Introduction
Post-traumatic epilepsy (PTE) is characterized by
the development of delayed unprovoked seizures after traumatic
brain injury (TBI) (1). It has been
estimated that approximately 20% of symptomatic epilepsies are
caused as a result of TBI, indicating that PTE is one of the most
common focal epilepsy types (2).
The incidence rate of PTE was found to range from 4.4-53%
worldwide, which accounts for 10-20% of symptomatic epilepsy cases
and for 5% of all epilepsy cases (3-5).
Despite significant efforts made to understand the pathogenesis and
improve the efficacy of PTE treatment, no effective treatment
option for PTE has been introduced (6). Thus, it is a crucial medical need to
further explore the pathogenesis of PTE to provide effective
therapeutic options.
Neuronal apoptosis has been previously reported to
occur during seizures (7,8). Moreover, repeated PTE seizures damage
neurons and cause neuronal death and secondary neuron loss
(9). Evidence has revealed the
typical apoptotic features in damaged regions and elevated
expression levels of caspase 3 and Bax after PTE (10). A recent study observed that cleaved
caspase-3 exerted an important role in chronic TBI pathologies and
neurodegenerative diseases (11).
All these findings suggest that neuronal apoptosis is involved in
PTE pathogenesis.
Apoptosis-antagonizing transcription factor (AATF)
is an RNA polymerase II-binding protein that was identified as an
interaction partner of the death-associated protein-like kinase
(12). Previous studies observed
that AATF inactivation or inhibition results in DNA damage
accumulation (13,14) and induces apoptosis (12,15-17).
Furthermore, AATF protects neurons against amyloid
β-peptide-induced oxidative damage (18), indicating that AATF may serve as a
novel cytoprotective factor against apoptotic insult in
neurological diseases. However, the role of AATF in PTE is still
poorly understood. Therefore, the present study aimed to explore
the role of AATF in the pathogenesis of PTE.
Materials and methods
Animals
A total of 54 male Sprague-Dawley rats (220-275 g,
6-8 weeks), obtained from the Experimental Animal Center of Nantong
University (Jiangsu, China), were maintained under a 12/12-h
light/dark cycle at 23±1˚C and received food and water ad
libitum. All animal care and surgical treatments and procedures
were carried out in accordance with the National Institutes of
Health (NIH) Guidelines for the Care and Use of Laboratory Animals
and the study protocol was approved by the Ethics Committee of
Nantong University. A PTE rat model was established as previously
described (19). In brief, after
anesthetization with 10% chloral hydrate (350 mg/kg,
intraperitoneal), the overlying bone of the rat was drilled with a
stereotaxic atlas (2.0 mm behind the left coronal suture and 2.0 mm
beside the sagittal suture). Then, using a microinjection pump, 5
µl of FeCl3 (0.1 mol/l) or sterile saline was injected
into the motor cortex via a trephine hole over a period of 5 min.
Afterwards, the trephine hole was covered by medical bone wax.
Suturing of the skin and the muscle were performed separately.
During the experimental procedure, no signs of peritonitis were
observed.
The behavioral changes of the mice were recorded 1 h
before and after modeling at 8-10 am every day, and assessed from
12 h to 28 days after the FeCl3 injection using the
Racine scale (20). The Racine
stages employed were as follows: Grade 0, no behaviors of epileptic
seizure; grade 1, face clonus, chewing, yawning; grade 2, nodding,
face clonus, neck muscle convulsion, in addition to the behaviors
of grade 1; grade 3, forelimb or hindlimb convulsion, in addition
to the behaviors of grade 2; grade 4, hindlimb standing, sudden
standing, in addition to the behaviors of grade 3; grade 5,
rhythmic convulsion of four limbs, hindlimb stiffness, and dorsal
flexure or convulsion, in addition to the behaviors of grade 4.
Rats at grade 4 or higher were selected as PTE models.
Rats injected with saline were classified as the
sham group. The rats in the experimental group (n=3/time point)
were sacrificed at 12 h or 1, 3, 5, 7, 14 and 28 days to extract
protein for western blot analysis. Rats in the control (n=3) and
sham groups (n=3) were sacrificed on day 5 according to the
preliminary western blot results (Fig.
1). Additional experimental rats were sacrificed at each time
point for further immunohistochemical analysis (n=3). The rats were
anesthetized by chloral hydrate (400 mg/kg), transcardially
perfused with 0.9% sterile saline, then the cortex tissues of the
injection area were collected and stored at -70˚C before
analysis.
Western blotting
Hypotonic lysis buffer (0.05% NP-40; 50 mmol/l
Tris-HCl; 5 mmol/l EDTA; 10 mM NaCl; pH 7.5) was used to separate
the nuclear and cytoplasmic fractions of the cortex tissues. The
protein concentration was detected by BCA protein assay (cat. no.
P0006; Beyotime Institute of Biotechnology). Next, a total of 20 µg
protein was separated by 10-12% SDS-Polyacrylamide gel
electrophoresis (PAGE) and transferred to PVDF membranes (EMD
Millipore). After blocking with 5% skim milk for 1 h at room
temperature, the membranes were incubated with the following
primary antibodies: Anti-AATF (1:2,000; cat. no. WH0026574M4;
Sigma-Aldrich; Merck KGaA), anti-cleaved caspase-3 (1:5,000; cat.
no. 9664; Santa Cruz Biotechnology, Inc.), anti-p53 (1:5,000; cat.
no. 2527; Santa Cruz Biotechnology, Inc.), anti-Bcl2 (1:5,000; cat.
no. 15071; Santa Cruz Biotechnology, Inc.), anti-Bax (1:1,000; cat.
no. 14-6999-37; Zymed; Thermo Fisher Scientific, Inc.) and
anti-GAPDH (1:5,000; cat. no. sc-365062; Santa Cruz Biotechnology,
Inc.), overnight at 4˚C. Then, the membranes were further incubated
with secondary antibodies (1:10,000; goat anti-mouse and goat
anti-rabbit; cat. nos. F0257 and F0382; Sigma-Aldrich; Merck KGaA).
