Introduction
Alzheimer's disease (AD) is the most common
neurodegenerative disorder characterized by various pathological
markers in the brain, such as large numbers of amyloid plaques
surrounded by neurons containing neurofibrillary tangles, vascular
damage and neuronal cell loss (1,2). The
pathogenesis of AD remains largely unknown and no effective
pharmacotherapy is available to date.
The amyloid cascade hypothesis suggests that the
deposition of amyloid plaques in the brain is the causative agent
for AD pathogenesis, and that the neurofibrillary tangles, cell
loss, vascular damage and dementia follow as a direct result of
this deposition (3). β-Amyloid is
generated from the sequential and proteolytic cleavage of the
amyloid precursor protein (APP) by β- and γ-secretases. Individual
amyloid β (Aβ)42 monomers form soluble oligomers of different
molecular weights, which further aggregate to form insoluble Aβ
fibrils and amyloid plaques (4).
In recent years, soluble Aβ aggregates have been found to cause
hippocampal synaptic plasticity impairment, induce progressive
memory loss and be associated with cognitive impairment, both in AD
mouse models and in humans (4,5).
Synaptic plasticity is the neurophysiological basis of learning and
memory. Abnormal hippocampal synaptic plasticity has been reported
to be a key biological basis for cognitive dysfunction in AD
(6). Therefore, the identification
of a biological reagent that can improve synaptic remodeling in the
hippocampus could serve as a therapeutic strategy for AD.
Dl-3-n-butylphthalide (NBP) is a synthetic compound
based on l-3-n-butylphthalide that is isolated from the seeds of
Apium graveolens. NBP was approved by the Food and Drug
Administration (FDA) of China for the treatment of ischemic stroke
in 2002(7). It has also been
approved by the US FDA to undergo a phase II trial for the
treatment of ischemic stroke (8).
Studies have shown that NBP can not only significantly ameliorate
the acute symptoms of stroke (9,10)
but can also play a strong neuroprotective role in improving the
recovery of stroke-related disabilities, as well as alleviating
cognitive impairment of vascular dementia and AD (11,12).
The neuroprotective effects of NBP may involve multiple mechanisms,
including improving microcirculation and ATP metabolism (13), decreasing oxidative damage
(14), attenuating inflammatory
responses (15) and reducing
neuronal apoptosis (16).
Striatal-enriched protein tyrosine phosphatase
(STEP) is a central nervous system (CNS)-enriched member of the
protein tyrosine phosphatase (PTP) family encoded by the PTP
non-receptor type 5 (PTPN5) gene and has two spliced isoforms;
STEP61 and STEP46. STEP61 targets
the endoplasmic reticulum and postsynaptic density of dendritic
spines and is an important regulator of synaptic function (17). Increased STEP61
expression and/or activity disrupts synaptic function and is
associated with a number of neuropsychiatric disorders, such as AD
(17). Studies have shown that the
application of Aβ oligomers activates STEP61, which
subsequently leads to the downregulation of N-methyl-D-aspartic
acid or N-methyl-D-aspartate receptor (NMDAR), and that ERK1/2
could be the basis of cognitive deficits in early AD (18). The results of our previous study
revealed that STEP61 negatively regulates the Aβ
protein-mediated ERK/cAMP-response element-binding protein (CREB)
signaling pathway, providing a theoretical basis for
STEP61 as an effective new target for AD (19). It was therefore hypothesized that
the STEP61/ERK1/2/CREB pathway may be involved in the
mechanism of NBP improving the cognitive function of AD mice.
In the present study, using APP/PS1 transgenic mice
as the AD model, the efficiency of NBP on the cognitive function of
AD mice was investigated and the possible molecular mechanism of
NBP regulating the STEP61/ERK1/2/CREB pathway was
explored in an AD animal model.
Materials and methods
Preparation of NBP
NBP (purity, >98%) was purchased from
Shijiazhuang Pharmaceutical Group Co., Ltd. and dissolved in
vegetable oil at a working concentration of 5 mg/ml.
Animals and treatment
A total of 30 male APP/PS1 transgenic mice (model
mice of AD) aged 12 months and weighing 30.0±0.5 g were purchased
from Beijing Huafukang Biotechnology Co., Ltd. A total of 10
age-matched male C57BL/6 mice weighing 30.0±0.5 g were purchased
from Beijing Vital River Laboratory Animal Technology Co., Ltd.
(wild-type controls). The mice were housed under a 12-h light/dark
cycle at 20-25˚C, 40-45% humidity and provided with free access to
food and water. All animal experiments were approved by the Animal
Care and Use Committee of Jinzhou Medical University (approval no.
20191110), and in accordance with the Guide for the Care and Use of
Laboratory Animals (20). All mice
were acclimated to the laboratory environment for 1 week prior to
the experiment.
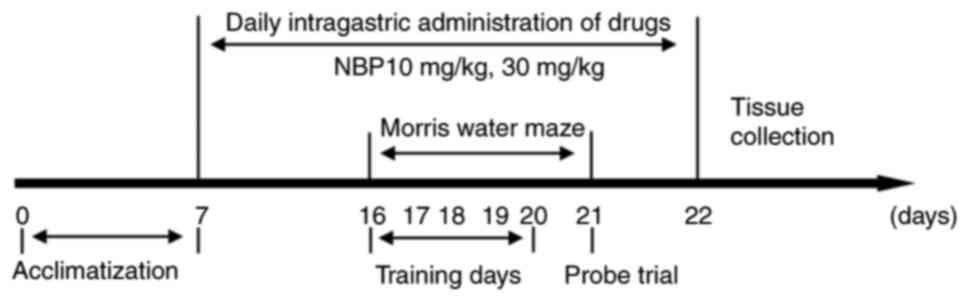
The experimental groups were as follows: C57BL/6
group (WT; n=10), APP/PS1 group (APP/PS1; n=10), NBP 10 mg/kg group
(NBP 10 mg/kg; n=10) and NBP 30 mg/kg group (NBP 30 mg/kg; n=10).
NBP was dissolved in vegetable oil. In the NBP 10 and 30 mg/kg
groups, the APP/PS1 mice underwent intragastric administration of
10 or 30 mg/kg NBP, respectively, once a day. The C57BL/6 and
APP/PS1 groups received equal amounts of vegetable oil in the same
manner. All mice were monitored daily for general health. The body
weight of each mouse was measured weekly. NBP was administered
continuously for 16 days, and behavioral experiments were performed
on days 10-15. Mice were administered NBP 30 min before the start
of the Morris water maze (MWM) training and euthanized on day 16
for histological examination. A schematic overview of the
experimental design is presented in Fig. 1.
