Introduction
The Bernard-Soulier syndrome (BSS) is an extremely
rare congenital disease that is characterized by
macrothrombocytopenia and defective binding of platelets to the von
Willebrand factor (vWF), which participates in the adherence of
platelets to the vascular subendothelial layer (1). BSS disease pathogenesis lies in the
expression of vWF receptor via the glycoprotein Ib-IX-V (GPIb-IX-V)
complex (2). The GPIb-IX-V complex
consists of four subunits, glycoprotein (GP)Ibα, GPIbβ, GPIX and
GPV, which are encoded by glycoprotein Ib platelet subunit alpha
(GP1BA), glycoprotein Ib platelet subunit beta (GP1BB), GP9 and
GP5(3). Mutations that occur in
GP1BA, GP1BB and GP9 may lead to BSS (4). The major subunit, GPIbα, holds an
essential ligand-binding site for vWF within its N-terminal domain,
and this features the leucine-rich repeat (LRR) (5,6).
Pathogenic mutations in its GP1BA gene are classically inherited as
a biallelic, autosomal recessive trait (4). A milder phenotype caused by
monoallelic autosomal dominant inheritance that shares characters
with BSS, including macrothrombocytopenia, has been previously
reported (6-12).
However, this aforementioned phenotype does not lead to severe
bleeding or serious surgical or obstetric hemorrhage that is
commonly seen in BSS. Therefore, the autosomal dominant variant of
BSS can be easily misdiagnosed as immune thrombocytopenia (ITP) and
managed with intravenous immunoglobulin and glucocorticoids, which
are used as a first-line treatment for ITP (13,14).
In the present case report, sequence analysis
performed on an 18-month-old girl identified an NM_000173
c.97T>A (p.Cys33Arg) (p.C33R) heterozygous missense mutation in
GP1BA. This specific mutation has not been previously reported and
may further expand the information regarding mutations in BSS.
Case report
An 18-month-old girl with thrombocytopenia consulted
the Hematology Oncology Center, Beijing Children's Hospital,
Capital Medical University (Beijing, China). A period of six months
prior to admission to the department, the patient underwent a
physical examination, which revealed a baseline platelet count of
31x109/l. Since this examination, the patient had no
bleeding symptoms, including easy bruising, epistaxis, gingival
bleeding and other symptoms as a result of internal bleeding. A
peripheral blood smear indicated giant platelets and a reduction in
platelet count. vWF antigen (vWF Ag) levels was 94.6%
(42.0-140.8%), aggregation response of platelets after adenosine
diphosphate (ADP) was 55% (55-90%), collagen was 62.1% (55-90%),
arachidonic acid was 82.6% (55-90%) and ristocetin was including
low concentration and [76.1% (55-90%); low concentation] and [2.6%
(<3%); standard concentration], all of which were within the
normal range. CBC, peripheral blood smear and platelet aggregation
were performed on the patients parents, and the patients father
shared a similar phenotype, including enlarged platelet size and
normal aggregation rate of platelet as follows: Aggregation
response of platelets after adenosine diphosphate (ADP) was 56.4%
(55-90%), collagen was 72.3% (55-90%), arachidonic acid was 85.7%
(55-90%) and ristocetin, including low concentration and standard
concentration, was 66.7% (55-90%) and 1.3% (<3%) respectively.
Considering the probability of inherited thrombocytopenia and
abnormal morphology of platelets, DNA samples for genetic analysis
were collected from her parents. Written consent from the patient's
parents was obtained at the time of sample collection. The present
study was performed with approval from the Ethics Committee of
Beijing Children's Hospital (Beijing, China; reference no.
2020-Z-113).