The bands were detected using an enhanced chemiluminescence system
(Amersham; Cytiva). GAPDH was used as an internal control. For
quantification of the protein bands, ImageJ software (v1.8.0;
National Institutes of Health) was utilized for densitometric value
evaluation.
Immunofluorescence staining
The cortex was fixed overnight using 4%
paraformaldehyde at 4˚C and dehydrated in 30% sucrose at 4˚C for
2-3 days. After embedding in optimal cutting temperature compound,
7-µm coronal sections were cut within 1-3 mm posterior to the
bregma and placed on gelatin-coated microscope slides. All sections
were frozen at -20˚C before use.
For immunofluorescence analysis, all sections were
blocked using 10% serum-blocking buffer (0.1% Triton X-100, 3%
(w/v) bovine serum albumin (BSA) and 0.05% Tween-20) for 2 h at
room temperature. Then, the sections were incubated with primary
antibodies against AATF (1:100; cat. no. WH0026574M4;
Sigma-Aldrich; Merck KGaA), NeuN (1:100; cat. no. SAB4300883;
Sigma-Aldrich; Merck KGaA), glial fibrillary acidic protein (GFAP;
1:100; cat. no. sc-33673; Santa Cruz Biotechnology, Inc.), ionized
Ca2+-binding adaptor molecule 1 (Iba-1; 1:100; cat. no.
sc-32725; Santa Cruz Biotechnology, Inc.) and cleaved-caspase-3
(1:100; cat. no. sc-3073; Santa Cruz Biotechnology, Inc.) overnight
at 4˚C. The sections were next incubated with FITC- and
Tetramethylrhodamine-conjugated secondary antibodies (1:1,000; cat.
no. F0257; Sigma-Aldrich; Merck KGaA) for 2 h at room temperature.
The sections were dehydrated and covered with coverslips, and the
slides were photographed under a 20x objective using a Leica
DM4000B fluorescence microscope (Leica Microsystems GmbH).
Terminal deoxynucleotidyl
transferase-mediated biotinylated-dUTP nick-end labeling
(TUNEL)
The animals were sacrificed by perfusion-fixation.
Dissected rat brains were postfixed in 4% paraformaldehyde solution
for another 24 h at 4˚C and dehydrated for 2 days at 4˚C. The
brains were then were immersed in 4% paraformaldehyde for 48 h,
embedded in optimal cutting temperature compound (OCT) for 10 min
in a freezing microtome (Leica Model CM 1900; Leica Microsystem
GmbH) at -25˚C. The OCT-embedded brain tissues were then sliced
into 30 µm sections. The sections were rinsed with PBS and then
were blocked with 3% goat serum (cat. no. 005-000-001; Jackson
ImmunoResearch Laboratories, Inc.) for 60 min at room temperature
and incubated with rabbit anti-NeuN antibody (1:100; cat. no.
SAB4300883; Sigma-Aldrich; Merck KGaA) overnight at 4˚C. The slides
were then probed using the in-situ Cell Death Detection kit
(cat. no. 11684795910; Roche Diagnostics) according to the
manufacturer's instructions. In brief, frozen tissue sections were
rinsed with PBS and then treated with 1% Triton X-100 for 2 min on
ice. The slides were next washed in PBS and incubated with 50 µl of
TUNEL reaction mixture for 60 min at 4˚C. Subsequently, the
sections were counterstained with DAPI (cat. no. C1006; Beyotime
Institute of Biotechnology) for 10 min at room temperature. Images
were acquired under a 20x objective using a Leica DM4000B
fluorescence microscope (Leica Microsystems GmbH). The negative
control sections were incubated for 60 min with a labeling
solution.
Lentiviral transfection of shRNA
targeting AATF
After FeCl3 induction, polybrene (5
µg/ml) was used to dilute the lentivirus (Invitrogen; Thermo Fisher
Scientific, Inc.) to 3x106 TU/µl. Rat brain cortex was
microinjected with lentivirus by the brain stereotaxic method, as
previously described (21). The
complementary oligonucleotides targeting AATF were obtained from
Shanghai GeneChem Co., Ltd. The targeted sequence for AATF was
5'-GGAGGAGATCTCAGAAGAT-3' and 5'-ATCTTCTGAGATCTCCACC-3'. The
sequence for negative control was 5'-TTCTCCGAACGTGTCACGT-3' and
5'-ACGTGACACGTTCGGAGAA-3'. These complementary oligonucleotides was
annealed and ligated to GV118 vector (Clontech Laboratories, Inc.).
The shRNA-carrying lentiviruses were produced in 293T cells and
subjected for microinjection, as were the negative controls. Five
days after microinjection, the rats in both the negative control
and AATF groups were sacrificed, then brain cortex was collected
and subjected for further analysis.
