Introduction
Acquired immunodeficiency syndrome (AIDS), caused by
human immunodeficiency virus (HIV), suppresses the human immune
system mainly through its effects on the CD4+ T-cell
compartment and has been extensively studied since the early 1980s
(1). The development of
antiretroviral therapy (ART) and highly active ART have markedly
reduced HIV-associated morbidity and mortality, but complete
eradication of this chronic infection is currently not feasible.
Increasing evidence has suggested that the gut microbiota has an
important role in the pathogenesis of AIDS, host deterioration and
viral transmission (2-4).
Studies have indicated increases in commensal bacteria, such as
Pseudomonas, in the feces of patients infected with HIV and
decreases in Lactobacilli and Bifidobacteria during
the early stages of HIV infection, elevating the risk of gut
dysbiosis (5). Furthermore,
Brenchley et al (6)
indicated that microbial translocation is a cause of systemic
immune activation in chronic HIV infection and that the levels of
certain bacterial products were elevated in HIV-infected
individuals.
The gut microbiota has attracted much attention due
to its newly discovered role in the maintenance of human health and
disease pathogenesis, particularly in association with the
gastrointestinal tract (7). Studies
have identified alterations in microbial diversity and bacterial
composition between the fecal microbiota and intestinal mucosal
microbiota of healthy individuals (3,8).
Deep-sequencing analysis of healthy volunteers has made it possible
to characterize differences between fecal microbiota and intestinal
mucosal microbiota (9-11).
These research studies have also demonstrated that the fecal
microbiota and intestinal mucosal microbiota are two distinct
microbial communities and that there is a requirement to further
investigate them to gain a better understanding of the significance
of alterations in the gut microbiota in different diseases.
However, most prior studies on the gut microbiota of patients with
HIV infection have focused on alterations in the fecal microbiota,
while the intestinal mucosal microbiota has been investigated
mostly in African and Caucasian populations.
In China, a national HIV epidemic has been declared
in 12 of the 31 provinces, and >10,000 individuals were living
with HIV or AIDS in 2014(12).
However, studies on the association between the gut microbiota and
HIV infection in China are currently limited. In a study performed
in Zhejiang province of eastern China, Ling et al (13) indicated that the α-diversity indices
were not significantly different between healthy controls and
patients infected with HIV, while the proportion of
Firmicutes/Bacteroidetes was significantly elevated in HIV
patients. Sun et al (14)
performed a study in Shanghai and determined that the microbiota of
individuals infected with HIV had a lower α-diversity, with
enrichment of Firmicutes and Proteobacteria at the phylum level and
suppression of the bacterial class Clostridia and the bacterial
families Ruminococcaceae and Lachnospiraceae. A previous study by
our group (15), in which stool
samples obtained from a population in Guangzhou, China, were
analyzed, also indicated that gut dysbiosis was more common among
patients with AIDS and was characterized by a lower α-diversity,
low mean counts of Bacteroidetes, Faecalibacterium,
Prevotella, Bacteroides vulgatus, Dialister
and Roseburia inulnivorans, as well as elevated mean counts
of Proteobacteria, Enterococcus,
Streptococcus, Lactobacillus,
Lachnoclostridium, Ruminococcus gnavus and
Streptococcus vestibularis. However, these studies only
analyzed fecal samples and did not explore the intestinal
mucosa.
In the present study, for further exploration, the
intestinal mucosal microbiota of healthy and HIV-infected
individuals from Guangzhou, China was examined. The intestinal
mucosa of HIV-infected patients was profiled by sequencing analysis
to obtain the composition of the microbiota and identify potential
universal and specific biomarkers for HIV infection. Furthermore,
differences in the characteristics of the microbiota between
patients who had been infected via different routes, i.e. sexual
transmission and intravenous drug abuse (IDA), were determined.
This investigation has the potential to improve the pathological
outcomes and characterize the intestinal mucosal microbiota of
HIV-infected individuals.
Materials and methods
Subjects
In total, 12 HIV-infected patients and 12 healthy
individuals were enrolled in the present study between March and
October 2015 at the Institute for Infectious Diseases, Guangzhou
No. 8 People's Hospital, Guangzhou Medical University (Guangzhou,
China), for cross-sectional comparison of their intestinal mucosal
microbiotas. The diagnosis of the HIV-infected individuals was
verified at the Guangzhou Center for Disease Control and Prevention
using PCR and HIV-1 antibody tests. According to the different
routes of HIV infection, patients were divided into IDA and sexual
transmission, and those who had a history/involvement in both
should be excluded. Nine patients were treatment-naïve and 3
patients with highly active antiretroviral therapy. Age-matched and
sex-matched healthy volunteers were recruited from the same
community (Table I). The exclusion
criteria used were an age of <18 years, use of antibiotics or
probiotics in the previous 4 weeks, a history of inflammatory bowel
disease, evidence of hepatitis B or C virus infection or other
chronic diseases, as well as pregnant or breastfeeding individuals
(16). This study was approved by
the Ethics Committee of Guangzhou No. 8 People's Hospital,
Guangzhou Medical University (Guangzhou, China). All individuals
enrolled provided written informed consent to participate.
 | Table IBaseline clinical characteristics for
each group of subjects. |
Table I
Baseline clinical characteristics for
each group of subjects.
| Item | HIV-infected
(n=12) | Healthy controls
(n=12) |
|---|
| Sex
(female/male) | 6/6 | 6/6 |
| Age (years) | 40.0±10.58 | 36.1±9.67 |
| CD4+
cell count (1/µl) | 94.92±41.439 | NA |
| Viral load (HIV-1
RNA copies/ml) |
5.65±2.384x105 | NA |
| Route of
transmission |
|
Intravenous
drug abuse | 3 | NA |
|
Sex | 9 | NA |
Sample collection and extraction of
bacterial genomic DNA
Terminal ileum mucosal biopsies were obtained during
colonoscopy using disposable flexible biopsy forceps. The samples
to be used for bacterial genomic DNA extraction were immediately
transferred to the laboratory and were stored at -80˚C. Total DNA
was extracted from samples using a DNeasy Blood and Tissue Kit
(Qiagen GmbH). DNA concentrations were determined using a NanoDrop
2000 Bioanalyzer at 260 nm (Thermo Fisher Scientific, Inc.). All
DNA samples were stored at -20˚C prior to PCR amplification and
Illumina sequencing was performed.