Approximately 2 ml peripheral blood of the patient,
parents and one sibling were collected in new EDTA containing tubes
(as anticoagulant) for genetic analysis. Exome sequencing of
germline DNA samples was performed using a QIAamp DNA Mini kit
(Qiagen, China Co., Ltd.), according to the manufacturer's
protocol. Selected targeted genes that were associated with
thrombocytopenia were analyzed using a gene capture strategy with a
GenCap custom enrichment kit (MyGenostics, Inc.), following the
manufacturer's protocol. The final enriched sequencing libraries
were sequenced for paired-end reads of 150 bp using Illumina
NovaSeq 6000 platform (Illumina, Inc.). The identified variants
were annotated using ANNOtate VARiation (ANNOVAR; http://annovar.openbioinformatics.org/en/latest/).
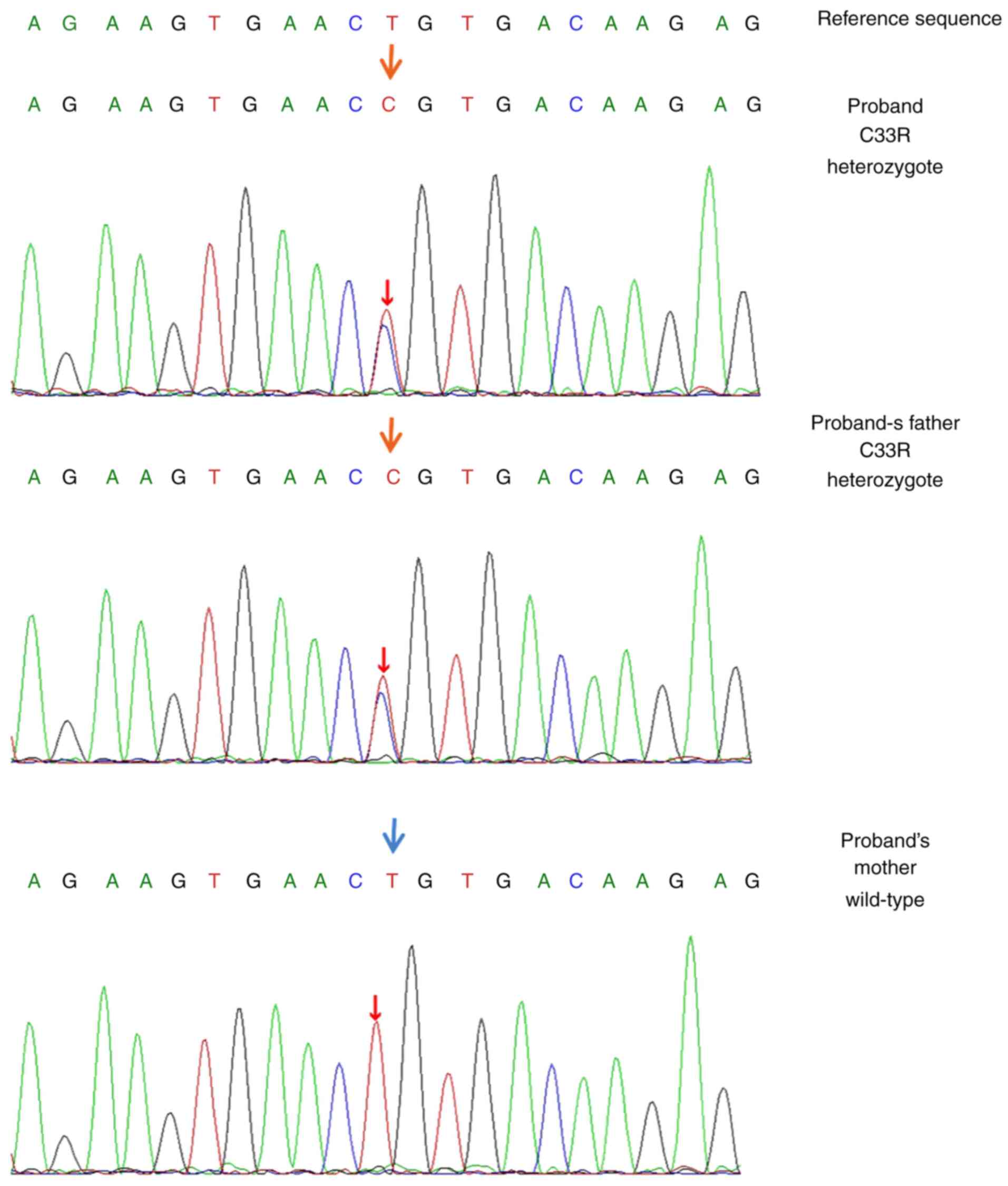
The sequencing analysis identified a heterozygous
nucleotide substitution in the GP1BA gene (NM_000173:
exon2:c.97T>G). An amino acid of cysteine 33 was substituted
with arginine in the N-terminal domain of GPIbα protein on the
platelet membrane. A validation of GP1BA sequencing was performed
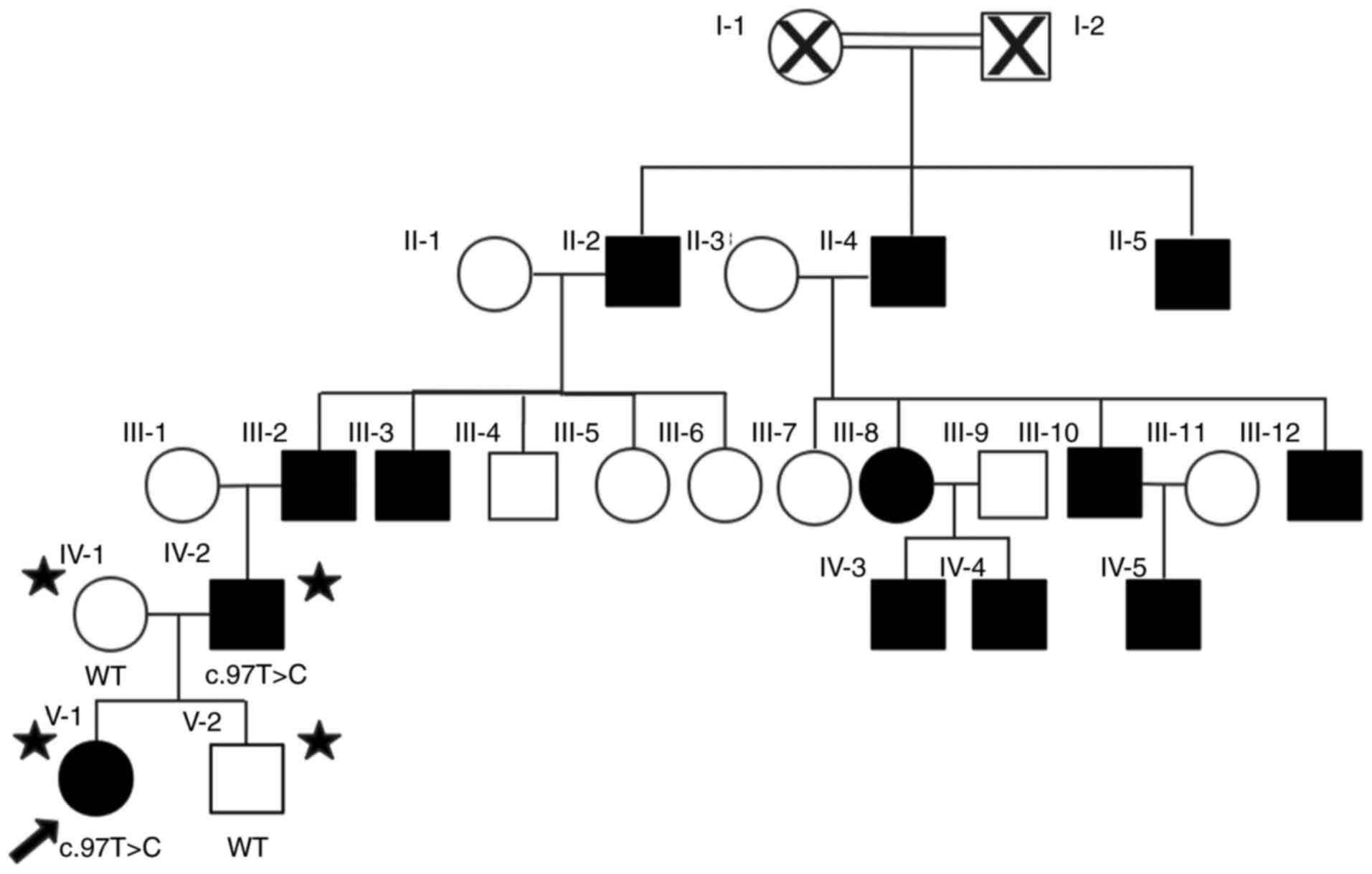
on the patient's parents and one sibling. The pedigree analysis
(Fig. 1) indicated an
autosomal-dominant mode of inheritance. The individuals father
carried a similar heterozygous p.C33R variant, while her mother
carried a wild-type sequence (Fig.