Statistical analysis
Data were expressed as mean ± standard error of the
mean (SEM). The statistical significance between groups was
determined by one-way ANOVA, followed by Tukey's post hoc multiple
comparison tests. All data were analyzed using SPSS 17.0 (SPSS,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
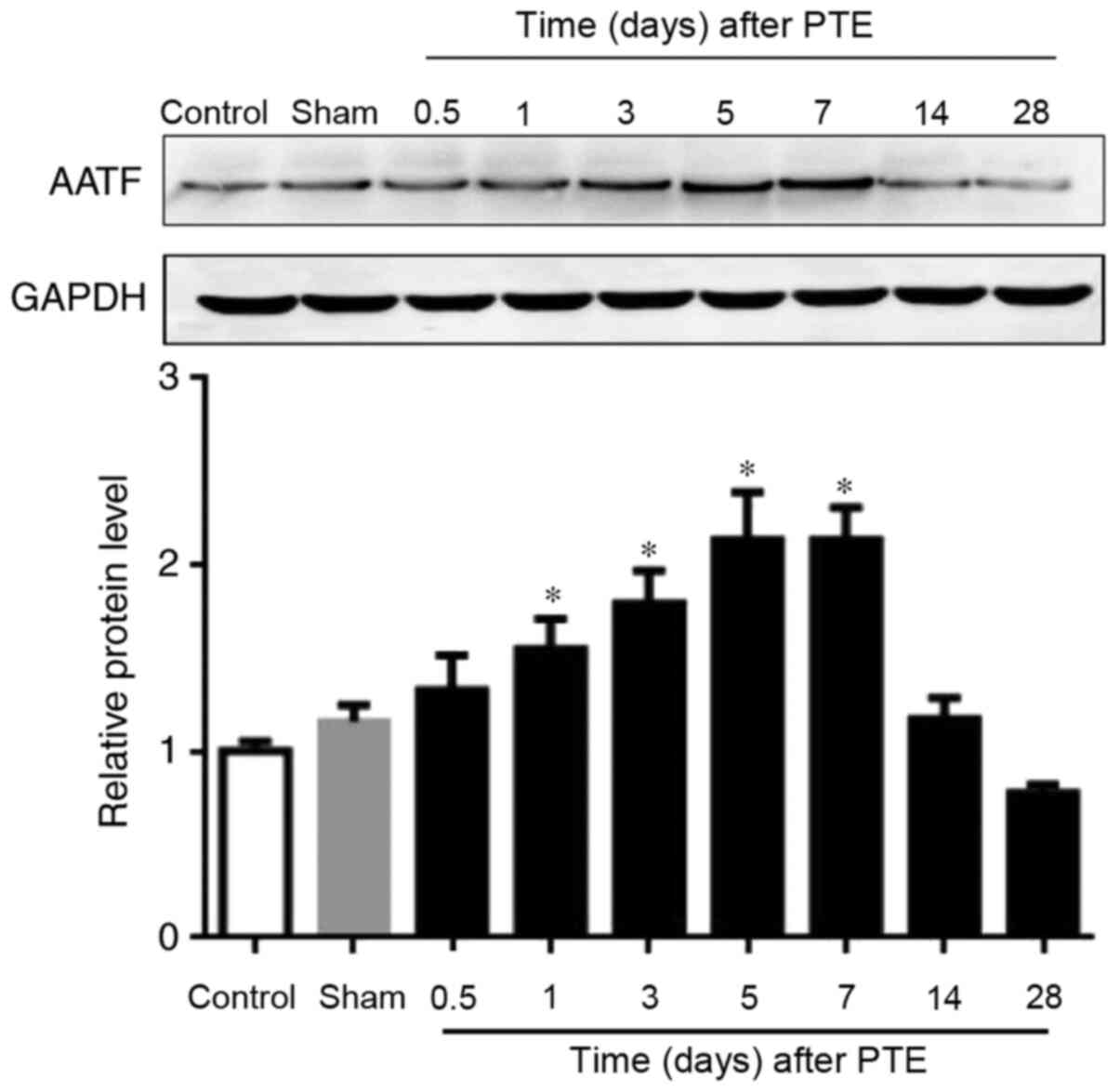
Expression of AATF is enhanced at the
early stage of PTE
Western blotting was employed to investigate the
expression pattern of AATF following PTE. The results demonstrated
that the expression levels of AATF in the sham and control groups
were low, but gradually elevated and peaked at day 5, and declined
thereafter (Fig. 1). These results
