Introduction
Cranioectodermal dysplasia (CED), which is also
known as Sensenbrenner syndrome, is a rare autosomal recessive
disorder. To date, IFT122 (WDR10), WDR35
(IFT121), IFT43 (C14orf179), WDR19
(IFT144), IFT52 (C20orf9) and IFT140
variants have been linked to CED (1-6).
The products of these genes are members of the intraflagellar
transport (IFT) system, which serves an essential role in the
assembly, maintenance and functioning of primary cilia (7). Mutations in the ciliary protein genes
cause defects in the structure and function of cilia, which can
lead to ‘ciliopathies’ (8).
Ciliopathies are a collection of complex developmental disorders
involving multiple organ systems (7-10).
Clinically, CED is highly heterogeneous and is
characterized by craniofacial, skeletal and ectodermal
abnormalities (8). Facial
dysmorphic features with frontal bossing and low set ears, hepatic
fibrosis, stunted growth and retinal dysfunction have also been
noted to vary across patients (11-13).
While there is currently no clear association between molecular
differences and clinical phenotypes, >60 patients diagnosed with
CED have been reported (11,14-16).
Additional reports on these gene variants and their phenotypes will
therefore be critical to the understanding of this condition. The
current study reports the compound heterozygous IFT122
(NM_052985.3) variants, c.366_376delAGGCCAAGGTG(p.Gly123Glufs*3)
and c.3879A>G (p.Ter1293Trpext), identified in a male Chinese
child with CED and additionally describes the patient's associated
clinical profile.
Case report
Participants
From the CED family, the proband, his parents and
two healthy siblings were recruited at Guangxi Maternal and Child
Health Hospital (Guangxi, China) on November 22, 2018. Physical and
imaging examinations of the proband were performed by an
experienced orthopedic surgeon. Detailed written informed consent
was obtained from the parents, with all study participants agreeing
to the genetic analysis of collected samples. All procedures in the
present study were approved by the Guangxi Maternal and Child
Health Hospital Ethics Committee.
Genetic analysis Whole-exome and
Sanger sequencing
Genomic DNA was extracted from 2 ml peripheral blood
of the proband and his family members using a Lab-Aid DNA kit
(Xiamen Zeesan Biotech Co., Ltd.) by standard methods, according to
the manufacturer's protocol. Whole-exome sequencing using
proband-derived genomic DNA was performed using an Agilent
SureSelect Human All Exon V5 kit (Agilent Technologies, Inc.) on a
Hiseq2500 platform (Illumina, Inc.), according to the
manufacturers' protocol. FASTQ files were generated and reads were
compared with the reference human genome (hg19) using
Burrows-Wheeler Aligner software (version 0.7.17; http://bio-bwa.sourceforge.net/bwa.shtml). A custom
pipeline, built predominantly using the Genome Analysis Tool Kit
GATK(https://wiki.rc.usf.edu/index.php/Genome_Analysis_ToolKit_),
was used for sequence data analysis and annotation. Identification
of causal variants by annotating the selected single-nucleotide
variants (SNVs) and indels was performed using TGex software
(LifeMap Sciences; Biotime, Inc.). The variants were defined as a
'rare deleterious' mutation if: i) The GnomAD database showed that
it had an allele frequency ≤0.5%; and ii) a stop-gain, stop-loss,
non-synonymous, frameshift or splice-site mutation was present.
When a potential novel mutation was considered following alignment
of the patient's genome sequence against the ClinVar (www.ncbi.nlm.nih.gov), HGMD (www.hgmd.cf.ac.uk/ac/), HPSD (liweilab.genetics.ac.cn/HPSD/) and SNP (www.ncbi.nlm.nih.gov/SNP) databases, and control
databases (ExAC and gnomAD), bioinformatics softwares PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2/), SIFT
(http://sift.bii.a-star.edu.sg/www/SIFT_seq_submit2.html),
the Human Splicing Finder website (http://www.umd.be/HSF/) and MutationTaster (http://www.mutationtaster.org) were used to predict
whether the novel variant(s) had any deleterious effects on the
protein(s). All the variants were classified according to American
College of Medical Genetics/Association of Molecular Pathology
guidelines (17).