PCR amplification and Illumina
sequencing
The primers, V4-515 forward,
5'-GTGCCAGCMGCCGCGGTAA-3' and V4-806 reverse,
5'-GGACTACHVGGGTWTCTAAT-3', were used to amplify the bacterial 16S
ribosomal RNA V4 fragments. PCR was performed using
Phusion® High-Fidelity PCR Master Mix (New England
Biolabs). The PCR products were combined with equal volumes of a
loading buffer (containing SYBR-Green) before electrophoresis was
performed on 2% agarose gel. DNA samples between 400 and 450 bp in
length were identified as bright strips and were selected for use
in subsequent experiments. Sequencing libraries were generated
using the TruSeq DNA PCR-Free Sample Preparation Kit (Illumina,
Inc.) following the manufacturer's protocol and index codes were
added. Library quality was assessed using a Qubit 2.0 Fluorometer
(Thermo Fisher Scientific, Inc.) and an Agilent Bioanalyzer 2100
system (Agilent Technologies). Thereafter, the library was
sequenced using the Illumina HiSeq 2500 platform (Illumina,
Inc.).
Data analysis
The sequences were analyzed using the QIIME software
(version 1.7.0) package (17).
In-house Perl scripts were used to analyze α-diversity within the
samples and α-diversity among the samples (18). First, reads were filtered out using
QIIME quality filters. Subsequently, the program
pick_de_novo_otus.py was used to select operational taxonomic units
(OTUs) by generating an OTU table. Sequences with ≥97% similarity
were assigned to the same OTU. A representative sequence of each
OTU was explored for further annotation. An RDP classifier
(19) was used to annotate
taxonomic information of each representative sequence. α-diversity
was determined through Shannon, Chao 1, Observed_species and
PD_whole_tree indices to analyze the complexity of species
diversity in each sample (computed with core_diversity_analysis.py
script within QIIME) (17,20). Wilcoxon rank-sum test was used to
compare the α-diversity differences between groups within SPSS
(version 22.0; IBM Corp.). Subsequently, β-diversity was generated
within QIIME by using weighted and unweighted Unifrac distance
matrices. UniFrac distances are appraised as the distance between
bacterial communities explaining phylogenetic relationships between
bacteria (21,22). Cluster analysis was performed prior
to principal component analysis (PCA), which was applied to reduce
the dimension of the original variables, using the FactoMineR
package and ggplot2 package in R software (version 2.15.3).
Significant differences between specific taxa were analyzed using