indicated that the expression levels of AATF was increased at the
early stage of PTE.
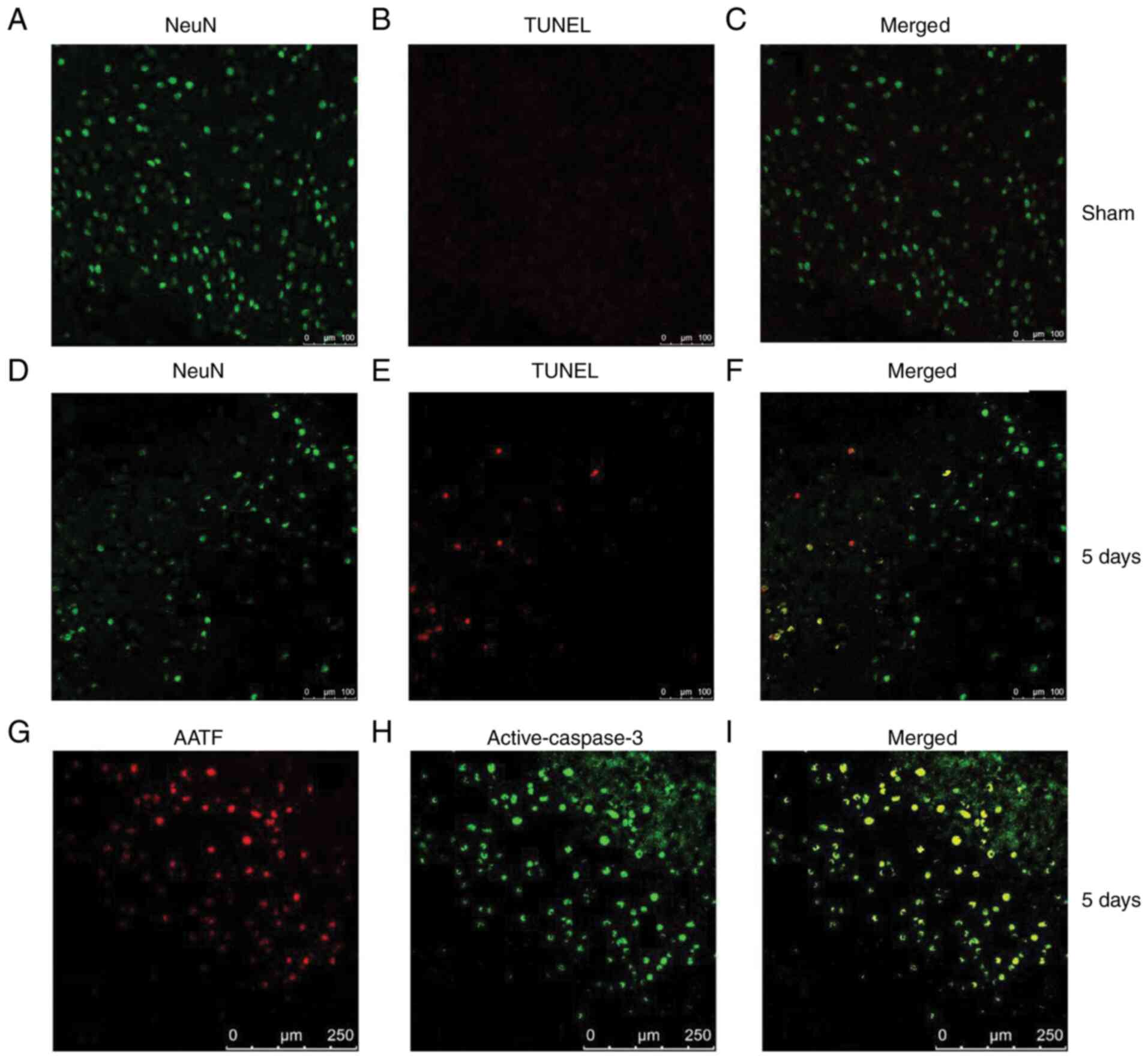
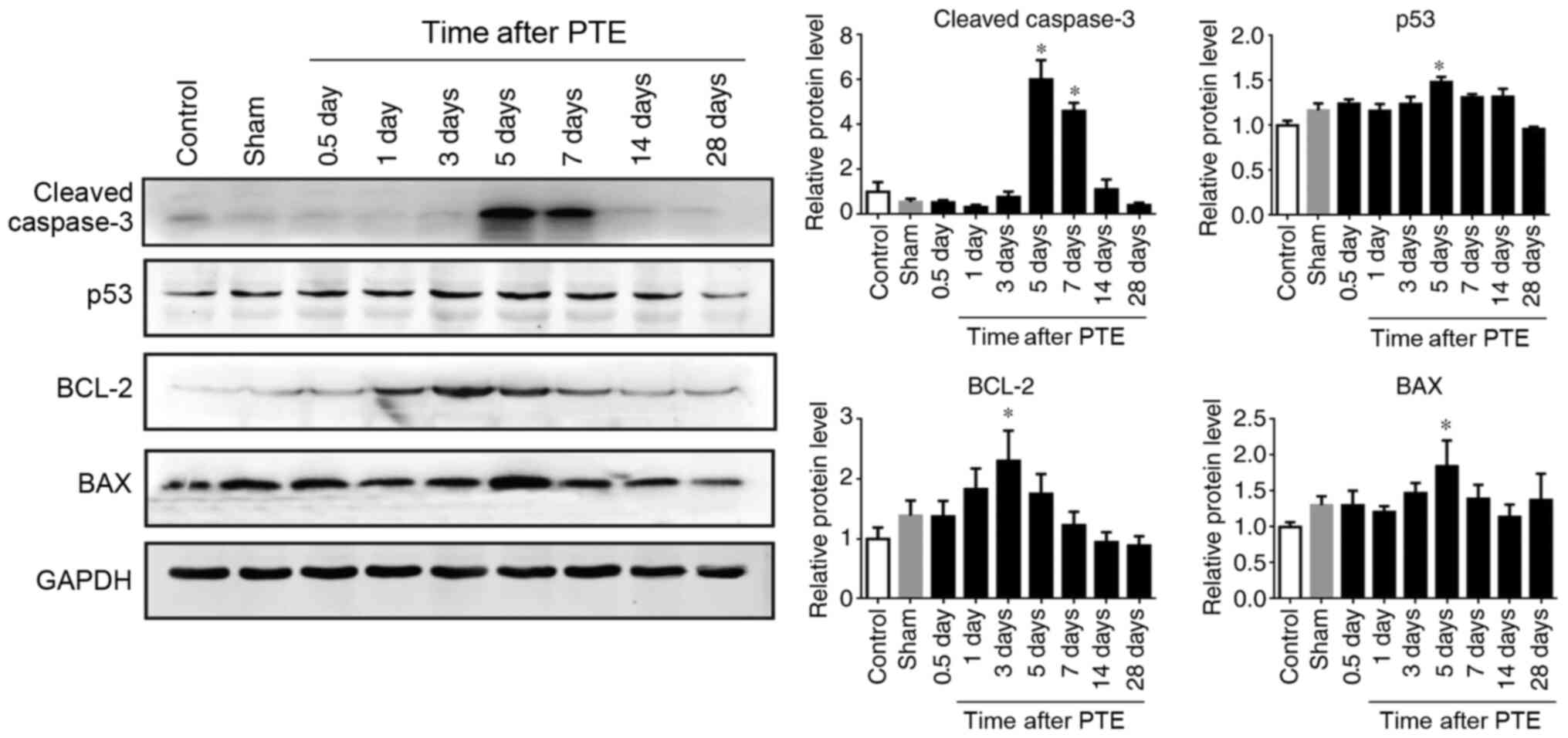
Apoptosis-related proteins have an
expression pattern similar to that of AATF after PTE
Since the expression levels of AATF peaked on day 5,
neural apoptosis 5 days post PTE was detected. TUNEL staining
demonstrated that the apoptosis of the neurons was increased 5 days
post PTE (Fig. 2A-F). Then, the
expression levels of apoptosis-related proteins were examined. The
expression levels of cleaved caspase-3 significantly increased
after PTE and peaked at day 5 (P<0.05; Fig. 3), which is consistent with the
expression profile of AATF (Fig.