Specific primers for amplification and sequencing
were designed using Primer3web version 4.1.0 (http://primer3.ut.ee/). PCR was performed using
genomic DNA of the proband and his family members followed by
Sanger sequencing using ABI3730xl Genetic analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. PCR was performed using Takara PrimeSTAR
Max DNA Polymerase (Takara Biotechnology Co., Ltd.) under the
following thermocycling conditions: initial denaturization at 95˚C
for 5 min, followed by 35 cycles of denaturation at 95˚C for 3 sec,
annealing at 60˚C for 30 sec and extension at 72˚C for 30 sec.
Final extension occurred at 72˚C for 5 min. IFT122
(NM_052985.3) variants were amplified and sequenced using two pairs
of IFT122-specific primers: IFT122 primer1 forward
(F; 5'-AGACGCCTTGCATGGTACA-3') and primer1 reverse (R;
5'-CCAGGACAGGACATCAGGAA-3') were used for exon 5 amplification; and
IFT122 primer2F (5'-GGCTTCAGCCTGGTCTTACA-3') and primer2R
(5'-CAAGGCATGAAATGCCATAA-3') were used for exon 31
amplification.
Results
Patient description
The third child born to healthy, non-consanguineous
parents originating from Guangxi, China was a 2-month-old infant
male proband who was admitted to Guangxi Maternal and Child Health
Hospital (Guangxi, China) for genetic counseling due to
macrocephaly, postaxial hand polydactyly and postaxial
polysyndactyly of the feet on November 22, 2018. The clinical
images were obtained when the proband was 3 months old (Fig. 1). The proband was born full term
with a birth weight of 3.1 kg, a birth length of 51 cm (51st
centile) and a head circumference of 37.5 cm (91st centile).
Typical CED-associated craniofacial features were observed
(Fig. 1). These features included:
Macrocephaly, dolichocephaly, a high forehead with frontal bossing,
long face, broad nasal bridge, long philtrum, telecanthus,
bilateral epicanthic folds, hypertelorism, esotropia, abnormality