analysis of variance followed by post-hoc t-tests, with Bonferroni
and Benjamini-Hochberg false discovery rate corrections applied for
multiple testing. Linear discriminant analysis (LDA) effect size
(LEfSe) analysis [http://huttenhower.sph.harvard.edu/lefse/] was used to
identify differences in taxa present between groups. First step,
the non-parametric factorial Kruskal-Wallis rank-sum test was used
to detect taxa with significant differential abundance.
Subsequently, biological consistency was investigated using a set
of pairwise tests among subclasses using the Wilcoxon rank-sum
test. Finally, the Linear Discriminant Analysis (LDA) was used to
estimate the effect size of each differentially abundant trait.
α-values of 0.05 were used for the Kruskal-Wallis sum-rank test and
a threshold of 3.6 was chosen for logarithmic LDA scores (23).
Results
Intestinal mucosal bacterial diversity
among HIV patients
Notably, α-diversity refers to bacterial diversity,
particularly the richness of each taxon within an environment/host,
while β-diversity compares similarities or differences in
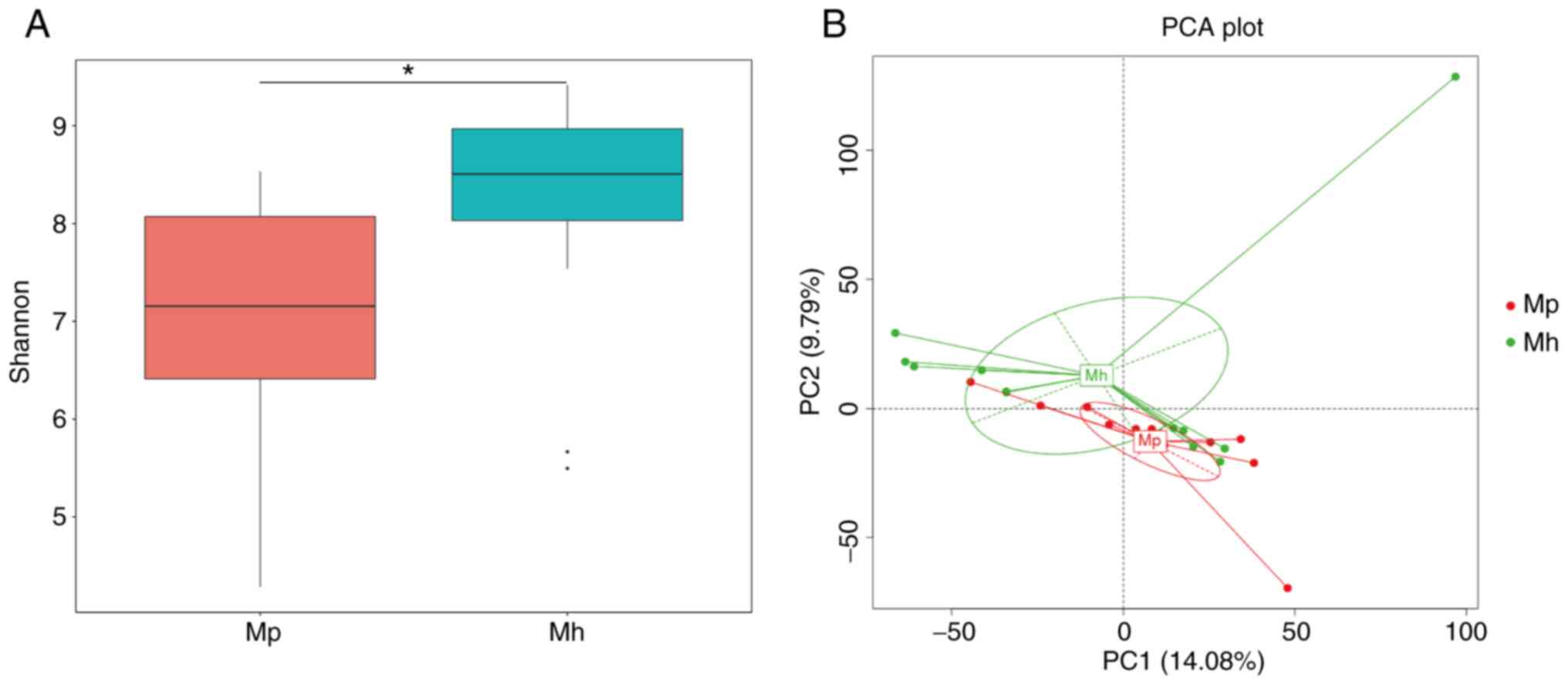
communities between environments/hosts. Shannon (P=0.02418), Chao 1
(P=0.03872), Observed_species (P=0.03324) and PD_whole_tree
(P=0.51370) indices were used to compare richness estimators and it
was indicated that the Shannon, Chao 1 and Observed_species indices
of the intestinal mucosal microbiota were significantly lower
compared with those of the healthy controls (Figs. 1A and S1). To reveal the effect of HIV infection
on the composition of the microbiota, β-diversity comparison (PCA)
was used to identify similarities in microbial community structure.
The results of the PCA suggested that the microbiota of patients
with HIV infection differed substantially from that of healthy
individuals (Fig. 1B).
Intestinal mucosal bacterial
composition in patients with HIV
Bacterial community structure was compared between
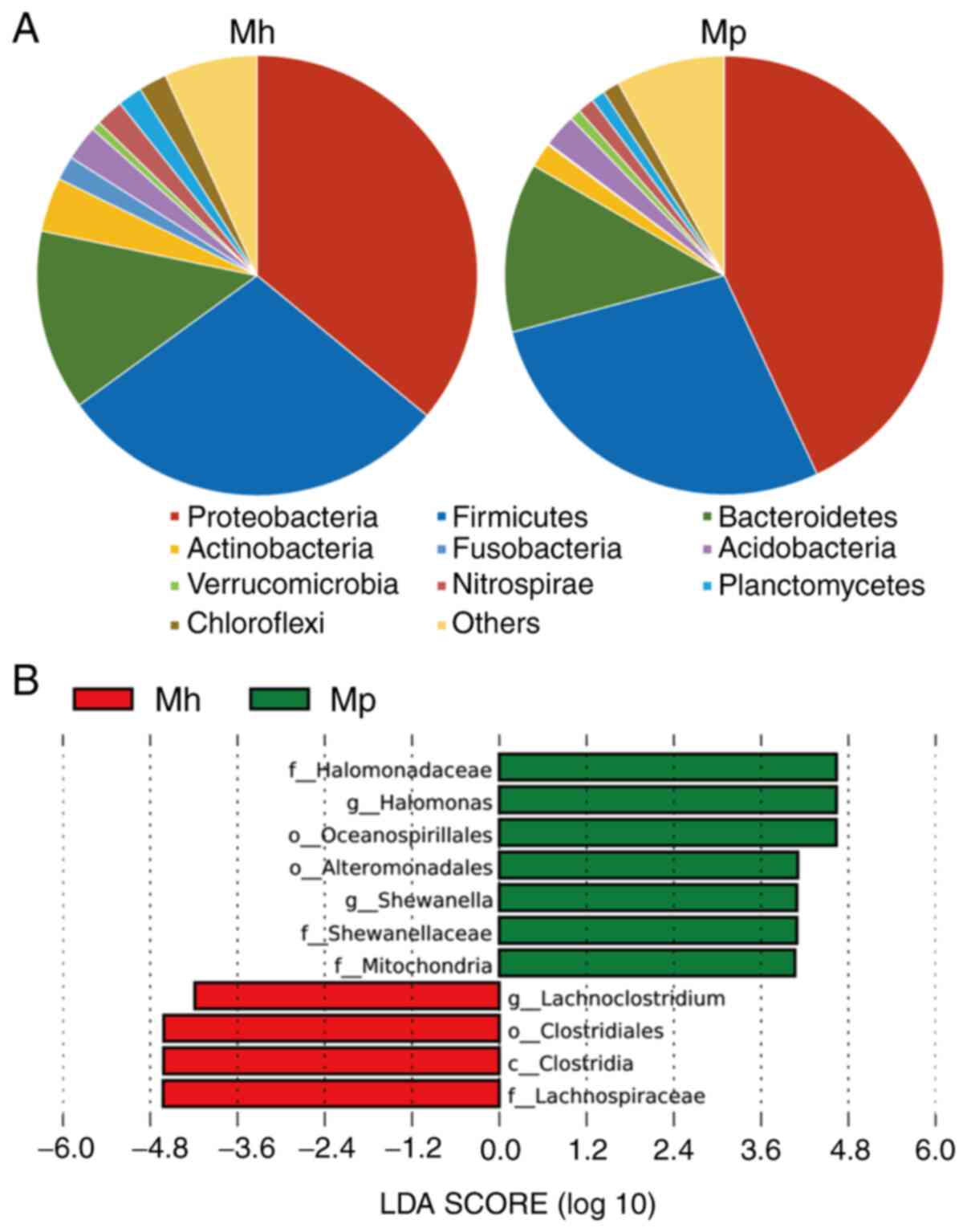
the HIV-infected group and the healthy group. Firmicutes,
Proteobacteria, Bacteroidetes and Actinobacteria were the most
predominant phyla, which constituted 82% of the intestinal mucosal
samples of HIV-infected patients and healthy controls (Fig. 2A). The proportions of Proteobacteria
(42.04 vs. 36.05%) and Bacteroidetes (17.58 vs. 13.23%) were
elevated in HIV-infected patients compared with those in the
controls, while the proportions of Firmicutes (22.35 vs. 28.97%)
and Actinobacteria (1.06 vs. 3.99%) were lower in HIV patients
compared with the controls.