1). Moreover, immunofluorescence staining demonstrated that
AATF and cleaved caspase-3 were co-localized (Fig. 2G-I). Additionally, the expression
levels of apoptosis-related proteins, such as p53 and Bax and
Bcl-2, increased gradually in the PTE group, with p53 and Bax
reaching their peak 5 days after PTE and Bcl-2 reaching its peak 3
days after PTE (P<0.05, Fig. 3).
These results suggested that AATF is associated with neuronal
apoptosis after PTE.
AATF is expressed primarily in the
neurons and astrocytes
To determine whether AATF was specifically expressed
in the neurons after PTE, double-immunofluorescence labeling was
employed to detect the co-localization of AATF (red) and NeuN
(Fig. 4A-C), GFAP (Fig. 4D-F) and Iba-1 (Fig. 4G-I). The results demonstrated that
AATF was expressed mainly in the neurons and astrocytes (Fig. 4), indicating that neural apoptosis
after PTE may be associated with AATF. However, no colocalization
between AATF and IBA-1 was detected (Fig. 4G-I).
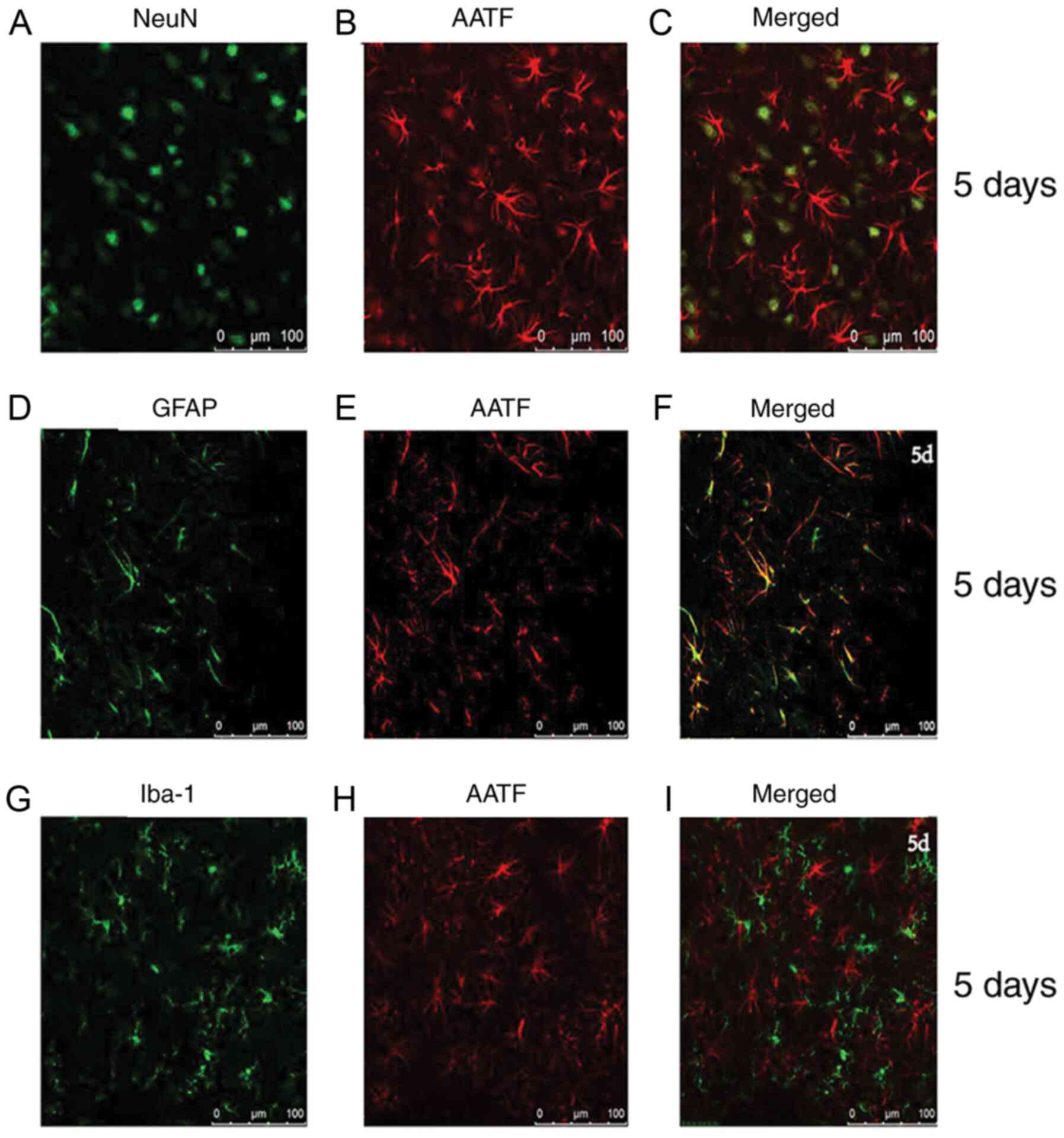 | Figure 4Localization of AATF in brain cortex.
