Introduction
Sepsis is a systemic inflammatory response that is
driven by a variety of pathogenic microorganisms including bacteria
and their derived products such as endotoxins (1). Sepsis-mediated inflammation may lead
to organ dysfunction or circulatory disorders (2). Globally, it's estimated that there are
31.5 million sepsis patients and potentially 5.3 million
mortalities every year (3); 51.1%
of sepsis patients receive intensive care and ~10% of all intensive
care unit patients have severe sepsis in the United States
(4,5). Several biomarkers of sepsis have been
identified, including phase proteins and inflammatory cytokines
such as interleukin (IL)-1, 6 and 10, as well as tumor necrosis
factor (TNF)-α (6). Unfortunately,
the outcome of patients with sepsis following treatment is often
unsatisfactory, due to inflammatory cytokine secretion during the
progression of sepsis (7,8). Therefore, it is essential to figure
out novel biomarkers for an early diagnosis of sepsis, and thus to
develop pharmacological therapy for sepsis by blocking the
inflammatory cascade.
High-mobility group box 1 (HMGB1) is a highly
conserved non-histone DNA-binding protein (9). It has been documented that HMGB, an
extracellularly released mediator, can regulate the inflammatory
response (10). Typically, the
production of pro-inflammatory cytokines such as TNF-α and IL-1β is
induced immediately once inflammation spreads into the blood
stream, which then triggers HMGB1 expression (11). HMGB1 is known as a late mediator of
inflammation (11). Existing
evidence indicates that extracellular HMGB1 may exhibit
pro-inflammatory activity in the pathogenesis of various
inflammatory diseases (12). HMGB1
is secreted by injured cells and innate immune cells including
macrophages (10,13). Moreover, in response to LPS
stimulation, HMGB1 can translocate from the nuclei to the cytoplasm
in macrophages (14). In the
clinic, plasma HMGB1 levels are suggested to be positively
correlated with organ dysfunction and death in patients with sepsis
(15). Therefore, HMGB1 has been
regarded as a promising therapeutic target in inflammatory
diseases, including sepsis (16).
However, the precise mechanism of HMGB1 involvement in sepsis
remains unknown.
MicroRNAs (miRNAs/miRs) are endogenous non-coding
transcripts of ~22 nucleotides in length that hinder protein
translation through direct base pairing to a broad range of
biological systems in animal cells (17). miRNAs serve as essential regulators
in inflammatory cytokine release and immune responses (18). In sepsis, it has been highlighted
that numerous miRNAs are differentially expressed (19). Moreover, several miRNAs have been
identified to be diagnostic or prognostic biomarkers in sepsis,
including miR-25 and miR-150 (20-22).
miR-23a-3p has been reported to contribute to pathology in cancer,
cardiac hypertrophy and muscular atrophy (23). miR-23a-3p is observed to be
upregulated in brain tissue, leukocytes and blood plasma during
focal cerebral ischemia (24).
Additionally, it has been reported that the downregulation of
miR-23a-3p in inflammation is associated with TNF-α-induced insulin
resistance, LPS-induced immune activation of rat testis and
sepsis-induced acute kidney injury (25-27).
Furthermore, circulating plasma miRNAs including miR-23a-3p can
differentiate human sepsis and systemic inflammatory response
syndrome (SIRS) (1). Macrophage
activation is involved in the host immune response and inflammatory
response to sepsis (28), and,
thus, the role of miR-23a-3p in macrophage inflammation needs to be
elucidated.
Although the role of HMGB1 in sepsis is well-known,
the underlying regulatory mechanism has yet to be fully uncovered
(29). The present study aimed to
provide a novel insight into the targeted regulation of HMGB1
expression in sepsis by studying the role of miR-23a-3p in
LPS-activated RAW264.7 macrophage cells and investigating the
regulatory relationship between miR-23a-3p and HMGB1.