For further analysis of changes at lower taxonomic
levels (genus level), the microbiota was compared between healthy
controls and HIV-infected patients using an LEfSe analysis. This
analysis revealed 11 discriminative features (|LDA score| >3.6).
Lachnoclostridium, a Firmicute, was abundant in the
healthy control samples, while Halomonas and
Shewanella, which are Proteobacteria, were abundant
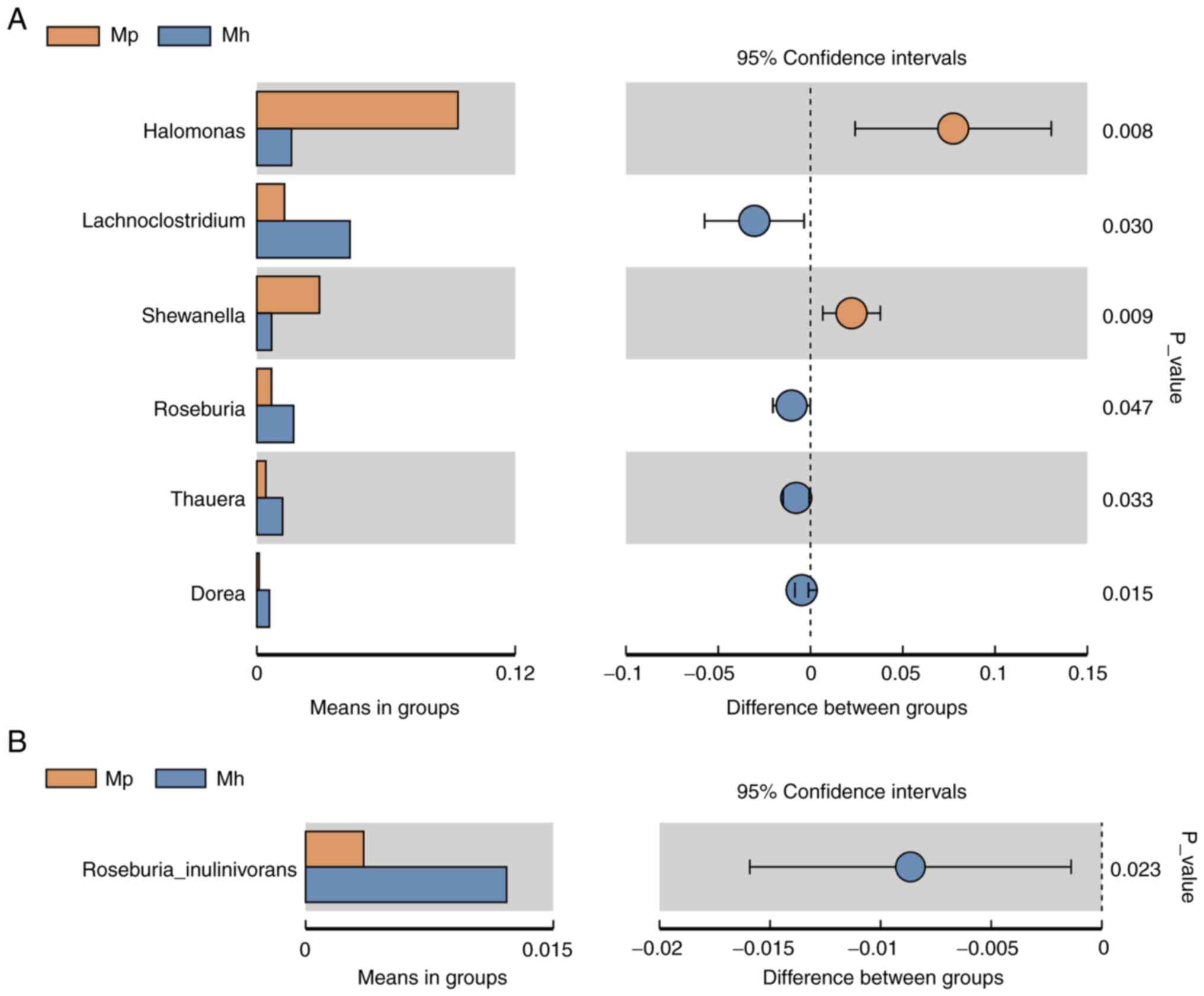
in the samples of HIV-infected patients. At the genus and species
levels, Halomonas (P=0.008) and Shewanella (P=0.009)
were the most abundant in the intestinal mucosal microbiota of HIV
patients compared with in the healthy controls, while the levels of
Lachnoclostridium (P=0.030), Roseburia (P=0.047),
Thauera (P=0.033), Dorea (P=0.015) and Roseburia
inulinivorans (P=0.023) were significantly lower in
HIV-infected patients than in the healthy controls (Figs. 2B and 3).