of the pinna, everted lower lip vermilion, abnormal and low-set
pinnae and micrognathia. The probands hair and eyebrows were
sparse, with hair that was also fine. Upper limb phocomelia and
postaxial polydactyly of both hands and feet were also observed
(Fig. 1). When the proband was
two-year-old, a renal ultrasound scan indicated normal-sized
kidneys and a normal renal function (estimated glomerular
filtration rate, 93 ml/min/1.73m2; serum creatinine
level, 51 µmol/l).
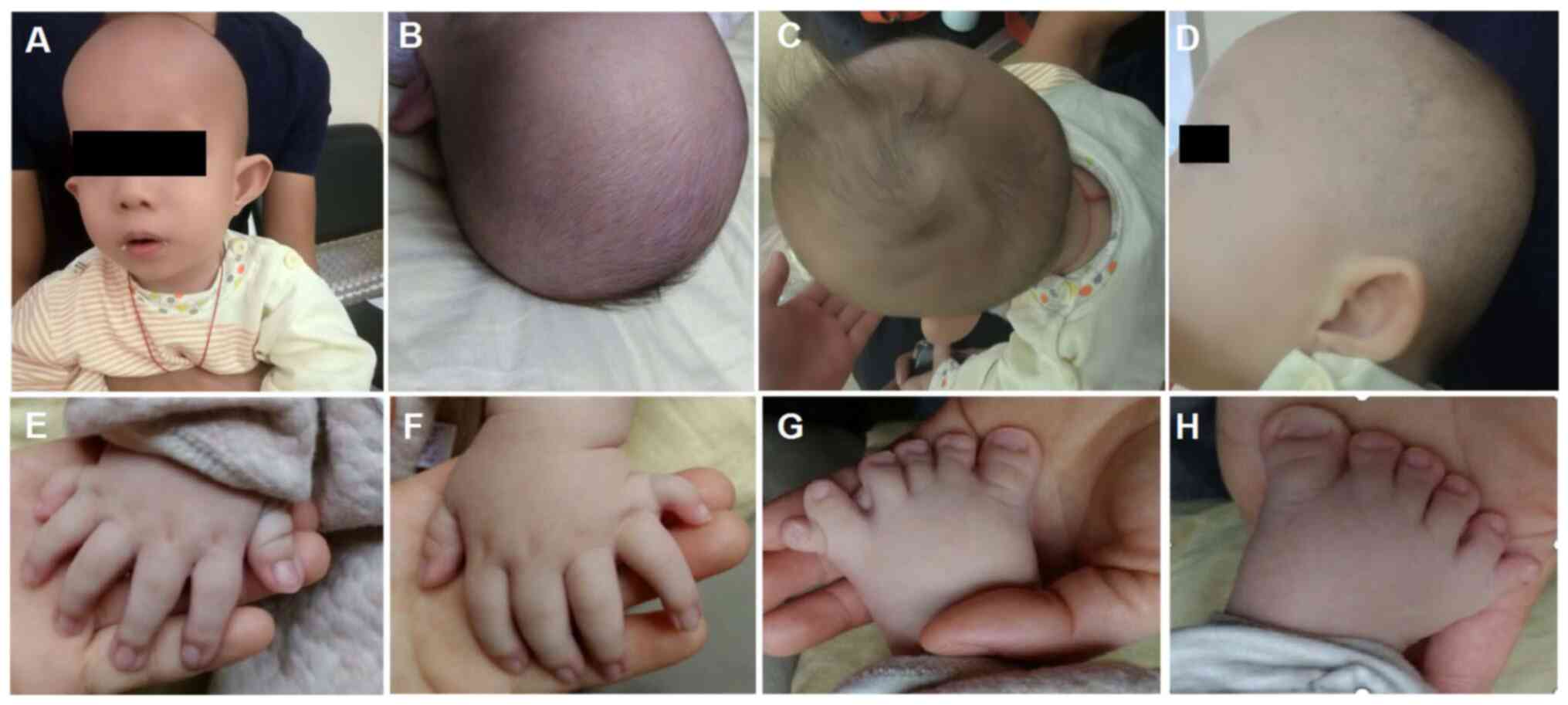 | Figure 1Images of the patient at the age of 3
months. (A and B) Macrocephaly, dolichocephaly, high forehead with
frontal bossing, long face, low set ears, broad nasal bridge, long
philtrum, telecanthus, bilateral epicanthic folds bilaterally,
sparse eyebrows, broad philtrum and prominent vermilion of the
lower lip. (C) Fine and sparse hair. (D) Abnormality of the pinnae.
(E-H) Postaxial hand polydactyly and postaxial polysyndactyly of
feet. |
Mutation analysis
With 20X coverage for >97% of the bases, a total
of >99% of reads were mapped to genomic targets. In the proband,
a total of 130,330 variants were identified in the proband and
included 17,560 variants (94.7%) classified with known SNP
identification numbers. Collectively, 121,448 SNVs, 3,951
insertions and 4,931 deletions were identified. After data analysis
using of TGex analysis software (https://tgex.genecards.cn/), two novel heterozygous
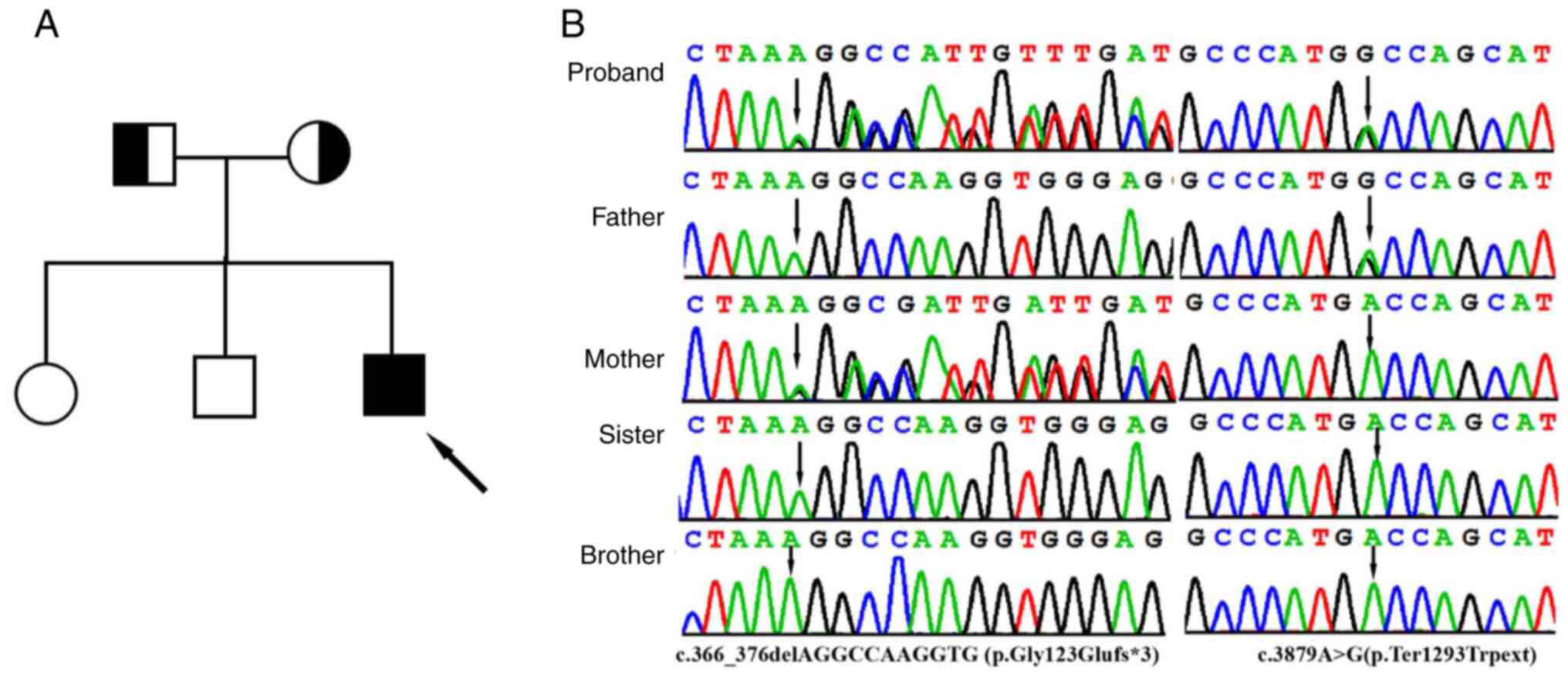
IFT122 variants, c.366_376delAGGCCAAGGTG (p.Gly123Glufs*3)
and c.3879A>G (p.Ter1293Trpext), were identified in the proband
(Fig. 2B). Sanger sequencing
further revealed that heterozygous c.366_376delAGGCCAAGGTG and
c.3879A>G variants were identified in the father and mother,
respectively, and that his sister and brother did not have either
variant (Fig. 2B).
These variants were not present in the Human Gene