MWM
Hippocampus-dependent spatial learning and memory
function were assessed using a MWM (21). The apparatus included a circular
pool (diameter, 120 cm; height, 50 cm) filled with opaque water to
a depth of 30 cm, which was maintained at 24±1˚C. The pool was
artificially divided into four equal quadrants. An escape platform
(diameter, 10 cm) was placed 1 cm below the water surface in the
center of one quadrant of the pool. The acquisition phase consisted
of 6 consecutive days of testing, with four trials per day. During
the first 5 days of training, the mice were given 60 sec per trial
to find the hidden platform. The time that the mice spent searching
for the platform was recorded as escape latency. If the mice failed
to find the platform within 60 sec, the training was terminated,
escape latency time was recorded as 60 sec, and the mice were
manually guided to the hidden platform. The escape latency, swim
paths and distance were recorded with a video camera (TOTA-450SIII)
and analyzed using the ANY-maze video tracking system (Stoelting
Co.). On day 6, a probe trial was performed with the platform
removed, in order to estimate spatial memory retention. The mice
were released into the pool directly opposite the platform location
and allowed to swim for 60 sec. The time spent in the target
quadrant and the number of crossings over the previous position of
the platform were recorded.
Sample preparation
Following the MWM training, all mice were
anesthetized through an intraperitoneal injection of 50 mg/kg
sodium pentobarbital (Sigma-Aldrich; Merck KGaA) and transcardially
perfused with 100 ml of 4% paraformaldehyde. The brain was quickly
removed and placed on an ice-cold glass dish and sagittally
bisected. The left hemisphere was routinely fixed in 4%
paraformaldehyde at 4˚C for 24 h, dehydrated with ethanol and
embedded in paraffin. The coronal sections were serially cut at a
thickness of 5 µm. The right cortex and hippocampus were separated
and stored at -80˚C until analysis.
Immunohistochemistry
Following dewaxing and rehydration of the paraffin
sections, antigen retrieval was carried out with citric acid buffer
(pH=6.0). The sections were incubated at room temperature with 3%
hydrogen peroxide solution for 30 min to block endogenous
peroxidase activity. Following rinsing with PBS, the slides were
incubated with 5% BSA (Beijing Solarbio Science & Technology
Co., Ltd.) blocking solution at a room temperature of 30 min to
remove the non-specific reaction. Following incubation with the
primary antibodies STEP61 (1:400; cat. no. ER1917-25;
Hangzhou Huaan Biotechnology Co., Ltd.), phosphorylated (p)-p44/42
MAPK (ERK1/2) (Tht202/Tyr204) (1:300; product no. 4370; Cell
Signaling Technology, Inc.) and p-CREB (Ser133) (1:400; product no.
9198; Cell Signaling Technology, Inc.) at 4˚C overnight, the
sections were incubated with a biotinylated secondary antibody
(1:200; product no. ZB2010; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd.) at 37˚C for 30 min. Peroxidase activity
was evaluated using 3,3'-diaminobenzidine and counterstained with
hematoxylin at room temperature for 10 min. Immunopositive
expression in the cortex and hippocampus was analyzed using an
Olympus BX53 light microscope (Olympus Corporation).
Immunofluorescence
Following dewaxing and rehydration of the paraffin
sections, antigen retrieval was carried out with citric acid buffer
(pH=6.0). The sections were incubated at room temperature with 3%
hydrogen peroxide solution for 30 min to block endogenous
peroxidase activity. Following rinsing with PBS, the slides were
incubated with 5% BSA (Beijing Solarbio Science & Technology
Co., Ltd.) blocking solution at room temperature for 1 h to remove
the non-specific binding. Following incubation with the primary
antibodies, NeuN (1:200; product code ab177487; Abcam),
STEP61 (1:400), p-ERK1/2 (1:300) and p-CREB (1:400), at
4˚C overnight, the sections were incubated with goat anti-mouse
secondary antibody conjugated with FITC (1:200; product no. ZF0312)
and goat anti-rabbit secondary antibody conjugated with
tetramethylrhodamine (1:200; product no. ZF0316; both from Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd.) at 37˚C for 30
min. Cell nuclei were counterstained with DAPI (0.005 mg/ml) for 5
min at 37˚C. The sections were mounted using anti-fluorescence
quenching sealing tablets, and observed, and images were captured
under a PerkinElmer Mantra™ Quantitative Pathology Imaging system
(PerkinElmer, Inc.). The mean number of positively stained cells
was counted from three visual fields in the cortex and hippocampus
of each section, and three discrete sections of each sample were
manually selected for statistical analysis.
Western blot analysis
The frozen brain samples were weighed and then
homogenized in RIPA lysis buffer (cat. no. R0020; Beijing Solarbio
Science & Technology Co., Ltd.) containing protease and
phosphatase inhibitor cocktails. Subsequently, the homogenate was
centrifuged at 12,000 x g for 30 min at 4˚C to collect the
supernatant. The protein concentrations were quantified using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). Equal amounts of total protein (30 µg/lane) were
separated on 10% SDS-PAGE gel electrophoresis and transferred to
PVDF membranes. The membranes were blocked against non-specific
binding for 1 h with 5% non-fat dry milk in TBS containing 0.1%
Tween-20 (TBST) at room temperature and incubated with the primary
antibodies STEP61 (1:2,000), p-ERK1/2 (1:2,000), p-CREB
(dilution, 1:2,000) and GAPDH (1:2,000; cat. no. AM1020b-400;
Abgent Biotech Co., Ltd.) at 4˚C overnight. They were then rinsed
with TBST three times for 10 min, followed by incubation with
HRP-labeled secondary antibody (1:5,000; product no. 7074; Cell
Signaling Technology, Inc.) at room temperature for 1 h.
Subsequently, the target protein expression was visualized by
SuperSignal West Pico PLUS chemiluminescence tsubstrate (Thermo
Fisher Scientific, Inc.) and analyzed using ImageJ software version
1.52a (National Institutes for Health). GAPDH was used as the
loading control. All experimental procedures were repeated at least
three times.