Association between the route of HIV
transmission and intestinal mucosal microbiota
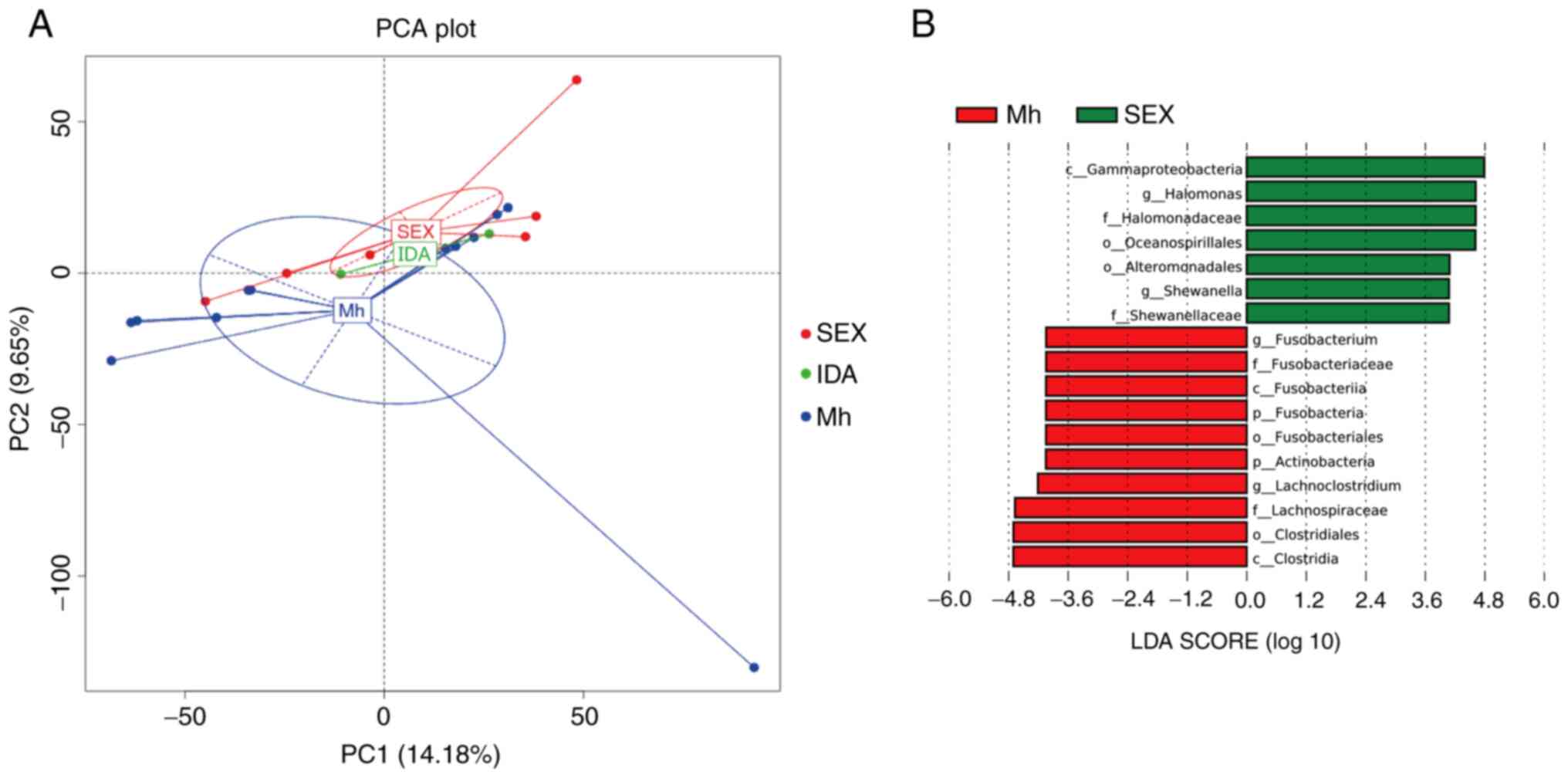
HIV-infected patients were divided into two groups
based on the route of HIV transmission: Sex or IDA. Subsequently,
the microbial community structures were compared between these two
groups and with the healthy control group. The results indicated
that the microbiota differed significantly between healthy group
and sexually transmitted HIV-infected group but there were no
significant differences between IDA HIV-infected group versus the
healthy group or sexually transmitted HIV-infected group (Fig. 4A). Furthermore, LEfSe analysis
revealed 17 discriminative features (|LDA score| >3.6) in the
gut microbiota of individuals with sexually transmitted HIV
infection compared with healthy controls and the relative
abundances of Fusobacterium and Lachnoclostridium
were significantly lower, while those of Halomonas and
Shewanella were significantly higher (Fig. 4B).
Discussion
The present study was the first to compare between
the intestinal mucosal microbiota of HIV-infected patients and
healthy individuals in a population from Guangzhou, China, using
16s-sequencing. The results demonstrated that the mucosal bacterial
communities of HIV-infected patients were suppressed, compared with
those of healthy controls. The gut microbiota of HIV-infected
patients contained higher proportions of potentially pathogenic
bacteria, including Halomonas and Shewanella. In
addition, the gut microbiota of HIV-infected patients was composed
of lower proportions of Lachnoclostridium, Roseburia,
Thauera, Dorea and Roseburia inulinivorans,
compared with that of healthy controls.
Compared to a traditional fecal microbiota analysis,
changes in the mucosal microbiota are able to more accurately
reflect changes in the gut microbiota of a patient's intestine
(24,25). The samples used in the present study
were collected from the end of the ileum and due to anatomical
particularities, different mucosal bacteria that were previously
underreported were identified. Traditional fecal sample sequencing
techniques are likely to be affected by dietary structure and air
exposure. Intestinal mucosa, as a part of human tissue, also
reflects ethnic differences, which may better reflect colonization
of gut microbiota in different populations and cultures. Dinh et
al (26) and Lozupone et
al (16) assessed American
subjects to determine the association between HIV infection and
fecal microbial changes. They indicated that the microbiota of
HIV-infected individuals in the US was similar to that of healthy
individuals from agrarian populations of Malawi and Venezuela.