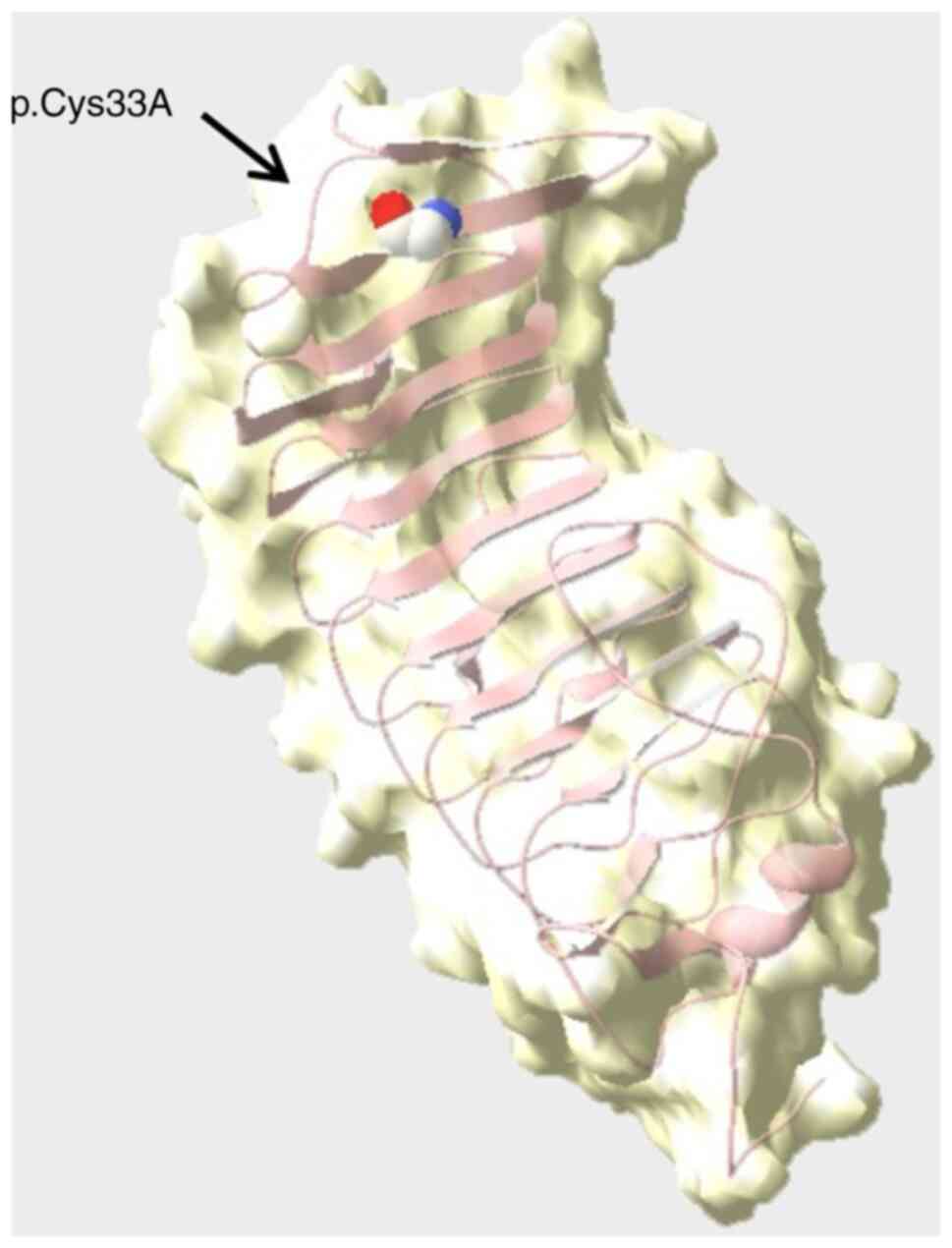
2). The structural effect of the sequence variant was predicted
to be deleterious via in-silico analysis (Fig. 3), using the following software with
default parameters: Mutation Taster (http://www.mutationtaster.org/), Sorting Intolerant
From Tolerant (http://sift.jcvi.org/), PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2/) and Genomic
Evolutionary Rate Profiling (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html).