The expression of AATF in different cell types in the adult rat
brain cortex within 5 mm of the lesion site on the 5th day after
PTE. (A) NeuN, green staining and (B) AAFT, red staining, as well
as (C) co-localization of NeuN and AATF. (D) GFAP green staining,
and (E) AAFT red staining, as well as (F) co-localization of GFAP
and AATF. (G) Iba-1 green staining and (H) AAFT red staining, as
well as (I) co-localization of Iba-1 and AATF. AATF, apoptosis
antagonizing transcription factor; GFAP, glial fibrillary acidic
protein; Iba-1, ionized Ca2+-binding adaptor molecule;
PTE, post-traumatic epilepsy. |
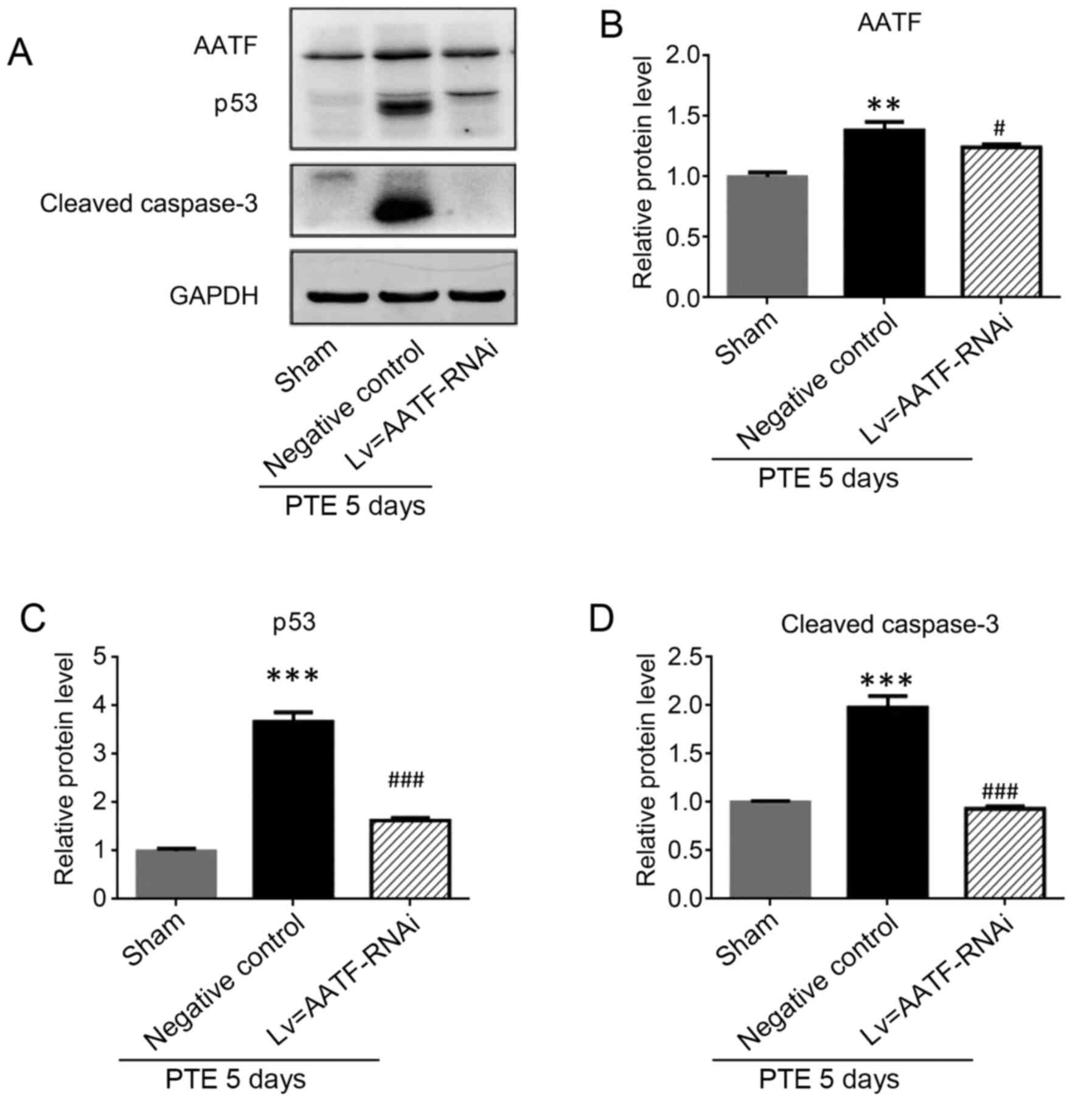
AATF inhibition attenuates the
expression of apoptosis-related proteins in the cortical
neurons
To elucidate the role of AATF in PTE, the expression
levels of AATF was significantly decreased following knockdown
using shRNA targeting AATF (Fig. 5A
and B). The results demonstrated
that the expression levels of p53 and cleaved caspase-3 were
significantly decreased in the AATF-knockdown group compared with
those of the sham group (P<0.05; Fig. 5A, C
and D), which supported the notion
that the intervention of AATF may have influenced neuronal
apoptosis after PTE.
Discussion
The results of the present study demonstrated that
the expression levels of AATF was gradually elevated and peaked at
the early phase of PTE, which was similar to the expression pattern
of apoptosis-related proteins, such as cleaved caspase-3, p53,
Bcl-2 and Bax. In addition, AATF was expressed primarily in the
neurons and was co-localized with cleaved caspase-3. These findings
suggested that AATF is involved in neural apoptosis after PTE and
may be a therapeutic target for PTE treatment.