Overall, this noticeable phenomenon indicated that the bacterial
composition is also dependent on the environment and diet.
The present study was the first to assess intestinal
mucosal samples in patients with HIV from Guangzhou (China). An
increased abundance of bacteria of the phylum Proteobacteria, as
well as decreased abundance of the phylum Firmicutes, was detected
in HIV-infected patients. Dillon et al (3) observed similar changes at the phylum
level in mucosal samples of HIV-infected patients. However, the
present study focused exclusively on Chinese subjects. The relative
abundances of the Proteobacteria, Halomonas and
Shewanella were indicated to be higher in the mucosal
microbiota of HIV-infected individuals than in healthy subjects.
Proteobacteria are known as conditional pathogens that mediate the
inflammatory response and may elicit an immune response. Of note,
the relative abundances of Halomonas and Shewanella,
which are extremely rare in the human gut, were significantly
elevated in patients infected with HIV. At present, there are no
other reports of these two bacterial strains in the gut microbiota
in HIV infected patients, to the best of our knowledge. These two
genera of bacteria tend to thrive in seawater and industrial
nitrification environments (27-29).
Therefore, Halomonas and Shewanella may be specific
biomarkers for HIV-infected individuals in Guangzhou, China,
particularly in coastal areas.
The present study also provided a comparison of the
gut microbiota between HIV-infected patients infected through two
different transmission routes to identify pivotal biomarkers that
may be associated with the transmission route of HIV infection. In
the largest study to date that examined the gut microbiota of
HIV-infected individuals, Noguera-Julian et al (30) indicated that a high
Prevotella/low Bacteroides enterotype in stool
specimens, was highly associated with males who have sex with
males, regardless of their HIV infection status. This may explain
the perceived association between this enterotype and HIV infection
status in prior studies that did not control for sexual behavior.
In the present study, the abundances of Fusobacterium and
Lachnoclostridium were significantly lower, while those of
Halomonas and Shewanella were significantly higher in
individuals with sexually transmitted HIV compared with patients
with IDA. However, the current results indicated that the
microbiota differed significantly between the healthy group and
sexually transmitted HIV-infected group but there were no
significant differences between the IDA HIV-infected group versus
healthy group or sexually transmitted HIV-infected group, the
reason is unknown and may be further elucidated in the future. The
present study was the first to attempt to compare mucosal
microbiota communities between two routes of transmission in a
Chinese HIV-infected population.
Several limitations of the present study should be
noted. First, the samples were collected from patients who had
already been diagnosed with HIV; therefore, the results did not
indicate whether the changes in the gut microbiota were a cause or
an outcome of HIV infection. HIV infection has been associated with
host-microbe interactions, which may trigger the mucosal immune
response, and which may have confounded the present results. Since
sampling was performed while each patient underwent colonoscopy,
the entire sample population was subjected to intestinal cleansing
through drinking laxative, which may have led to changes in the gut
microbiota. Based on previous studies (31-33),
intestinal cleansing may affect the structure of the gut
microbiota, leading to changes including decreases in abundances at
the family level, causing changes in the abundances of
Latobacillaceae and Enterobacteriaceae, as well as at
the genus level, causing changes in the abundances of
Blautia, Butyricicaccus and Mucispirillum. The
results of the present study were not significantly different from
those of previous studies. However, the effect of intestinal
cleansing on the intestinal microbiota requires to be addressed in
future studies. Furthermore, alterations of the gut microbiota are
dynamic, and due to a lack follow-up data and a more diverse
patient population, it was not possible to evaluate long-term gut
microbial changes or include more mucosal samples. A previous study
by our group reported that the abundance of
Lachnoclostridium was elevated in stool samples of patients
with HIV infection (15); however,
in the present study, the relative abundance of
Lachnoclostridium was lower. The reasons for this difference
may be as follows: i) Different sources of samples (feces vs.
terminal ileum mucosal biopsies); ii) influence of intestinal
preparation (use of laxatives prior to colonoscopy); iii)
anti-viral drugs for HIV may have a large influence on the gut
microbiota. Villanueva-Millán et al (34) indicated that different anti-viral
drugs for HIV produced different effects on the α-diversity of HIV
patients and these differences were closely associated with changes
at lower taxonomic levels (genus level and species level) rather
than at the phylum level. Nowak et al (35) reported that anti-AIDS drugs may
decrease the levels of the most abundant bacteria,
Prevotella, and increase the levels of pathogenic bacteria,
including Peptoniphilus, Finegoldia,
Anaerococcus and Campylobacter, compared with those
in untreated HIV-infected individuals. Therefore, longitudinal and
functional studies are required to better understand the role of
the gut microbiota of patients with HIV at different treatment
stages.
Overall, changes to the gut microbiota during HIV
infection are influenced by factors including ethnicity, sex, age,
geography, dietary habits, lifestyle, operational factors, sample
type and treatment (36-39).
In the present study, alterations in the intestinal mucosal
microbiota and dysbiosis were identified in HIV-infected patients
from Guangzhou, China. The results confirmed that the gut
microbiota contains promising biomarkers for non-invasive
evaluation of routes of HIV transmission and they may help to
develop principles that guide HIV management. Increased abundance
of the phylum Proteobacteria and decreased abundance of the phylum
Firmicutes are important characteristics of the intestinal mucosal
microbiota of patients with HIV. The determination of elevated
levels of Halomonas and Shewanella may lay a
foundation for establishing a set of microbiota-based biomarkers
for the diagnosis of HIV. In the future, larger, long-term studies
that include metabolomic approaches and culturomics may help
identify the association between a specific gut microbiota
composition and HIV infection.