According to the ACMG guidelines (15), the variant was classified as ‘likely
pathogenic’, which confirmed BSS.
Discussion
In the current case report, a novel heterozygous
c.97T>A substitution was reported in the GP1BA gene, which
causes a Cys33Arg amino acid substitution. The mutation was
annotated with ANNOVAR and the results demonstrated that it has not
been recorded in the GnomAD (https://gnomad.broadinstitute.org/), 1000 Genomes
(http://www.1000genomes.org/), ExAC
database (16), the human gene
mutation database (http://www.biobase-international.com/product/hgmd;
http://www.hgmd.cf.ac.uk/ac), OMIM
(https://www.omim.org/) or ClinVar databases
(https://www.ncbi.nlm.nih.gov/clinvar/). To date, ~60
variants of GP1BA encoding GPIBα protein have been identified, and
the majority of these are homozygous and contribute to symptoms of
BSS (6,17). However, the symptoms of individuals
in the pedigree of the current study seemed milder than the
symptoms of individuals with classic homozygous BSS. Platelet
aggregation of the present patients with heterozygous BSS
(including the response to the low concentration of ristocetin)
appeared normal despite giant platelets being identified in the
results of the peripheral blood smear.
Mutations in BSS may be missense, nonsense,
frameshift, insertions or deletions, and these mutations can cause
abnormality in GPIBα protein expression (18,19).
The dominant inheritance of BSS remains relatively rare as only six
variants have been previously identified, including the Bolzano
variant (p. Ala172Val), which is the most frequent type in Italy
(12). The remaining four
heterozygous mutations were described in single families (6-12).
The phenotype of monoallelic BSS, which is the same
as that of the biallelic BSS in the degree of symptom presentation,
may vary based on different genotypes and relevant changes in the
expression and function of GPIbα (2). Mutations in the patient outlined in
the current case report and her father were located in the
conserved N-terminal flanking domain. The domain lies ahead of the
LRR region of GP Ibα, which serves a role in the orientation of
charged residues (6). All
previously reported variants (p.Ala172Val, p.Asn57His, p.Tyr70Asp,
p.Leu73Phe, p.Asn150Ser and p.Leu59Arg), which are located in the
LRR region (amino acids 35-200), may lead to macrothrombocytopenia.
This is due to the abnormal interaction of the cytoplasmic end of
GPIbα protein with the actin-binding protein, which controls the
normal process of megakaryocyte maturation, demarcation membrane
system production and the size of the platelets (20,21).
While the conserved N-terminal flanking region of GPIbα, where the
novel mutation was discovered in the pedigree outlined the present
study, may interact with C-terminal flanking (amino acid 201-268)
and exhibit a weak influence on the binding of GPIbα to vWF that is
induced by ristocetin or botrocetin, the mutations within the LRR
caused a more significant change in the formation of GPIbα as well
as a pronounced disturbance in ligand binding (22).
