Notch signaling pathway through
its interaction with the deacetylase SIRT1 in cell cultures
(24). The knockdown of LSD1
expression in CD4+T cells obtained from active RA
synovial fluids has been demonstrated to inhibit cell proliferation
and proinflammatory cytokine secretion (20). Consistent with the results in a
previous study, the expression of LSD1 was downregulated by the
knockdown of CoREST in the present study, and then the
proliferation of FLSs was inhibited, in which the Notch signaling
pathway may play a regulatory role in RA-FLSs. However, further
study is necessary to elucidate the detailed molecular mechanisms
through which CoREST participates in RA progression.
The present study had a number of limitations.
Primary cells derived from synovial tissues of knee joints were
used, and in order to prevent phenotypic drift, cells at a low
passage were cultured in medium without growth factors. However,
the gene expression profile of synovial cells may have been altered
due to adaptation of the cells in the culture conditions.
Furthermore, this study was performed by using RA synovial tissues
and a TNF-α-induced cell model, and an experimental design that
more accurately mimics RA-FLFs should be considered in future
studies.
The present study revealed that CoREST expression
was upregulated in RA-FLSs. TNF-α stimulation increased CoREST
expression, which could also increase the proliferation, migration
of FLSs through cooperating with LSD1. However, in vivo
experiments are required to further verify the function of CoREST
in RA and explore its potential applications in clinical
practice.
Supplementary Material
Inhibition of CoREST expression
following transfection with siCoREST. Western blotting was
performed to determine that CoREST expression was knocked down in
the siCoREST group compared with the siNC group.
**P<0.01 vs. siNC. CoREST, corepressor element-1
silencing transcription factor; si, small interfering RNA; NC,
negative control.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
Jiangsu Six-One Project (grant no. LGY2020047), the Science &
Technology Bureau of Nantong (grant no. MA2019005), the Health
Commission of Nantong (grant no. JC2019075) and the Health
Commission of Nantong (grant no. MB2020005).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WL, ZY and FC contributed to the design of the work.
ZY, FC, HL, JF, XD, XZhu, SC, HY, XZhou and YH carried out the
specific experiments. ZY, FC, HL and JF contributed to the
acquisition and analysis of data, and WL gave the final approval of
the version to be published. ZY and FC confirm the authenticity of
all the raw data. All authors have read and approved the final
manuscript, and guaranteed the integrity of the work.
Ethics approval and consent to
participate
The procedures and processes of the present study
were reviewed and approved by the institutional medical ethics
committee of The Second Affiliated Hospital of Nantong University
(Nantong, China). Patients provided written informed consent to
participate before they received any treatment.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McInnes IB and Schett G: Pathogenetic
insights from the treatment of rheumatoid arthritis. Lancet.
389:2328–2337. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Rossini M, Caimmi C, Bernardi D, Rossi E,
Viapiana O, Rosa MD and Adami S: Epidemiology and hospitalization
rate of rheumatoid arthritis patients in real world setting in
Italy. Ann Rheumatic Diseases. 72 (Suppl 3):A409. 2013.
|
|
3
|
Rossini M, Rossi E, Bernardi D, Viapiana
O, Gatti D, Idolazzi L, Caimmi C, Derosa M and Adami S: Prevalence
and incidence of rheumatoid arthritis in Italy. Rheumatol Int.
34:659–664. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sung YK, Cho SK, Choi CB and Bae SC:
Prevalence and incidence of rheumatoid arthritis in South Korea.
Rheumatol Int. 33:1525–1532. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Klareskog L, Gregersen PK and Huizinga TW:
Prevention of autoimmune rheumatic disease: State of the art and
future perspectives. Ann Rheum Dis. 69:2062–2066. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Essouma M and Noubiap JJ: Is air pollution
a risk factor for rheumatoid arthritis? J Inflamm (Lond).