Materials and methods
Clinical specimens
A total of 20 patients (male:female, 12:8; age,
37-60 years) with severe sepsis were recruited from Jingzhou
Central Hospital of Hubei Province (Jingzhou, China) during July
2018-December 2018 in the present study, together with 12 healthy
controls (male:female, 7:5; age, 33-60 years). Peripheral venous
blood (4 ml) was collected from all participants. Sepsis was
defined according to the Sepsis-3 criteria (30). The excluding criteria for healthy
controls: Pregnant women, bone marrow or organ transplant
recipients, patients with cancer, individuals suffering with
neutropenia, leukopenia or acquired immune deficiency syndrome. The
present study was approved by the Ethics Committee of Jingzhou
Central Hospital of Hubei, and informed written consent was
obtained from all participants prior to study commencement.
Isolation of serum and peripheral
blood mononuclear cells (PBMCs)
Blood freshly collected in sodium citrate was used
for isolation. For serum preparation, 2 ml blood was kept at -4˚C
overnight, then centrifuged at 800 x g for 10 min at 4˚C. The
supernatant was collected as serum samples and stored at -80˚C
until further use. For PBMC isolation, 2 ml blood was centrifuged
at 400 x g for 30 min at 18˚C on Ficoll-Paque PREMIUM (GE
Healthcare; Cytiva) according to the manufacturer's instructions.
The PBMC layer was collected and washed, and the cell pellet was
re-suspended in Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1% antibiotics
(100 U/ml penicillin and 100 µg/ml streptomycin; Invitrogen; Thermo
Fisher Scientific, Inc.).
Cell culture and lipopolysaccharide
(LPS) treatment
The murine macrophage cell line RAW264.7 was
acquired from the American Type Culture Collection and cultured in
DMEM supplemented with 10% FBS at 37˚C in 5% CO2. To
induce inflammation in vitro, LPS (cat. no. L4516) was
purchased from Sigma-Aldrich (Merck KGaA). RAW264.7 cells were
exposed to 100 ng/ml LPS at 37˚C for 48 h prior to collection of
cells and supernatants and RAW264.7 cells without LPS treatment
served as the control.
Cell transfection
RAW264.7 cells were transferred to a six-well plate
(Corning, Inc.) and incubated overnight. The pcDNA3.1+
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to
overexpress HMGB1 via molecular cloning technology.
Oligonucleotides (30 nM) and pcDNA-HMGB1 plasmids (2 µg) were
transfected into RAW264.7 cells using Lipofectamine™ 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. GMR-miR™ miR-23a-3p mimic
(miR-23a-3p; 5'-AUCACAUUGCCAGGGAUUU-3'), miR-23a-3p inhibitor
(in-miR-23a-3p; 5'-AAAUCCCUGGCAAUGUGAU-3') and the indicated
negative controls (miR-NC; 5'-CAGUACUUUUGUGUAGUACAA-3') and
in-miR-NC (5'-UUGUACUACACAAAAGUACUG-3') were acquired from Shanghai
GenePharma Co., Ltd. Transfected cells were cultured for an
additional 48 h prior to further studies, among which transfected
cells were treated LPS (100 ng/ml at 37˚C for 48 h).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted with TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). For
examination of the mRNAs (including IL-6, TNF-α and HMGB1),
first-strand cDNA was synthesized using All-In-One 5X RT MasterMix
(Abcam), and the reaction thermal profile was 37˚C for 15 min, 60˚C
for 10 min and 95˚C for 3 min. For examination of the miRs, a
miScript Reverse Transcription kit (Qiagen GmbH) was used, and the
reaction thermal profile was 37˚C for 60 min, and 95˚C for 5 min.