Neuronal death following a seizure may be triggered
by apoptosis (22), and neuronal
apoptosis may also cause repeated antiepileptic events (23). Thus, a comprehensive understanding
of the underlying mechanism of neuronal apoptosis after epileptic
seizures is needed to improve the efficacy of epilepsy treatment.
Neuronal apoptosis is reportedly accompanied by a change in the
expression pattern of apoptosis-elated proteins, such as p53, Bcl-2
and Bax (24,25). Morrison et al (26) knocked out p53 in the PTE rat model
and reported decreased apoptosis in the hippocampus of epileptic
rats compared with the control group. Bruno et al (27) observed a significant increase in
apoptosis-related protein expression levels in patients with
refractory temporal lobe epilepsy compared with control patients.
The results of the present study demonstrated significantly
increased expression levels of apoptosis-related proteins, which is
consistent with the aforementioned earlier findings, suggesting
that altering the neuronal apoptosis pattern may be a potential
therapeutic strategy for PTE treatment.
AATF is associated with gene transcription
regulation, cell proliferation, DNA damage response and apoptosis
(27). However, the association
between AATF and central nervous system function following epilepsy
is still not clearly understood. The results of the present study
demonstrated that the expression levels of AATF were increased 5
days after PTE, and the apoptosis of the neurons was also
significantly increased. Moreover, AATF and cleaved caspase-3 were
co-localized in the cortex. Additionally, the expression levels of
p53, cleaved caspase-3 and Bax exhibited a trend similar to that of
AATF after traumatic brain injury. In addition, immunofluorescence
staining evidenced that AATF was expressed predominantly in the
neurons and astrocytes. Notably, this study demonstrated that AATF
was highly expressed in the astrocytes after PTE and that
astrocytes also served an important role in PTE. These results
strongly suggested the involvement of AATF in the neuronal
apoptosis of PTE.
Recent studies have reported that certain molecules
or drugs can alleviate epilepsy by reducing neuronal apoptosis
(28-30).
For example, it has been found that ferulic acid ameliorates
pentylenetetrazol-induced seizures through reducing neuron cell
death (28). Furthermore, AATF was
previously demonstrated to be involved in Alzheimer's disease, and
could be considered a novel cytoprotective factor against apoptosis
(18,31). In the present study, AAT expression
was inhibited and the results demonstrated that AATF silencing
significantly reduced the expression levels of p53 and cleaved
caspase-3 compared with those of the control group. These findings
suggested that AATF may represent a potential target for
therapeutic application.
The present study is not without limitations. Though
cleaved caspase-3 could reflect the apoptosis state of cells, this
study failed to detect the expression levels of caspase-3, but
since the expression levels of p53 were also altered post AATF
silencing it could be inferred that the conclusion was robust.
In conclusion, neuronal apoptosis serves a crucial
role in epilepsy pathogenesis and may be involved in AATF
expression changes. The inhibition of AATF may represent a
potential therapeutic strategy for PTE treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Nantong
Science and Technology Planning Project (grant nos. MS32016020 and
YYZ17019).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WW conceived and coordinated the study, designed,
performed and analyzed the experiments and wrote the study. YMM,
ZLJ, ZWG and WGC performed the data collection and data analysis
and revised the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Nantong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lucke-Wold BP, Nguyen L, Turner RC,
Logsdon AF, Chen YW, Smith KE, Huber JD, Matsumoto R, Rosen CL,
Tucker ES and Richter E: Traumatic brain injury and epilepsy:
Underlying mechanisms leading to seizure. Seizure. 33:13–23.
2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Piccenna L, Shears G and O'Brien TJ:
Management of post-traumatic epilepsy: An evidence review over the
last 5 years and future directions. Epilepsia Open. 2:123–144.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Christensen J: The epidemiology of
posttraumatic epilepsy. Semin Neurol. 35:218–222. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ferguson PL, Smith GM, Wannamaker BB,
Thurman DJ, Pickelsimer EE and Selassie AW: A population-based
study of risk of epilepsy after hospitalization for traumatic brain
injury. Epilepsia. 51:891–898. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Elghazouani F, Aarab C, Faiz F, Midaoui A,
Barrimi M, Elrhazi K, Berraho A, Belahssen MF, Rammouz I and
Aalouane R: Psychiatric disorders and associated factors in
patients with epilepsy in Fez, Morocco. Encephale. 41:493–498.
2015.PubMed/NCBI View Article : Google Scholar : (In French).
|
|
6
|
Saletti PG, Ali I, Casillas-Espinosa PM,
Semple BD, Lisgaras CP, Moshé SL and Galanopoulou AS: In search of
antiepileptogenic treatments for post-traumatic epilepsy. Neurobiol
Dis. 123:86–99. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu JT, Wu SX, Zhang H and Kuang F:
Inhibition of MyD88 signaling skews microglia/macrophage
polarization and attenuates neuronal apoptosis in the hippocampus
after status Epilepticus in Mice. Neurotherapeutics. 15:1093–1111.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Teocchi MA and D'Souza-Li L: Apoptosis
through death receptors in temporal lobe epilepsy-associated
hippocampal sclerosis. Mediators Inflamm.
2016(8290562)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Webster KM, Sun M, Crack P, O'Brien TJ,
Shultz SR and Semple BD: Inflammation in epileptogenesis after
traumatic brain injury. J Neuroinflammation. 14(10)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ghadiri T, Vakilzadeh G, Hajali V and
Khodagholi F: Progesterone modulates post-traumatic epileptogenesis
through regulation of BDNF-TrkB signaling and cell survival-related
pathways in the rat hippocampus. Neurosci Lett.