Statistical analysis
GraphPad Prism 8 software (GraphPad software, Inc.)
was used to carry out the statistical analysis of the experimental
data, which are all presented as the mean ± SEM. One- or two-way
ANOVA was performed. LSD and Tukey's post hoc tests were used for
comparisons between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
NBP improves spatial learning and
memory in APP/PS1 transgenic mice with AD
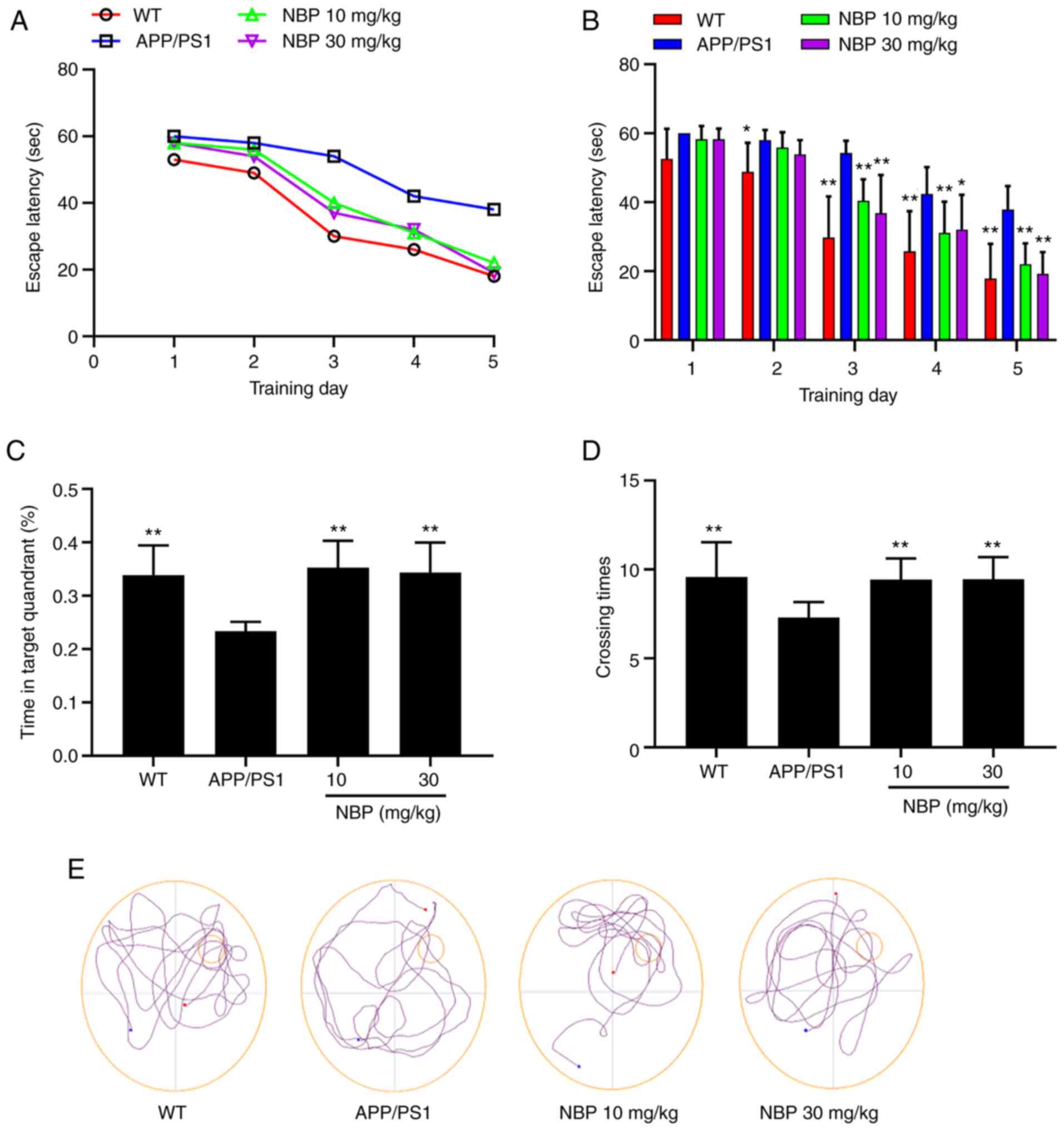
To investigate the effects of NBP on spatial
learning and memory function in APP/PS1 transgenic mice, their
behavior was assessed using the MWM. Following 5 training days in
the MWM, the escape latency to find the platform was significantly
increased in the APP/PS1 group compared with the C57BL/6 group. The
results revealed that the spatial learning of APP/PS1 transgenic
mice was clearly impaired in the water maze. In addition, compared
with the APP/PS1 group, the escape latency in the NBP 10 and 30
mg/kg groups had significantly declined (P<0.01; Fig. 2A and B), indicating that NBP alleviated the
spatial learning impairment in APP/PS1 transgenic mice. In the
probe trial of the MWM, mice from the APP/PS1 group spent
significantly less time in the target quadrant and had a lower
number of crossings, which was significantly different from the
C57BL/6 group. However, an obvious improvement was observed
following NBP treatment. As compared with the APP/PS1 group, mice
from the NBP 10 and 30 mg/kg groups spent considerably more time in
the target quadrant and had a higher number of crossings
(P<0.01; Fig. 2C and D). Representative swim paths from all
groups during the probe trial are presented in Fig. 2E. There was no significant
difference in the average swimming speed among all groups,
indicating that the APP/PS1 transgenic mice exhibited no impairment
in motor ability (P>0.05).
These results indicated that the spatial learning
and memory impairment in the APP/PS1 mice could be effectively
ameliorated following NBP treatment, but no significant difference
was observed between the NBP 10 and 30 mg/kg groups.