In conclusion, the present study indicated that the
intestinal mucosal microbiota of patients with HIV had an increased
abundance of Proteobacteria and a decreased abundance of Firmicutes
bacteria in a population from Guangzhou, China. Certain bacteria,
such as Halomonas and Shewanella, may be potential
biomarkers for HIV infection in this population. Alterations to the
intestinal mucosal microbiota during HIV infection were indicated
to be associated with the route of HIV transmission.
Supplementary Material
Different α-diversity indexes of
healthy individuals and HIV.infected individuals. Values are
expressed as the median ± IQR. (A) Chao 1; (B) observed_species;
(C) PD_whole_tree. *P<0.05. Groups: Mp, HIV patients; Mh,
healthy controls. HIV, human immunodeficiency virus.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by grants from the National
Natural Science Foundation of China (grant nos. 81700487 and
81871905), the Guangdong Medical Science and Technology Research
Fund (grant no. A2019243), the Guangzhou Planned Project of Science
and Technology (grant no. 202002030288), the Guangzhou High
Technology Project (grant no. 2019GX05), the Fundamental Research
Funds for the Central Universities of SCUT (grant no. 2018MS82) and
the Guangzhou General Science and Technology Project of Medicine
and Health (grant no. 20171A011244).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HX and ZO designed the study, collected the clinical
data and wrote the manuscript; YJZ interpreted the results; YL and
JX collected and carried out the experiments; HH, HZ and ML
performed data interpretation and coordinated the revision of the
paper. YLZ and YN planned and directed the project and interpreted
the results. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The protocol of the present study was reviewed and
approved by the Committee of Guangzhou No. 8 People's Hospital
(Guangzhou, China). Written informed consent was obtained from all
patients before the research study was conducted.
Patient consent for publication
No applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zilberman-Schapira G, Zmora N, Itav S,
Bashiardes S, Elinav H and Elinav E: The gut microbiome in human
immunodeficiency virus infection. BMC Med. 14(83)2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Cohen CR, Lingappa JR, Baeten JM, Ngayo
MO, Spiegel CA, Hong T, Donnell D, Celum C, Kapiga S, Delany S, et
al: Bacterial vaginosis associated with increased risk of
female-to-male HIV-1 transmission: A prospective cohort analysis
among African couples. PLoS Med. 9(e1001251)2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dillon SM, Lee EJ, Kotter CV, Austin GL,
Dong Z, Hecht DK, Gianella S, Siewe B, Smith DM, Landay AL, et al:
An altered intestinal mucosal microbiome in HIV-1 infection is
associated with mucosal and systemic immune activation and
endotoxemia. Mucosal Immunol. 7:983–994. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Doerflinger SY, Throop AL and
Herbst-Kralovetz MM: Bacteria in the vaginal microbiome alter the
innate immune response and barrier properties of the human vaginal
epithelia in a species-specific manner. J Infect Dis.
209:1989–1999. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gori A, Tincati C, Rizzardini G, Torti C,
Quirino T, Haarman M, Ben Amor K, van Schaik J, Vriesema A, Knol J,
et al: Early impairment of gut function and gut flora supporting a
role for alteration of gastrointestinal mucosa in human
immunodeficiency virus pathogenesis. J Clin Microbiol. 46:757–758.
2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brenchley JM, Price DA, Schacker TW, Asher
TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann
D, et al: Microbial translocation is a cause of systemic immune
activation in chronic HIV infection. Nat Med. 12:1365–1371.
2006.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Blumberg R and Powrie F: Microbiota,
disease, and back to health: A metastable journey. Sci Transl Med.
4(137rv7)2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zoetendal EG, von Wright A,
Vilpponen-Salmela T, Ben-Amor K, Akkermans AD and de Vos WM:
Mucosa-associated bacteria in the human gastrointestinal tract are
uniformly distributed along the colon and differ from the community
recovered from feces. Appl Environ Microbiol. 68:3401–3407.
2002.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Durbán A, Abellán JJ, Jiménez-Hernández N,
Salgado P, Ponce M, Ponce J, Garrigues V, Latorre A and Moya A:
Structural alterations of faecal and mucosa-associated bacterial
communities in irritable bowel syndrome. Environ Microbiol Rep.
4:242–247. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ringel Y, Maharshak N, Ringel-Kulka T,
Wolber EA, Sartor RB and Carroll IM: High throughput sequencing
reveals distinct microbial populations within the mucosal and
luminal niches in healthy individuals. Gut Microbes. 6:173–181.
2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tap J, Derrien M, Törnblom H, Brazeilles
R, Cools-Portier S, Doré J, Störsrud S, Le Nevé B, Öhman L and
Simrén M: Identification of an intestinal microbiota signature
associated with severity of irritable bowel syndrome.
Gastroenterology. 152:111–123.e8. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang L, Chow EP, Jing J, Zhuang X, Li X,
He M, Sun H, Li X, Gorgens M, Wilson D, et al: HIV prevalence in
China: Integration of surveillance data and a systematic review.
Lancet Infect Dis. 13:955–963. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ling Z, Jin C, Xie T, Cheng Y, Li L and Wu
N: Alterations in the fecal microbiota of patients with HIV-1
infection: An Observational Study in A Chinese Population. Sci Rep.
6(30673)2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sun Y, Ma Y, Lin P, Tang YW, Yang L, Shen
Y, Zhang R, Liu L, Cheng J, Shao J, et al: Fecal bacterial
microbiome diversity in chronic HIV-infected patients in China.
Emerg Microbes Infect. 5(e31)2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhou Y, Ou Z, Tang X, Zhou Y, Xu H, Wang
X, Li K, He J, Du Y, Wang H, et al: Alterations in the gut
microbiota of patients with acquired immune deficiency syndrome. J
Cell Mol Med. 22:2263–2271. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Lozupone CA, Li M, Campbell TB, Flores SC,
Linderman D, Gebert MJ, Knight R, Fontenot AP and Palmer BE:
Alterations in the gut microbiota associated with HIV-1 infection.