709(134384)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Glushakova OY, Glushakov AO, Borlongan CV,
Valadka AB, Hayes RL and Glushakov AV: Role of caspase-3-mediated
apoptosis in chronic caspase-3-cleaved tau accumulation and
blood-brain barrier damage in the corpus callosum after traumatic
brain injury in rats. J Neurotrauma. 35:157–173. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hopker K, Hagmann H, Khurshid S, Chen S,
Hasskamp P, Seeger-Nukpezah T, Schilberg K, Heukamp L, Lamkemeyer
T, Sos ML, et al: AATF/Che-1 acts as a phosphorylation-dependent
molecular modulator to repress p53-driven apoptosis. EMBO J.
31:3961–3975. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Jain M, Kaiser RWJ, Bohl K, Hoehne M,
Göbel H, Bartram MP, Habbig S, Müller RU, Fogo AB, Benzing T, et
al: Inactivation of apoptosis antagonizing transcription factor in
tubular epithelial cells induces accumulation of DNA damage and
nephronophthisis. Kidney Int. 95:846–858. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ling XX, Liu JX, Yun L, DU YJ, Chen SQ,
Chen JL, Tang HW and Liu LH: Poly(ADP-ribosyl)ation of apoptosis
antagonizing transcription factor involved in hydroquinone-induced
DNA damage response. Biomed Environ Sci. 29:80–84. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Benakanakere MR, Zhao J, Finoti L,
Schattner R, Odabas-Yigit M and Kinane DF: MicroRNA-663 antagonizes
apoptosis antagonizing transcription factor to induce apoptosis in
epithelial cells. Apoptosis. 24:108–118. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sharma M: Apoptosis-antagonizing
transcription factor (AATF) gene silencing: Role in induction of
apoptosis and down-regulation of estrogen receptor in breast cancer
cells. Biotechnol Lett. 35:1561–1570. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ishigaki S, Fonseca SG, Oslowski CM,
Jurczyk A, Shearstone JR, Zhu LJ, Permutt MA, Greiner DL, Bortell R
and Urano F: AATF mediates an antiapoptotic effect of the unfolded
protein response through transcriptional regulation of AKT1. Cell
Death Differ. 17:774–786. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Xie J and Guo Q: AATF protects neural
cells against oxidative damage induced by amyloid beta-peptide.
Neurobiol Dis. 16:150–157. 2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li Q, Li QQ, Jia JN, Sun QY, Zhou HH, Jin
WL and Mao XY: Baicalein exerts neuroprotective effects in
FeCl3-induced posttraumatic epileptic seizures via
suppressing ferroptosis. Front Pharmacol. 10(638)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Phelan KD, Shwe UT, Williams DK,
Greenfield LJ and Zheng F: Pilocarpine-induced status epilepticus
in mice: A comparison of spectral analysis of electroencephalogram
and behavioral grading using the Racine scale. Epilepsy Res.
117:90–96. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gu J, Bao Y, Chen J, Huang C, Zhang X,
Jiang R, Liu Q, Liu Y, Xu X and Shi W: The expression of NP847 and
Sox2 after TBI and its influence on NSCs. Front Cell Neurosci.
10(282)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lee SH, Choi BY, Lee SH, Kho AR, Jeong JH,
Hong DK and Suh SW: Administration of protocatechuic acid reduces
traumatic brain injury-induced neuronal death. Int J Mol Sci.
18(2510)2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lee SH, Choi BY, Kho AR, Jeong JH, Hong
DK, Lee SH, Lee SY, Lee MW, Song HK, Choi HC and Suh SW: Protective
effects of protocatechuic acid on seizure-induced neuronal death.
Int J Mol Sci. 19(187)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tenorio JR, Santana T, Queiroz SI, de
Oliveira DH and Queiroz LM: Apoptosis and cell cycle aberrations in
epithelial odontogenic lesions: An evidence by the expression of
p53, Bcl-2 and Bax. Med Oral Patol Oral Cir Bucal. 23:e120–e125.
2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yang LY, Greig NH, Huang YN, Hsieh TH,
Tweedie D, Yu QS, Hoffer BJ, Luo Y, Kao YC and Wang JY:
Post-traumatic administration of the p53 inactivator
pifithrin-alpha oxygen analogue reduces hippocampal neuronal loss
and improves cognitive deficits after experimental traumatic brain
injury. Neurobiol Dis. 96:216–226. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Morrison RS, Wenzel HJ, Kinoshita Y,
Robbins CA, Donehower LA and Schwartzkroin PA: Loss of the p53
tumor suppressor gene protects neurons from kainate-induced cell
death. J Neurosci. 16:1337–1345. 1996.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bruno T, Iezzi S and Fanciulli M:
Che-1/AATF: A critical cofactor for both wild-type- and mutant-p53
proteins. Front Oncol. 6(34)2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang SH, Liu D, Hu Q, Zhu J, Wang S and
Zhou S: Ferulic acid ameliorates pentylenetetrazol-induced seizures
by reducing neuron cell death. Epilepsy Res.
156(106183)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li X, Giri V, Cui Y, Yin M, Xian Z and Li
J: LncRNA FTX inhibits hippocampal neuron apoptosis by regulating
miR-21-5p/SOX7 axis in a rat model of temporal lobe epilepsy.
Biochem Biophys Res Commun. 512:79–86. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yu X, Guan Q, Wang Y, Shen H, Zhai L, Lu X
and Jin Y: Anticonvulsant and anti-apoptosis effects of salvianolic
acid B on pentylenetetrazole-kindled rats via AKT/CREB/BDNF
signaling. Epilepsy Res. 154:90–96. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Raina A and Kaul D: LXR-alpha genomics
programmes neuronal death observed in Alzheimer's disease.
Apoptosis. 15:1461–1469. 2010.PubMed/NCBI View Article : Google Scholar
|