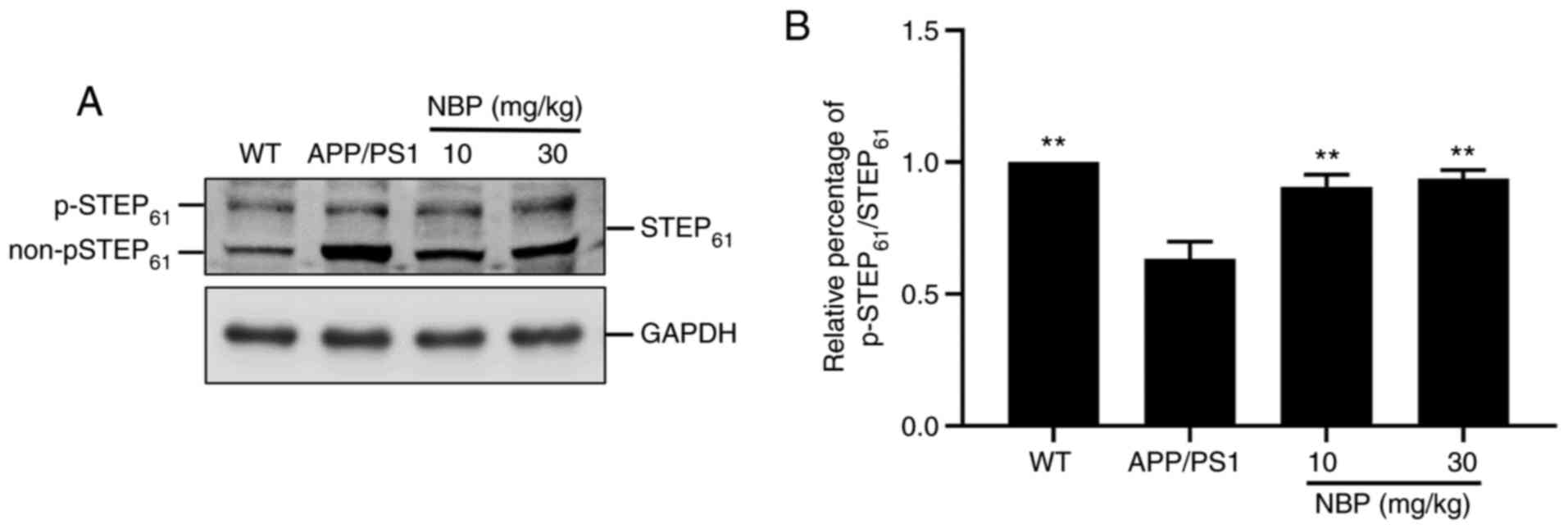
NBP increases STEP61
phosphorylation in APP/PS1 transgenic mice
STEP61 is expressed in neurons of the
hippocampus, cerebral cortex and striatum and its activity is
modulated by the phosphorylation/dephosphorylation of a serine
residue in the kinase-interacting motif domain, which affects the
affinity of STEP61 towards its substrate. The
dephosphorylation of the serine residue, referred to as the ‘active
form’, causes STEP61 to bind to its substrates, thus
inhibiting their activities, while the phosphorylation of the
serine residue prevents the binding of STEP61 to its
substrates (22). To evaluate
whether NBP impacted STEP61 activity in APP/PS1 mice,
cell lysates were analyzed by western blot analysis using an
anti-STEP61 antibody (Fig.
3A). The predominant isoform of STEP61 expressed in
neurons was mainly exhibited as a doublet, with the top band
representing the STEP61 phosphorylated form and the
bottom band representing the STEP61 non-phosphorylated
(or active) form (22). The
intensity of p-STEP61 is presented as a percentage of
the total intensity of p-STEP61 and
non-p-STEP61. Western blot analysis demonstrated a
marked decrease in the level of STEP61 phosphorylation
in the APP/PS1 group compared with that in the C57BL/6 group. By
contrast, the level of STEP61 phosphorylation was
significantly increased following NBP treatment compared with that
in the APP/PS1 group (P<0.01; Fig.
3A and B), particularly in the
NBP 30 mg/kg group.
STEP61 is a brain-specific phosphatase
that modulates key signaling molecules involved in synaptic
plasticity and neuronal function (23). An increase in STEP61
activity is considered to interfere with synaptic strengthening,
resulting in cognitive and behavioral deficits in AD (24). Western blot analysis suggested that
the decrease in STEP61 activity in both the hippocampus
and the cortex of APP/PS1 mice was restored by NBP treatment.
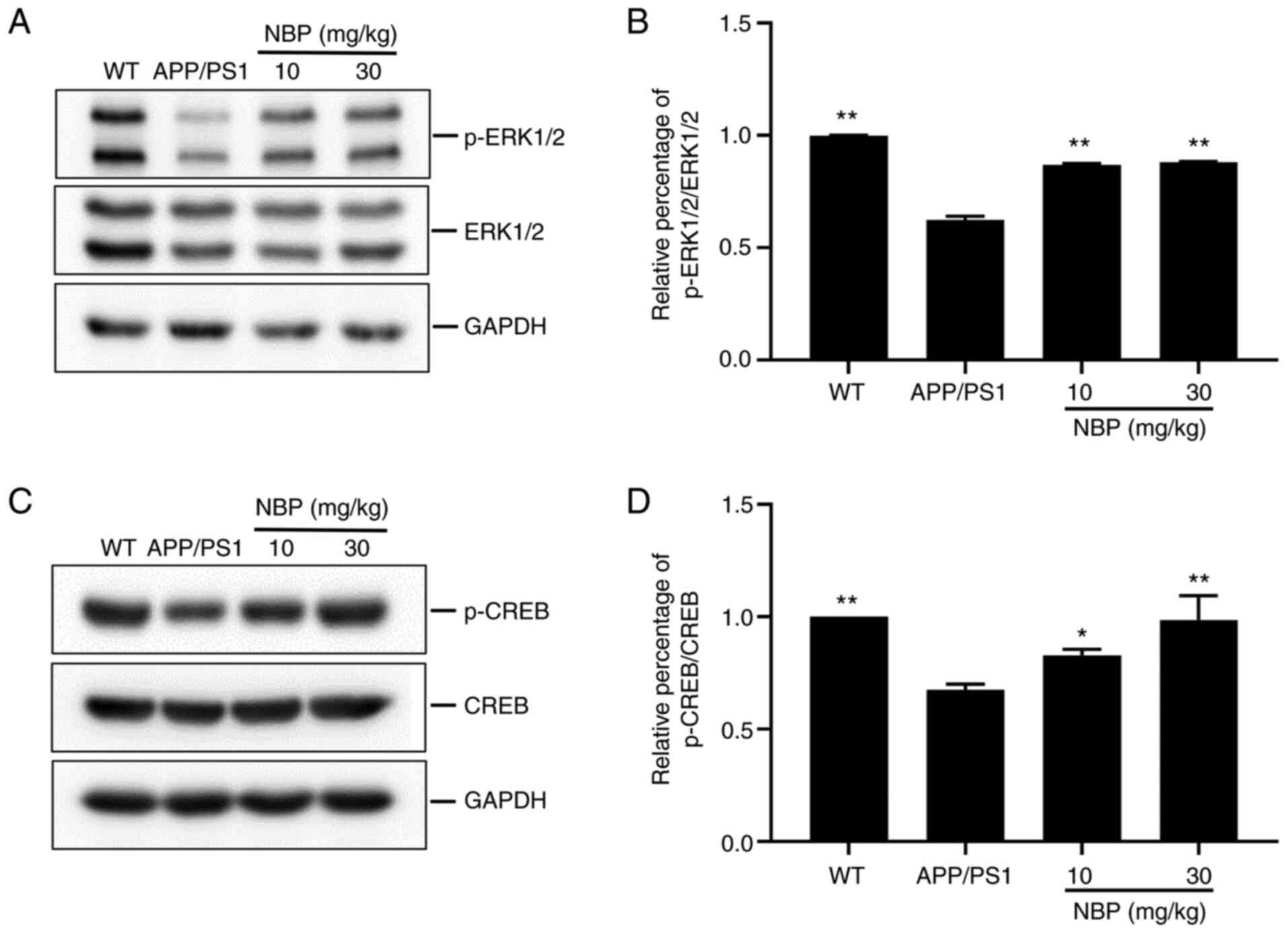
NBP increases the phosphorylation
level of ERK1/2 and CREB
Among the downstream targets of STEP61,