BSS is commonly misdiagnosed with ITP and is
subsequently treated with futile therapies, including
glucocorticoids and immunoglobulins (14). BSS is a rare bleeding disorder that
is accompanied by thrombocytopenia, and symptoms are less
pronounced in the monoallelic phenotype of the disease (6,8). BSS
shares similar clinical characteristics with ITP, such as
mucocutaneous bleeding, negative family history and normal platelet
aggregation (14). However, the
identification of giant platelets in a peripheral blood smear may
help clinicians make an accurate diagnosis. In the current case
report, the patient and her father shared the same mutation and
similar mild manifestation. Her mother carried a wild-type genotype
and did not exhibit macrothrombocytopenia and bleeding. For those
suspected of having BSS, gene sequencing should be performed, and
symptoms such as abnormal levels of bleeding should be investigated
throughout the family to encourage early diagnosis and
subsequently, the establishment of effective treatment.
In the present study, platelet immunophenotype and
vWF-cofactor activity analysis (binding of VWF to recombinant
mutated GPIb) could not be performed. Therefore, whether this novel
dominant variant contributes to the reduced expression of GPIb-V-IX
and whether the inactive interaction between platelets and vWF was
indicated, which is commonly identified in classic BSS, was not
confirmed. Identification of the effect of specific mutations on
the structure and function of GPIbα protein and the pathogenesis to
a milder phenotype is required in future studies.
A novel autosomal dominant inheritance mutation in
GP1BA was identified in a family with mild bleeding symptoms and
macrothrombocytopenia. This monoallelic mutation that occurs in the
N-terminal flanking domain has, to the best of our knowledge, not
been previously reported. Due to common misdiagnosis, gene
sequencing is considered essential and urgent for diagnosing
suspected patients who exhibit a blood smear showing giant
platelets, with BSS.
Acknowledgements
The authors would like to thank Dr Lulu Mao and Mr.
Xinwen Fu from MyGenostics, Inc. for technical support in NGS work
and drawing the crystal structure.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant no. 81970111), the Beijing
Natural Science Foundation of China (grant no. 7192064), the
Pediatric Medical Coordinated Development Center of Beijing
Municipal Administration of Hospitals (grant no. XTZD20180205) and
the National Science and Technology Key Projects (grant no.
2017ZX09304029004).
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JM was involved in the collection and critical
analysis of clinical data and wrote the manuscript. RW contributed
to the designing the study, supervision and critical review of the
manuscript. ZC contributed to the supervision and critical analysis
of laboratory data, including the interpretation of NGS results. GL
performed the platelet aggregation. HG was involved in the
collection and sorting of the clinical data. RW and ZC confirmed
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Beijing Children's Hospital (Beijing, China; reference
no. 2020-Z-113) and written consent from the patient's parents was
obtained.
Patient consent for publication
The patient's parents have provided consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
López JA, Andrews RK, Afshar-Kharghan V
and Berndt MC: Bernard-Soulier syndrome. Blood. 91:4397–4418.
1998.PubMed/NCBI
|
|
2
|
Savoia A, Pastore A, De Rocco D, Civaschi
E, Di Stazio M, Bottega R, Melazzini F, Bozzi V, Pecci A, Magrin S,
et al: Clinical and genetic aspects of Bernard-Soulier syndrome:
Searching for genotype/phenotype correlations. Haematologica.
96:417–423. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Andrews RK, Gardiner EE, Shen Y, Whisstock
JC and Berndt MC: Glycoprotein Ib-IX-V. Int J Biochem Cell Biol.
35:1170–1174. 2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Savoia A, Kunishima S, De Rocco D, Zieger
B, Rand ML, Pujol-Moix N, Caliskan U, Tokgoz H, Pecci A, Noris P,
et al: Spectrum of the mutations in Bernard-Soulier syndrome. Hum
Mutat. 35:1033–1045. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Othman M and Emsley J: Gene of the issue:
GP1BA gene mutations associated with bleeding. Platelets.