Cell Host Microbe. 14:329–339. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Caporaso JG, Kuczynski J, Stombaugh J,
Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich
JK, Gordon JI, et al: QIIME allows analysis of high-throughput
community sequencing data. Nat Methods. 7:335–336. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kuczynski J, Stombaugh J, Walters WA,
Gonzalez A, Caporaso JG and Knight R: Using QIIME to analyze 16S
rRNA gene sequences from microbial communities. Curr Protoc
Bioinformatics. Chapter 10: Unit 10.7:2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang Q, Garrity GM, Tiedje JM and Cole JR:
Naive Bayesian classifier for rapid assignment of rRNA sequences
into the new bacterial taxonomy. Appl Environ Microbiol.
73:5261–5267. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li B, Zhang X, Guo F, Wu W and Zhang T:
Characterization of tetracycline resistant bacterial community in
saline activated sludge using batch stress incubation with
high-throughput sequencing analysis. Water Res. 47:4207–4216.
2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lozupone C and Knight R: UniFrac: A new
phylogenetic method for comparing microbial communities. Appl
Environ Microbiol. 71:8228–8235. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nagpal R, Shively CA, Appt SA, Register
TC, Michalson KT, Vitolins MZ and Yadav H: Gut Microbiome
Composition in Non-human Primates Consuming a Western or
Mediterranean Diet. Front Nutr. 5(28)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Segata N, Izard J, Waldron L, Gevers D,
Miropolsky L, Garrett WS and Huttenhower C: Metagenomic biomarker
discovery and explanation. Genome Biol. 12(R60)2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lo Presti A, Zorzi F, Del Chierico F,
Altomare A, Cocca S, Avola A, De Biasio F, Russo A, Cella E, Reddel
S, et al: Fecal and mucosal microbiota profiling in irritable bowel
syndrome and inflammatory bowel disease. Front Microbiol.
10(1655)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mira-Pascual L, Cabrera-Rubio R, Ocon S,
Costales P, Parra A, Suarez A, Moris F, Rodrigo L, Mira A and
Collado MC: Microbial mucosal colonic shifts associated with the
development of colorectal cancer reveal the presence of different
bacterial and archaeal biomarkers. J Gastroenterol. 50:167–179.
2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dinh DM, Volpe GE, Duffalo C, Bhalchandra
S, Tai AK, Kane AV, Wanke CA and Ward HD: Intestinal microbiota,
microbial translocation, and systemic inflammation in chronic HIV
infection. J Infect Dis. 211:19–27. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Gasperotti AF, Revuelta MV, Studdert CA
and Herrera-Seitz MK: Identification of two different chemosensory
pathways in representatives of the genus Halomonas. BMC Genomics.
19(266)2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Janda JM and Abbott SL: The genus
Shewanella: From the briny depths below to human pathogen. Crit Rev
Microbiol. 40:293–312. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Long MR, Zhang DF, Yang XY, Zhang XM,
Zhang YG, Zhang YM, Zhu H and Li WJ: Halomonas nanhaiensis sp.
nov., a halophilic bacterium isolated from a sediment sample from
the South China Sea. Antonie van Leeuwenhoek. 103:997–1005.
2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Noguera-Julian M, Rocafort M, Guillén Y,
Rivera J, Casadellà M, Nowak P, Hildebrand F, Zeller G, Parera M,
Bellido R, et al: Gut microbiota linked to sexual preference and
HIV infection. EBioMedicine. 5:135–146. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Drago L, Toscano M, De Grandi R, Casini V
and Pace F: Persisting changes of intestinal microbiota after bowel
lavage and colonoscopy. Eur J Gastroenterol Hepatol. 28:532–537.
2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Vich Vila A, Collij V, Sanna S, Sinha T,
Imhann F, Bourgonje AR, Mujagic Z, Jonkers DMAE, Masclee AAM, Fu J,
et al: Impact of commonly used drugs on the composition and
metabolic function of the gut microbiota. Nat Commun.
11(362)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tropini C, Moss EL, Merrill BD, Ng KM,
Higginbottom SK, Casavant EP, Gonzalez CG, Fremin B, Bouley DM,
Elias JE, et al: Transient osmotic perturbation causes long-term
alteration to the gut microbiota. Cell. 173:1742–1754.e1717.
2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Villanueva-Millán MJ, Pérez-Matute P,
Recio-Fernández E, Lezana Rosales JM and Oteo JA: Differential
effects of antiretrovirals on microbial translocation and gut
microbiota composition of HIV-infected patients. J Int AIDS Soc.
20(21526)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nowak RG, Bentzen SM, Ravel J, Crowell TA,
Dauda W, Ma B, Liu H, Blattner WA, Baral SD and Charurat ME:
TRUSTRV368 Study Group: Rectal microbiota among HIV-uninfected,
untreated HIV, and treated HIV-infected in Nigeria. AIDS.
31:857–862. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ashuro AA, Lobie TA, Ye DQ, Leng RX, Li
BZ, Pan HF and Fan YG: Review on the alteration of gut microbiota:
The role of HIV infection and old age. AIDS Res Hum Retroviruses.
36:556–565. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Bayigga L, Kateete DP, Anderson DJ,
Sekikubo M and Nakanjako D: Diversity of vaginal microbiota in
sub-Saharan Africa and its effects on HIV transmission and
prevention. Am J Obstet Gynecol. 220:155–166. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hager CL and Ghannoum MA: The mycobiome in
HIV. Curr Opin HIV AIDS. 13:69–72. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Tuddenham S, Koay WL and Sears C: HIV,
sexual orientation, and gut microbiome interactions. Dig Dis Sci.
65:800–817. 2020.PubMed/NCBI View Article : Google Scholar
|