Mutation Database (http://www.hgmd.cf.ac.uk/ac/), HPSD (http://liweilab.genetics.ac.cn/HPSD/)
and the dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) databases, nor were
they present in ExAC and gnomAD). The
c.366_376delAGGCCAAGGTG(p.Gly123Glufs*3) variant in exon 5, which
was inherited from the mother, is an 11 bp deletion that causes a
frameshift alteration after codon 123 (Glycine) and leads to a
premature termination codon that is located at codon 126. The
c.3879A>G (p.Ter1293Trpext) variant was inherited from the
father and is located in exon 31. Changing the length of the
protein, this variant results in the substitution of a TGA stop
codon to a missense TGG codon. It is predicted that the novel
variant likely has a deleterious effect by damaging the function of
the IFT122 protein. According to the ACMG standards and guidelines
(17) for the interpretation of
sequence variants, the novel variants are likely to be
pathogenic.
Discussion
Defects in the IFT122 gene have been
identified in patients with type IV short-rib polydactyly (SRP) and
type 1 CED (6,18). While SRP is characterized by
multiple skeletal anomalies that include short ribs, shortening of
the tubular bones, acetabula, irregular metaphyses and post-axial
polydactyly, CED is considered to be a mild form of SRP (19). The current study performed
whole-exome sequencing and identified compound heterozygosity novel
variants in the IFT122 gene of a male Chinese infant.
Consistent with previously reported CED patient presentation
(20-22),
the child manifested fine and sparse hair, macrocephaly, dysmorphic
facial features, upper limb phocomelia and postaxial polydactyly of
both hands and feet. Therefore, the patient was diagnosed with
CED.
In previous reports, the phenotypic severity of
ciliary disorders has been indicated to be determined by a
combination of missense and truncating mutations (21-23).