12(48)2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hu Y, Costenbader KH, Gao X, Al-Daabil M,
Sparks JA, Solomon DH, Hu FB, Karlson EW and Lu B: Sugar-sweetened
soda consumption and risk of developing rheumatoid arthritis in
women. Am J Clin Nutr. 100:959–967. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
van der Linden MP, le Cessie S, Raza K,
van der Woude D, Knevel R, Huizinga TW and van der Helm-van Mil AH:
Long-term impact of delay in assessment of patients with early
arthritis. Arthritis Rheum. 62:3537–3546. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bartok B and Firestein GS: Fibroblast-like
synoviocytes: Key effector cells in rheumatoid arthritis. Immunol
Rev. 233:233–255. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pap T, Meinecke I, Müller-Ladner U and Gay
S: Are fibroblasts involved in joint destruction? Ann Rheum Dis. 64
(Suppl 4):iv52–iv54. 2005.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Müller-Ladner U, Pap T, Gay RE, Neidhart M
and Gay S: Mechanisms of disease: The molecular and cellular basis
of joint destruction in rheumatoid arthritis. Nat Clin Pract
Rheumatol. 1:102–110. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Huber LC, Distler O, Tarner I, Gay RE, Gay
S and Pap T: Synovial fibroblasts: Key players in rheumatoid
arthritis. Rheumatology (Oxford). 45:669–675. 2006.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Guo X, Zhang D, Zhang X, Jiang J, Xue P,
Wu C, Zhang J, Jin G, Huang Z, Yang J, et al: Dyrk1A promotes the
proliferation, migration and invasion of fibroblast-like
synoviocytes in rheumatoid arthritis via down-regulating Spry2 and
activating the ERK MAPK pathway. Tissue Cell. 55:63–70.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Malemud CJ: Matrix metalloproteinases
(MMPs) in health and disease: An overview. Front Biosci.
11:1696–1701. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Mor A, Abramson SB and Pillinger MH: The
fibroblast-like synovial cell in rheumatoid arthritis: A key player
in inflammation and joint destruction. Clin Immunol. 115:118–128.
2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu J, Fei D, Xing J and Du J:
MicroRNA-29a inhibits proliferation and induces apoptosis in
rheumatoid arthritis fibroblast-like synoviocytes by repressing
STAT3. Biomed Pharmacother. 96:173–181. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Coulson JM: Transcriptional regulation:
Cancer, neurons and the REST. Curr Biol. 15:R665–R668.
2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Majumder S: REST in good times and bad:
Roles in tumor suppressor and oncogenic activities. Cell Cycle.
5:1929–1935. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Andrés ME, Burger C, Peral-Rubio MJ,
Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N and
Mandel G: CoREST: A functional corepressor required for regulation
of neural-specific gene expression. Proc Natl Acad Sci USA.
96:9873–9878. 1999.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Liu W, Fan JB, Xu DW, Zhu XH, Yi H, Cui
SY, Zhang J and Cui ZM: Knockdown of LSD1 ameliorates the severity
of rheumatoid arthritis and decreases the function of CD4 T cells
in mouse models. Int J Clin Exp Pathol. 11:333–341. 2018.PubMed/NCBI
|
|
21
|
Ceballos-Chávez M, Rivero S,
García-Gutiérrez P, Rodríguez-Paredes M, García-Domínguez M,
Bhattacharya S and Reyes JC: Control of neuronal differentiation by
sumoylation of BRAF35, a subunit of the LSD1-CoREST histone
demethylase complex. Proc Natl Acad Sci USA. 109:8085–8090.
2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Maiques-Diaz A and Somervaille TC: LSD1:
Biologic roles and therapeutic targeting. Epigenomics. 8:1103–1116.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Shi YJ, Matson C, Lan F, Iwase S, Baba T
and Shi Y: Regulation of LSD1 histone demethylase activity by its
associated factors. Mol Cell. 19:857–864. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mulligan P, Yang F, Di Stefano L, Ji JY,
Ouyang J, Nishikawa JL, Toiber D, Kulkarni M, Wang Q,
Najafi-Shoushtari SH, et al: A SIRT1-LSD1 corepressor complex
regulates notch target gene expression and development. Mol Cell.