ERK1/2, which regulates memory and synaptic plasticity, was of
particular interest in the present study. In addition, p-ERK1/2
activates the transcription factor CREB, which regulates LTP
induction and maintenance, and synaptic plasticity (25). Thus, the levels of p-ERK1/2 and
p-CREB were evaluated. Western blot analysis demonstrated a marked
decrease in the levels of p-ERK1/2 and p-CREB in both the
hippocampus and cortex of the APP/PS1 group compared with that in
the C57BL/6 group. Conversely, the levels of p-ERK1/2 and p-CREB
were significantly increased following NBP treatment compared with
those in the APP/PS1 group (P<0.01, Fig. 4A and B; P<0.05, Fig. 4C and D), particularly in the NBP 30 mg/kg
group, suggesting that NBP can increase the levels of p-ERK1/2 and
p-CREB in APP/PS1 transgenic mice.
Localization, distribution and
expression changes of STEP61, p-ERK1/2 and p-CREB in the
brain of APP/PS1 transgenic mice
To investigate the effects of NBP on the
localization, distribution, and expression of STEP61,
p-ERK1/2 and p-CREB in APP/PS1 transgenic mouse brains, mouse brain
sections from four experimental groups were analyzed using
immunohistochemistry and immunofluorescence.
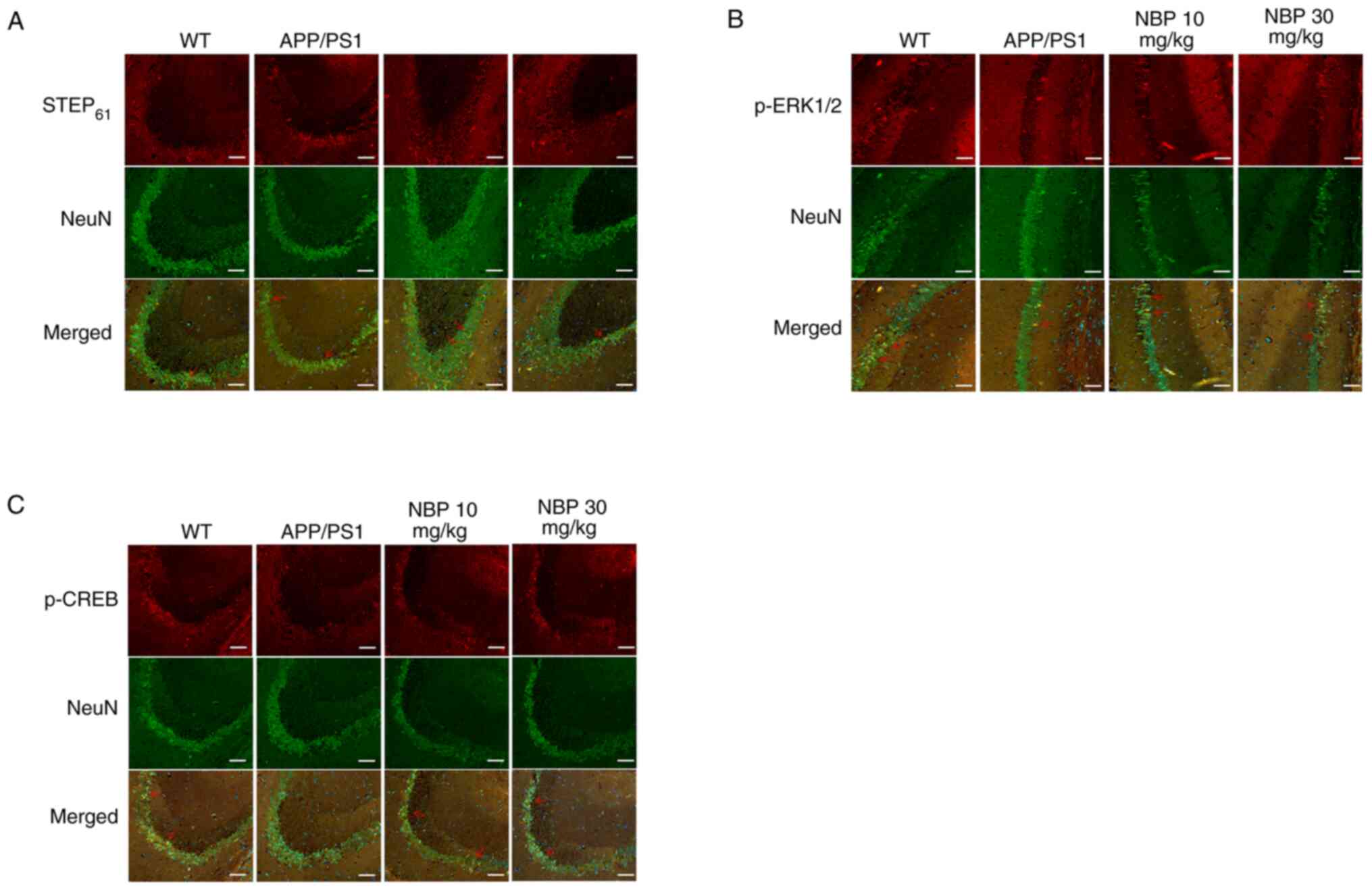
Immunofluorescence staining for STEP61,
p-ERK1/2 and p-CREB was performed using a PerkinElmer Mantra™
Quantitative Pathology Imaging system. Sections were co-stained
with the neuronal marker NeuN (green) and anti-STEP61,
anti-p-ERK1/2 and anti-p-CREB antibodies (red), respectively
(Fig. 5). STEP61
(Fig. 5A), p-ERK1/2 (Fig. 5B) and p-CREB (Fig. 5C) staining was observed within
hippocampal neurons. The results showed that compared with the
C57BL/6 group, the fluorescence intensity of STEP61 in
the APP/PS1 group was significantly increased, while the
fluorescence intensity of p-ERK1/2 and p-CREB in the APP/PS1 group
was significantly decreased. After treatment with NBP, the
fluorescence intensity of STEP61 decreased, which was
close to the level of the C57BL/6 group, and particularly in the
NBP 30 mg/kg group, the fluorescence intensity of p-ERK1/2 and
p-CREB was significantly increased.