In the patient described in the present study, the biallelic
IFT122 variants identified were a combination of frameshift
and stop-loss variants. The IFT122 gene contains 31 exons
and encodes a member of the WD repeat protein family that is
comprised of 1,292 amino acids and seven WD40 functional domains
(6). These domains are involved in
the formation of β-propeller structures and act as platforms for
the association of binding partners (24). In the present study, the
c.366_376delAGGCCAAGGTG (p.Gly123Glufs*3) variant was indicated to
be a novel frameshift variant located in the third WD domain. This
may subsequently result in a truncated protein, and a loss of
function. It can be hypothesized that the p.Gly123Glufs*3 variant
acts similarly to other truncating IFT122 variants like
p.Tyr1077Valfs*10 and p.Tyr1077Valfs*11 that have been reported. It
may consequently prevent the retrograde transport of cilia and
affect the assembly and maintenance of eukaryotic cilia and
flagella, thereby causing abnormal development (18). The c.3879A>G (p.Ter1293Trpext)
variant is a novel stop-loss variant that results in a
missense-derived TGA stop codon substitution to a TGG codon. The
variant changes the length of the protein, which may cause
alteration of the overall structure the protein. This leads to the
partial loss of function of IFT122, preventing the formation of the
IFT-A complex and interrupting IFT122-related intraflagellar
transport. According to the ACMG standards and guidelines (17), the novel c.366_376delAGGCCAAGGTG
(p.Gly123Glufs*3) variant is likely pathogenic according to PVS1,
PM2 and PP4 criteria, while the novel c.3879A>G
(p.Ter1293Trpext) variant is likely pathogenic according to PM2,
PM3, PM4 and PP4. To date, ~16 patients with CED or lethal SRP type
IV have been linked to IFT122 variants and described in the
literature. It should be noted that of all mutations in these
patients, none were biallelic nonsense, deletion or null mutations
(6,20,21,25).
Of those mutations described in living patients, the biallelic
IFT122, WDR35, IFT43 and WDR19 variants
were missense mutations or a combination of missense and truncated
mutations (13). The combination of
a missense and nonsense variant can lead to lethal phenotypes
(25,26). This suggests that the phenotype is
dependent on the variable degree of functional IFT122 impairment.
In the current study, the patient presented with a typical CED
phenotype and was identified to carry a frameshift and a stop-loss
variant. It appears that p.Gly123Glufs*3 is associated with a
similar truncating variant to those IFT122 variants
reported, while the c.3879A>G (p.Ter1293Trpext) stop-loss
variant may result in partial loss-of-function of the IFT122
protein. A future functional study of the variants would improve
understanding of the disease and its mechanism, which require
further study.
Although polydactyly is often a component of
ciliopathies, it has infrequently been reported in patients with
CED and has only described in individuals with WDR35 and IFT43
variants (18,27). Notably, polydactyly has only been
identified in patients with IFT122 variants that have also
been diagnosed with SRP syndrome (27). In the present study, while
presenting with phenotypes typically associated with CED, to the
best of our knowledge, the current study is the first reported
patient with CED with IFT122 variants and with polydactyly.
Polydactyly is related to the impairment of the sonic hedgehog
(SHH) signaling pathway (28).
Previous studies have demonstrated that the alterations of limb
patterning in IFT122-null mouse embryos are associated with
the inhibition of SHH signaling (29,30).
IFT122 affects the expression of multiple genes in the SHH
pathway, suggesting that the heterogeneity of polydactyly may be
associated with the degree of IFT122 deficiency (31,32).
Functional studies are necessary to demonstrate the cause of
IFT122 variants and determine their associated disease
phenotypes. The role of IFT122 variants in polydactyly also
requires further study.