28:832–836. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Trizuljak J, Kozubík KS, Radová L, Pešová
M, Pál K, Réblová K, Stehlíková O, Smejkal P, Zavřelová J, Pacejka
M, et al: A novel germline mutation in GP1BA gene N-terminal domain
in monoallelic Bernard-Soulier syndrome. Platelets. 29:827–833.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ghalloussi D, Saut N, Bernot D, Pillois X,
Rameau P, Sébahoun G, Alessi MC, Raslova H and Baccini V: A new
heterozygous mutation in GP1BA gene responsible for
macrothrombocytopenia. Br J Haematol. 183:503–506. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Noris P, Perrotta S, Bottega R, Pecci A,
Melazzini F, Civaschi E, Russo S, Magrin S, Loffredo G, Di Salvo V,
et al: Clinical and laboratory features of 103 patients from 42
Italian families with inherited thrombocytopenia derived from the
monoallelic Ala156Val mutation of GPIbα (Bolzano mutation).
Haematologica. 97:82–88. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Vettore S, Scandellari R, Moro S, Lombardi
AM, Scapin M, Randi ML and Fabris F: Novel point mutation in a
leucine-rich repeat of the GP1Balpha chain of the platelet von
Willebrand factor receptor, GPIb/IX/V, resulting in an inherited
dominant form of Bernard-Soulier syndrome affecting two unrelated
families: The N41H variant. Haematologica. 93:1743–1747.
2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kunishima S, Imai T, Hamaguchi M and Saito
H: Novel heterozygous missense mutation in the second leucine rich
repeat of GP1Balpha affects GPIb/IX/V expression and results in
macrothrombocytopenia in a patient initially misdiagnosed with
idiopathic thrombocytopenic purpura. Eur J Haematol. 76:348–355.
2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Miller JL, Lyle VA and Cunningham D:
Mutation of leucine-57 to phenylalanine in a platelet glycoprotein
Ib alpha leucine tandem repeat occurring in patients with an
autosomal dominant variant of Bernard-Soulier disease. Blood.
79:439–446. 1992.PubMed/NCBI
|
|
12
|
Savoia A, Balduini CL, Savino M, Noris P,
Del Vecchio M, Perrotta S, Belletti S, Poggi and Iolascon A:
Autosomal dominant macrothrombocytopenia in Italy is most
frequently a type of heterozygous Bernard-Soulier syndrome. Blood.
97:1330–1335. 2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Balduini CL, Savoia A and Seri M:
Inherited thrombocytopenias frequently diagnosed in adults. J
Thromb Haemost. 11:1006–1019. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Reisi N: Bernard-Soulier syndrome or
idiopathic thrombocytopenic purpura: A case series. Caspian J
Intern Med. 11:105–109. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Proulle V, Strassel C, Perrault C, Baas
MJ, Moog S, Mangin P, Nurden P, Nurden A, Adam F, Bryckaert M, et
al: A novel missense mutation in a leucine-rich repeat of GPIbα in
a Bernard-Soulier variant reduces shear-dependent adherence on von
Willebrand factor. Br J Haematol. 186:e184–e187. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li R and Emsley J: The organizing
principle of the platelet glycoprotein Ib-IX-V complex. J Thromb
Haemost. 11:605–614. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Simon D, Kunicki T and Nugent D: Platelet
function defects. Haemophilia. 14:1240–1249. 2010.
|
|
20
|
López JA: The platelet glycoprotein Ib-IX
complex. Blood Coagul Fibrinolysis. 5:97–119. 1994.PubMed/NCBI
|
|
21
|
Mhawech P and Saleem A: Inherited giant
platelet disorders. Classification and literature review. Am J Clin
Pathol. 113:176–190. 2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cauwenberghs N, Vanhoorelbeke K, Vauterin
S and Deckmyn H: Structural determinants within platelet
glycoprotein Ibalpha involved in its binding to von Willebrand
factor. Platelets. 11:373–378. 2000.PubMed/NCBI View Article : Google Scholar
|