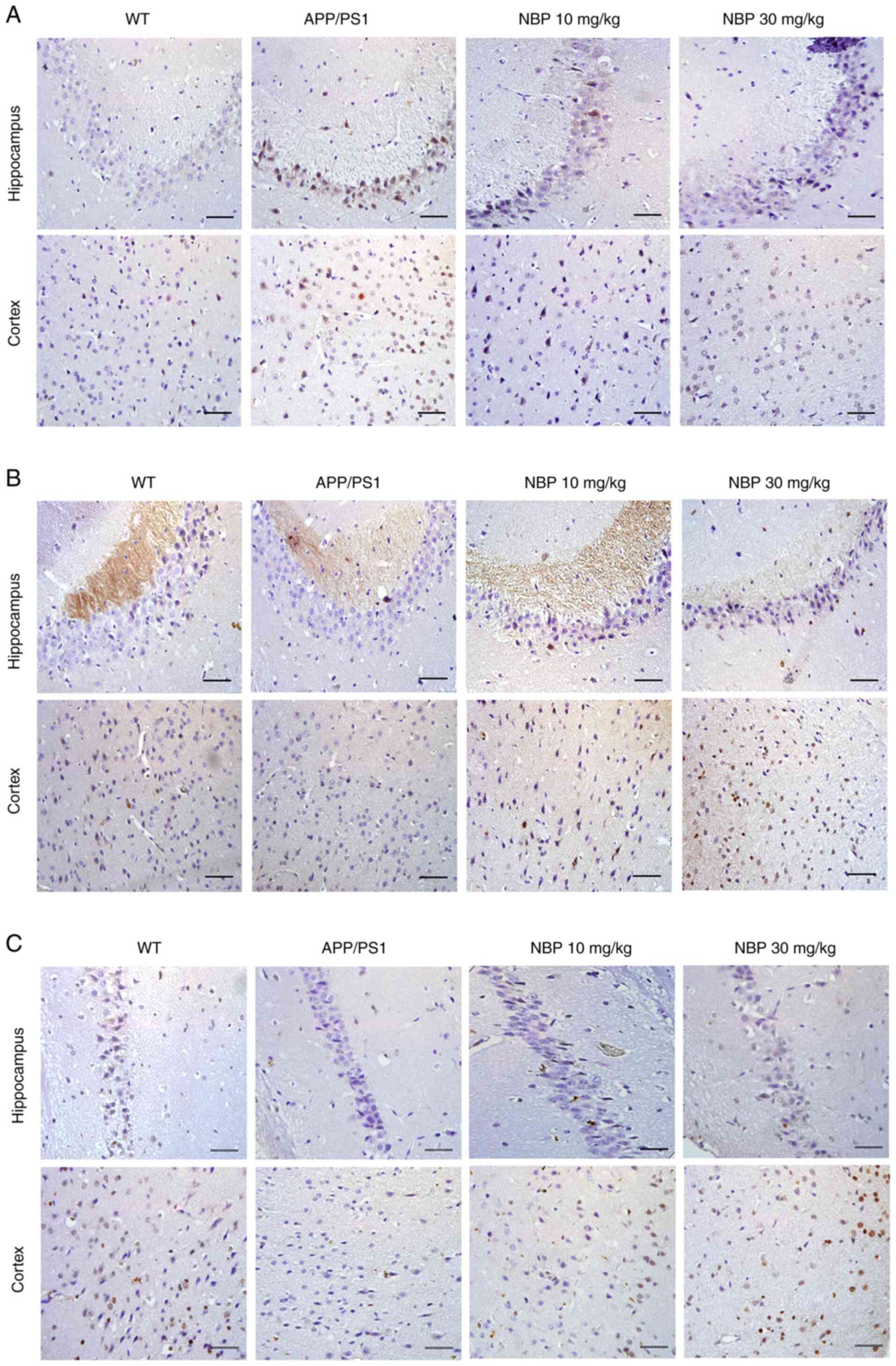
Immunohistochemistry revealed a positive
distribution of STEP61 in the cerebral cortex and
hippocampal CA1 region of mice in the APP/PS1 group, and the
expression of STEP61 was markedly increased in both the
cortex and hippocampus of the APP/PS1 group compared with the
C57BL/6 group, but not in the NBP 10 and 30 mg/kg groups (Fig. 6A). p-ERK1/2 and p-CREB expression
was similar (Fig. 6B and C). Immunohistochemistry indicated that
p-ERK1/2 and p-CREB immunoproducts were decreased in the cerebral
cortex and hippocampus of APP/PS1 mice compared with those of WT
mice. In addition, the decrease in p-ERK1/2 and p-CREB was restored
following NBP treatment, particularly in the NBP 30 mg/kg group. It
was further demonstrated that NBP reduced amyloid-induced
STEP61 levels, while increasing p-ERK1/2 and p-CREB
levels.