In conclusion, to the best of our knowledge, the
current study is the first instance in which a combination of
frameshift and stop-loss variants in a patient with CED have been
identified. The current study provided new insights into phenotypes
caused by different variant combinations. The molecular
confirmation of CED in this patient expands the clinical profile of
patients with CED as well as the CED-associated IFT122
variant spectrum.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Project of
Yu-Miao (grant no. GXWCH-YMJH-2017006) and Guangxi Medical and
Health Appropriate Technology Development and Promotion Application
(grant no. S2020060), which was used for the patient recruitment
and determining genetic variants.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QY and JL designed the study and drafted the
manuscript. QZ, QY, FC and ShaY performed the data analysis and the
experiments. XX analyzed the clinical data. ML, SheY and XX
performed analyzed the experimental results. QY and XX revised the
manuscript. QY and QZ confirmed the authenticity of all the raw
data. All authorsread and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All procedures in this study were approved by the
Ethics Committee of Guangxi Maternal and Child Health Hospital.
Detailed written informed consent was obtained from all
participants. For the patients who are underage, written informed
consent for participation in this study was obtained from the
patients' parents or guardians.
Patient consent for publication
Parents of the children (under 18) provided written
informed consent for the publication of any associated data and
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arts HH, Bongers EM, Mans DA, van Beersum
SE, Oud MM, Bolat E, Spruijt L, Cornelissen EA, Schuurs-Hoeijmakers
JH, de Leeuw N, et al: C14ORF179 encoding IFT43 is mutated in
Sensenbrenner syndrome. J Med Genet. 48:390–395. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bayat A, Kerr B and Douzgou S: DDD Study.
The evolving craniofacial phenotype of a patient with Sensenbrenner
syndrome caused by IFT140 compound heterozygous mutations. Clin
Dysmorphol. 26:247–251. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bredrup C, Saunier S, Oud MM, Fiskerstrand
T, Hoischen A, Brackman D, Leh SM, Midtbø M, Filhol E, Bole-Feysot
C, et al: Ciliopathies with skeletal anomalies and renal
insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum
Genet. 89:634–643. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gilissen C, Arts HH, Hoischen A, Spruijt
L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant
SG, et al: Exome sequencing identifies WDR35 variants involved in
Sensenbrenner syndrome. Am J Hum Genet Sep. 87:418–423.
2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Girisha KM, Shukla A, Trujillano D,
Bhavani GS, Hebbar M, Kadavigere R and Rolfs A: A homozygous
nonsense variant in IFT52 is associated with a human skeletal
ciliopathy. Clin Genet. 90:536–539. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Walczak-Sztulpa J, Eggenschwiler J, Osborn
D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi
M, Tzschach A, et al: Cranioectodermal dysplasia, Sensenbrenner
syndrome, is a ciliopathy caused by mutations in the IFT122 gene.
Am J Hum Genet. 86:949–956. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Prevo B, Scholey JM and Peterman EJ:
Intraflagellar transport: Mechanisms of motor action, cooperation,
and cargo delivery. FEBS J. 284:2905–2931. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Baker K and Beales PL: Making sense of
cilia in disease: The human ciliopathies. Am J Med Genet C Semin
Med Genet. 151C:281–295. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yuan X, Serra RA and Yang S: Function and
regulation of primary cilia and intraflagellar transport proteins
in the skeleton. Ann NY Acad Sci. 1335:78–99. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Oud MM, Lamers IJ and Arts HH:
Ciliopathies: Genetics in Pediatric Medicine. J Pediatr Genet.
6:18–29. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Arts H and Knoers N: Cranioectodermal
Dysplasia. GeneReviews [Internet]. Adam MP, Ardinger HH and Pagon
RA (eds). University of Washington Seattle, WA, 2013.
|
|
12
|
Zhang W, Taylor SP, Ennis HA, Forlenza KN,
Duran I, Li B, Sanchez JA, Nevarez L, Nickerson DA, Bamshad M, et
al: University of Washington Center for Mendelian Genomics:
Expanding the genetic architecture and phenotypic spectrum in the
skeletal ciliopathies. Hum Mutat. 39:152–166. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Handa A, Voss U, Hammarsjö A,
Grigelioniene G and Nishimura G: Skeletal ciliopathies: A pattern
recognition approach. Jpn J Radiol. 38:193–206. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lin AE, Traum AZ, Sahai I, Keppler-Noreuil
K, Kukolich MK, Adam MP, Westra SJ and Arts HH: Sensenbrenner
syndrome (Cranioectodermal dysplasia): Clinical and molecular
analyses of 39 patients including two new patients. Am J Med Genet
A. 161A:2762–2776. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Walczak-Sztulpa J, Posmyk R,
Bukowska-Olech EM, Wawrocka A, Jamsheer A, Oud MM, Schmidts M, Arts
HH, Latos-Bielenska A and Wasilewska A: Compound heterozygous
IFT140 variants in two Polish families with Sensenbrenner syndrome
and early onset end-stage renal disease. Orphanet J Rare Dis.