42:689–699. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang SC: PCNA: A silent housekeeper or a
potential therapeutic target? Trends Pharmacol Sci. 35:178–186.
2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Aaltomaa S, Lipponen P and Syrjänen K:
Proliferating cell nuclear antigen (PCNA) immunolabeling as a
prognostic factor in axillary lymph node negative breast cancer.
Anticancer Res. 13:533–538. 1993.PubMed/NCBI
|
|
27
|
Aletaha D, Neogi T, Silman AJ, Funovits J,
Felson DT, Bingham CO III, Birnbaum NS, Burmester GR, Bykerk VP,
Cohen MD, et al: 2010 rheumatoid arthritis classification criteria:
An American college of rheumatology/European league against
rheumatism collaborative initiative. Arthritis Rheum. 62:2569–2581.
2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Alsaleh G, François A, Knapp AM, Schickel
JN, Sibilia J, Pasquali JL, Gottenberg JE, Wachsmann D and
Soulas-Sprauel P: Synovial fibroblasts promote immunoglobulin class
switching by a mechanism involving BAFF. Eur J Immunol.
41:2113–2122. 2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chen Z, Lin CX, Song B, Li CC, Qiu JX, Li
SX, Lin SP, Luo WQ, Fu Y, Fang GB, et al: Spermidine activates RIP1
deubiquitination to inhibit TNF-α-induced NF-κB/p65 signaling
pathway in osteoarthritis. Cell Death Dis. 11(503)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Peng M, Qiang L, Xu Y, Li C, Li T and Wang
J: IL-35 ameliorates collagen-induced arthritis by promoting
TNF-α-induced apoptosis of synovial fibroblasts and stimulating M2
macrophages polarization. FEBS J. 286:1972–1985. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhang X, Zhang D, Wang Q, Guo X, Chen J,
Jiang J, Li M, Liu W, Gao Y, Zhang Q, et al: Sprouty2 inhibits
migration and invasion of fibroblast-like synoviocytes in
rheumatoid arthritis by down-regulating ATF2 expression and
phosphorylation. Inflammation. 44:91–103. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Rooprai HK and McCormick D: Proteases and
their inhibitors in human brain tumours: A review. Anticancer Res.
17:4151–4162. 1997.PubMed/NCBI
|
|
34
|
Zhang G, Liao Y, Yang H, Tao J, Ma L and
Zuo X: Irigenin reduces the expression of caspase-3 and matrix
metalloproteinases, thus suppressing apoptosis and extracellular
matrix degradation in TNF-α-stimulated nucleus pulposus cells. Chem
Biol Interact. 349(109681)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Taghadosi M, Adib M, Jamshidi A, Mahmoudi
M and Farhadi E: The p53 status in rheumatoid arthritis with focus
on fibroblast-like synoviocytes. Immunol Res. 69:225–238.
2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Mousavi MJ, Karami J, Aslani S, Tahmasebi
MN, Vaziri AS, Jamshidi A, Farhadi E and Mahmoud M: Transformation
of fibroblast-like synoviocytes in rheumatoid arthritis; from a
friend to foe. Auto Immun Highlights. 12(3)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhang Y, Wu T, Wang Y, Zhao X, Zhao B,
Zhao X, Zhang Q, Jin Y, Li Z and Hu X: The R251Q mutation of LSD1
promotes invasion and migration of luminal breast cancer cells. Int
J Biol Macromol. 164:4000–4009. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lan F, Nottke AC and Shi Y: Mechanisms
involved in the regulation of histone lysine demethylases. Curr
Opin Cell Biol. 20:316–325. 2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Brennan FM, Maini RN and Feldmann M:
TNFα-A pivotal role in rheumatoid arthritis? Rheumatology.
31:293–298. 1992.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Muller-Ladner U, Gay RE and Gay S:
Activation of synoviocytes. Curr Opin Rheumatol. 12:186–194.
2000.PubMed/NCBI View Article : Google Scholar
|