Discussion
AD is one of the most common age-linked
neurodegenerative diseases causing dementia. The aggregation of Aβ
peptides in the brain is the predominant pathological hallmark of
AD (26). APP/PS1 AD model mice
were double-transfected with the human APP695swe and the human
PS1dE9 mutation, both of which are associated with early-onset AD
(27). The mouse/human APP695swe
and PS1dE9 double transgene allowed mice to secrete human-amyloid
oligopeptides. In the early stages of AD physiopathology, soluble
Aβ oligomers interfere with synaptic function by controlling the
synaptic levels of the NMDAR (28). STEP, encoded by the PTPN5 gene, is
a CNS-enriched member of the PTP family that regulates tyrosine
residues on NMDARs through dephosphorylation, resulting in the
internalization of these receptor complexes (29). The protective effect of NBP on
synaptic function has been previously reported (12,30).
In the present study, the mechanism through which NBP protects
synaptic function by inhibiting STEP61 activity, thus
improving cognitive function in the AD mouse model, was
investigated.
The activity of STEP61 was modulated due
to the altered STEP61 phosphorylation/dephosphorylation
level, leading to a correlative change in the phosphorylation level
of its substrate. The MAPK family member ERK1/2 is the substrate of
STEP61. The ERK1/2 cascade plays a key role in
maintaining and inducing synaptic plasticity. p-ERK1/2 initiates
transcription by activating the transcription factor CREB in the
nucleus, while the STEP61-mediated dephosphorylation of
ERK1/2 at TYR204/187 inactivates ERK1/2, thereby limiting the
duration of ERK1/2 activity following NMDAR stimulation (23). CREB is a transcription factor and a
downstream target of ERK1/2. Our previous study has shown that
STEP61 affects the ERK/CREB signaling pathway in animal
and cell models of AD (19).
In the present study, APP/PS1 transgenic mice were
used to study the effects of NBP on cognitive impairment. Cognitive
function was evaluated using a MWM. Furthermore, the mechanism of
NBP therapy in APP/PS1 transgenic mice was studied by detecting the
levels of p-STEP61, p-ERK1/2 and p-CREB in the cortex
and hippocampus using western blot analysis and immunostaining. In
the MWM, the APP/PS1 group revealed a clear impairment in spatial
learning and memory compared with the C57BL/6 group, which
manifested as prolonged escape latency, shorter swimming time spent
in the target quadrant and a lower number of platform crossings in
the probe trial. NBP administration markedly decreased escape
latency, and increased the percentage of target quadrant search
time and number of platform location crossings in APP/PS1
transgenic mice. The results revealed that NBP clearly alleviated
learning and memory functions in APP/PS1 transgenic mice. In the
present study, it was also revealed that the inhibition of ERK1/2
and CREB phosphorylation in the cortex and hippocampus of APP/PS1
transgenic mice was accompanied by an increase in STEP61
dephosphorylation. However, the continuous administration of 10 or
30 mg/kg NBP significantly reduced the levels of activated
STEP61 and increased those of p-ERK1/2 and p-CREB, but
no significant differences were observed between the NBP groups (10
vs. 30 mg/kg; P>0.05). NBP concentrations were set to 10 and 30
mg/kg according to the published literature (7,31).
In conclusion, NBP alleviated amyloid-induced learning and memory
impairment, and decreased Aβ-induced activated STEP61
levels and promote ERK1/2 and CREB phosphorylation. Therefore, it
was hypothesized that NBP improved learning and cognitive defects
in APP/PS1 transgenic mice, likely through the regulation of the
STEP/ERK/CREB pathway. The results showed that, as a multi-target
drug, NBP may exert a neuroprotective effect. NBP may serve as an
effective treatment for AD. These effects will help elucidate and
possibly underlie the development of effective drugs to improve
cognitive function in patients with AD. Further study is required
to determine the mechanism underlying the effect of NBP on
phosphorylation level of STEP61.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Liaoning Province, China (grant no. 2019-ZD-0603) and
the Taizhou Science and Technology Support Plan of Jiangsu Province
(Social Development; grant no. TS202014).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ, JY and HRZ designed the study. WQY and LY
performed experiments and collected all experimental data. YZ and
JY drafted the manuscript and contributed substantially to its
revision. YZ and JY performed the statistical analysis. JY
contributed reagents and materials. YZ and LZ confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Care and Use Committee of Jinzhou Medical University (approval no.
20191110), and in accordance with ‘The detailed rules for the
implementation of the management of medical laboratory animals’
(Ministry of Health of China, 1998).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Masters CL, Bateman R, Blennow K, Rowe CC,
Sperling RA and Cummings JL: Alzheimer's disease. Nat Rev Dis
Primers. 1(15056)2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hardy JA and Higgins GA: Alzheimer's
disease: The amyloid cascade hypothesis. Science. 256:184–185.
1992.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang X, Perumalsamy H, Kwon HW, Na YE and
Ahn YJ: Effects and possible mechanisms of action of acacetin on
the behavior and eye morphology of Drosophila models of Alzheimer's
disease. Sci Rep. 5(16127)2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Tao CC, Cheng KM, Ma YL, Hsu WL, Chen YC,
Fuh JL, Lee WJ, Chao CC and Lee EHY: Galectin-3 promotes Aβ
oligomerization and Aβ toxicity in a mouse model of Alzheimer's
disease. Cell Death Differ. 27:192–209. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
De S, Whiten DR, Ruggeri FS, Hughes C,
Rodrigues M, Sideris DI, Taylor CG, Aprile FA, Muyldermans S,
Knowles TPJ, et al: Soluble aggregates present in cerebrospinal
fluid change in size and mechanism of toxicity during Alzheimer's
disease progression. Acta Neuropathol Commun. 7(120)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gu X, Wu H, Xie Y, Xu L, Liu X and Wang W:
Caspase-1/IL-1β represses membrane transport of GluA1 by inhibiting
the interaction between Stargazin and GluA1 in Alzheimer's disease.
Mol Med. 27(8)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang S, Ma F, Huang L, Zhang Y and Peng Y,
Xing C, Feng Y, Wang X and Peng Y: Dl-3-n-butylphthalide (NBP): A
promising therapeutic agent for ischemic stroke. CNS Neurol Disord
Drug Targets. 17:338–347. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cheng X, Wang H, Liu C, Zhong S, Niu X,
Zhang X, Qi R, Zhao S, Zhang X, Qu H and Zhao C:
Dl-3-n-butylphthalide promotes remyelination process in cerebral
white matter in rats subjected to ischemic stroke. Brain Res.