15(36)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Schmidts M: Clinical genetics and
pathobiology of ciliary chondrodysplasias. J Pediatr Genet.
3:46–94. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: ACMG
Laboratory Quality Assurance Committee: Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Silveira KC, Moreno CA and Cavalcanti DP:
Beemer-Langer syndrome is a ciliopathy due to biallelic mutations
in IFT122. Am J Med Genet A. 173:1186–1189. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Huber C and Cormier-Daire V: Ciliary
disorder of the skeleton. Am J Med Genet C Semin Med Genet.
160C:165–174. 2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Alazami AM, Seidahmed MZ, Alzahrani F,
Mohammed AO and Alkuraya FS: Novel IFT122 mutation associated with
impaired ciliogenesis and cranioectodermal dysplasia. Mol Genet
Genomic Med. 2:103–106. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Moosa S, Obregon MG, Altmüller J, Thiele
H, Nürnberg P, Fano V and Wollnik B: Novel IFT122 mutations in
three Argentinian patients with cranioectodermal dysplasia:
Expanding the mutational spectrum. Am J Med Genet A.
170A:1295–1301. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cardenas-Rodriguez M and Badano JL:
Ciliary biology: Understanding the cellular and genetic basis of
human ciliopathies. Am J Med Genet C Semin Med Genet. 151C:263–280.
2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bacino CA, Dhar SU, Brunetti-Pierri N, Lee
B and Bonnen PE: WDR35 mutation in siblings with Sensenbrenner
syndrome: A ciliopathy with variable phenotype. Am J Med Genet A.
158A:2917–2924. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Orlicky S, Tang X, Willems A, Tyers M and
Sicheri F: Structural basis for phosphodependent substrate
selection and orientation by the SCFCdc4 ubiquitin ligase. Cell.
112:243–256. 2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tsurusaki Y, Yonezawa R, Furuya M,
Nishimura G, Pooh RK, Nakashima M, Saitsu H, Miyake N, Saito S and
Matsumoto N: Whole exome sequencing revealed biallelic IFT122
mutations in a family with CED1 and recurrent pregnancy loss. Clin
Genet. 85:592–594. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Xu Y, Sun S, Li N, Yu T, Wang X, Wang J
and Bao N: Identification and analysis of the genetic causes in
nine unrelated probands with syndromic craniosynostosis. Gene.
641:144–150. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Walczak-Sztulpa J, Wawrocka A,
Sobierajewicz A, Kuszel L, Zawadzki J, Grenda R, Swiader-Lesniak A,
Kocyla-Karczmarewicz B, Wnuk A, Latos-Bielenska A, et al:
Intrafamilial phenotypic variability in a Polish family with
Sensenbrenner syndrome and biallelic WDR35 mutations. Am J Med
Genet A. 173:1364–1368. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bouldin CM, Gritli-Linde A, Ahn S and
Harfe BD: Shh pathway activation is present and required within the
vertebrate limb bud apical ectodermal ridge for normal autopod
patterning. Proc Natl Acad Sci USA. 107:5489–5494. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tsao CC and Gorovsky MA: Tetrahymena
IFT122A is not essential for cilia assembly but plays a role in
returning IFT proteins from the ciliary tip to the cell body. J
Cell Sci. 121:428–436. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pedersen LB and Rosenbaum JL:
Intraflagellar transport (IFT) role in ciliary assembly, resorption
and signalling. Curr Top Dev Biol. 85:23–61. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Qin J, Lin Y, Norman RX, Ko HW and
Eggenschwiler JT: Intraflagellar transport protein 122 antagonizes
Sonic Hedgehog signaling and controls ciliary localization of
pathway components. Proc Natl Acad Sci USA. 108:1456–1461.
2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Brooks ER, Islam MT, Anderson KV and
Zallen JA: Sonic hedgehog signaling directs patterned cell
remodeling during cranial neural tube closure. eLife.
9(e60234)2020.PubMed/NCBI View Article : Google Scholar
|