1717:167–175. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chong ZZ and Feng YP:
DI-3-n-butylphthalide improves regional cerebral blood flow after
experimental subarachnoid hemorrhage in rats. Zhongguo Yao Li Xue
Bao. 20:509–512. 1999.PubMed/NCBI
|
|
10
|
Wu F, Xu K, Xu K, Teng C, Zhang M, Xia L,
Zhang K, Liu L, Chen Z, Xiao J, et al: Dl-3n-butylphthalide
improves traumatic brain injury recovery via inhibiting
autophagy-induced blood-brain barrier disruption and cell
apoptosis. J Cell Mol Med. 24:1220–1232. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xiong Z, Lu W, Zhu L, Zeng L, Shi C, Jing
Z, Xiang Y, Li W, Tsang CK, Ruan Y and Huang L:
Dl-3-n-butylphthalide treatment enhances hemodynamics and
ameliorates memory deficits in rats with chronic cerebral
hypoperfusion. Front Aging Neurosci. 9(238)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lv C, Ma Q, Han B, Li J, Geng Y, Zhang X
and Wang M: Long-term DL-3-n-butylphthalide treatment alleviates
cognitive impairment correlate with improving synaptic plasticity
in SAMP8 mice. Front Aging Neurosci. 10(200)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen XQ, Qiu K, Liu H, He Q, Bai JH and Lu
W: Application and prospects of butylphthalide for the treatment of
neurologic diseases. Chin Med J (Engl). 132:1467–1477.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu CY, Zhao ZH, Chen ZT, Che CH, Zou ZY,
Wu XM, Chen SG, Li YX, Lin HB, Wei XF, et al: DL-3-n-butylphthalide
protects endothelial cells against advanced glycation end
product-induced injury by attenuating oxidative stress and
inflammation responses. Exp Ther Med. 14:2241–2248. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang G, Zhang J, Hu X, Zhang L, Mao L,
Jiang X, Liou AK, Leak RK, Gao Y and Chen J: Microglia/macrophage
polarization dynamics in white matter after traumatic brain injury.
J Cereb Blood Flow Metab. 33:1864–1874. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liao D, Xiang D, Dang R, Xu P, Wang J, Han
W, Fu Y, Yao D, Cao L and Jiang P: Neuroprotective effects of
dl-3-n-butylphthalide against doxorubicin-induced
neuroinflammation, oxidative stress, endoplasmic reticulum stress,
and behavioral changes. Oxid Med Cell Longev.
2018(9125601)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
He RJ, Yu ZH, Zhang RY and Zhang ZY:
Protein tyrosine phosphatases as potential therapeutic targets.
Acta Pharmacol Sin. 35:1227–1246. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jang SS, Royston SE, Lee G, Wang S and
Chung HJ: Seizure-induced regulations of amyloid-β, STEP61, and
STEP61 substrates involved in hippocampal synaptic plasticity.
Neural Plast. 2016(2123748)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang L, Xie JW, Yang J and Cao YP:
Tyrosine phosphatase STEP61 negatively regulates amyloid β-mediated
ERK/CREB signaling pathways via α7 nicotinic acetylcholine
receptors. J Neurosci Res. 91:1581–1590. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press, Washington, DC, 2011.
|
|
21
|
Zhuang XX, Zang X, Zheng GY, Hua N, Sun Y,
Hu YH and He L: Polyprenols mitigate cognitive dysfunction and
neuropathology in the APP/PS1 mouse. Phytother Res. 32:1098–1107.
2018.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Poddar R, Rajagopal S, Shuttleworth CW and
Paul S: Zn2+-dependent activation of the Trk signaling pathway
induces phosphorylation of the brain-enriched tyrosine phosphatase
STEP: Molecular basis for ZN2+-induced ERK MAPK activation. J Biol
Chem. 291:813–825. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Goebel-Goody SM, Baum M, Paspalas CD,
Fernandez SM, Carty NC, Kurup P and Lombroso PJ: Therapeutic
implications for striatal-enriched protein tyrosine phosphatase
(STEP) in neuropsychiatric disorders. Pharmacol Rev. 64:65–87.
2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Paul S, Nairn AC, Wang P and Lombroso PJ:
NMDA-mediated activation of the tyrosine phosphatase STEP regulates
the duration of ERK signaling. Nat Neurosci. 6:34–42.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
Lugo JN, Brewster AL, Spencer CM and
Anderson AE: Kv4.2 knockout mice have hippocampal-dependent
learning and memory deficits. Learn Mem. 19:182–189.
2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Owona BA, Zug C, Schluesener HJ and Zhang
ZY: Amelioration of behavioral impairments and neuropathology by
antiepileptic drug topiramate in a transgenic Alzheimer's disease
model mice, APP/PS1. Int J Mol Sci. 20(3003)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Huang P, Yang YH, Chang YH, Chang SL, Chou
MC, Lai CL, Liu CK and Chen HY: Association of early-onset
Alzheimer's disease with germline-generated high affinity
self-antigen load. Transl Psychiatry. 10(146)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bush AI and Tanzi RE: Therapeutics for
Alzheimer's disease based on the metal hypothesis.
Neurotherapeutics. 5:421–432. 2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kamceva M, Benedict J, Nairn AC and
Lombroso PJ: Role of striatal-enriched tyrosine phosphatase in
neuronal function. Neural Plast. 2016(8136925)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Huang L, Lan J, Tang J, Kang Y, Feng X, Wu
L and Peng Y: L-3-n-butylphthalide improves synaptic and dendritic
spine plasticity and ameliorates neurite pathology in Alzheimer's
disease mouse model and cultured hippocampal neurons. Mol
Neurobiol. 58:1260–1274. 2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhang Y, Huang LJ, Shi S, Xu SF, Wang XL
and Peng Y: L-3-n-butylphthalide rescues hippocampal synaptic
failure and attenuates neuropathology in aged APP/PS1 mouse model
of Alzheimer's disease. CNS Neurosci Ther. 22:979–987.
2016.PubMed/NCBI View Article : Google Scholar
|