To determine the levels of mRNAs and mature miR-23a-3p, qPCR was
performed using SYBR Green qPCR Mix (Abcam) and miScript SYBR Green
PCR Kit (Qiagen GmbH), respectively on an ABI PRISM 7500 Real-time
PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The thermocycling conditions were 95˚C for 15 min, and 35 cycles of
94˚C for 15 sec, 55˚C for 30 sec and 70˚C for 30 sec. GAPDH and U6
small nuclear RNA were used as internal controls for mRNA and
miRNA, respectively. We chose 2-ΔΔCq method for
evaluation of data (31). The
reactions were performed in triplicate for each sample. The primers
involved were listed as follows: IL-6 forward (F),
5'-ACGGCCTTCCCTACTTC-3', and reverse (R),
5'-GCTGGACTGTTTCTAATGC-3'; TNF-α F, 5'-GGGTGTTCATCCATTCTC-3', and
R, 5'-GGAAAGCCCATTTGAGT-3'; HMGB1 F, 5'-GGAGTGGCTTTTGTCCCTCAT-3',
and R, 5'-TGCCTCTCGGCTTTTTAGGA-3'; GAPDH F,
5'-GGTTGTCTCCTGCGACTTCA-3', and R, 5'-GGTGGTCCAGGGTTTCTTACT-3'; U6
F, 5'-CTCGCTTCGGCAGCACA-3', and R, 5'-AACGCTTCACGAATTTGCGT-3'.
These primer sequences were supported by a previous literature
(32). Primers for hsa-miR-23a-3p
and mmu-miR-23a-3p were purchased from Exiqon A/S (Qiagen AB).
Western blotting
Total protein was extracted in RIPA lysis buffer
(Beyotime Institute of Biotechnology). After determining the
protein concentration using a Bradford protein assay (Bio-Rad
Laboratories, Inc.), 20 µg samples were loaded for the standard
procedures of western blotting. In brief, 15% sodium dodecyl
sulfate polyacrylamide gel electrophoresis gel and polyvinylidene
fluoride membrane (MilliporeSigma) were used, and the blocking was
performed in 5% skim milk for 1 h at 25˚C. β-actin served as a
loading control to normalize protein levels. Primary antibodies
including HMGB1 (cat. no. ab92310; 1:2,000; Abcam) and β-actin
(cat. no. ab179467; 1:10,000; Abcam) were incubated overnight at
4˚C, and secondary antibody goat anti-rabbit IgG H&L
(HRP-conjugated; cat. no. ab6721; 1:20,000; Abcam) was incubated at
25˚C for 1 h. Then, protein blot signals were detected by enhanced
chemiluminescence (MilliporeSigma) and quantified using Image Lab™
v3.0 Software (Bio-Rad Laboratories, Inc.).
ELISA
The protein levels of IL-6 and TNF-α in the culture
supernatants or serum were measured using mouse IL-6 ELISA kit
(cat. no. EM004-48; Genetimes Technology, Inc.) and mouse TNF-α
ELISA kit (cat. no. EM008-96; Genetimes Technology, Inc.) according
to the instructions of the manufacturer. The serum supernatant was
obtained from whole bloods after clot formation and culture
supernatant was collected from cell culture media after LPS
treatment by centrifuging at 800 x g for 10 min at 4˚C. The
reactions were repeated for three times for each sample.
Dual luciferase reporter assay
DianaTools (Diana Lab, University of Thessaly,
Thessaly, Greece) with microT-CDS algorithm (33) determined that there was a potential
target binding site for miR-23a-3p in the 3'untranslated region
(3'UTR) of HMGB1. For determination, the fragment of the HMGB1
3'UTR containing the putative sequence (positions 649-669) was
mutated by replacing the AAU…GC…UGUGAU of the complementary
sequence. Then, the wild type and mutant of HMGB1 3'UTR (HMGB1
WT/MUT) were cloned into a pGL3 vector (Invitrogen; Thermo Fisher
Scientific, Inc.). RAW264.7 cells were co-transfected with HMGB1
WT/MUT (2 µg) and miR-23a-3p/NC mimic (30 nM), or co-transfected
with HMGB1 WT/MUT (2 µg) and in-miR-23a-3p/NC (30 nM) using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.).
pRL-TK plasmids were used as an internal control and co-transfected
with those at a dose of 50 ng. Every transfection group was carried
out in triplicate. After 48 h, transfected cells were collected to
measure Firefly and Renilla luciferase activity using
a dual-luciferase reporter assay system (Promega Corporation), and
relative luciferase activity was the ratio of Firefly and
Renilla.
Statistical analysis
Data are presented as the mean ± standard error of
mean. Statistical analyses were performed using GraphPad Prism 5.0
(GraphPad Software, Inc.). Differences between groups were
evaluated using one-way analysis of variance followed by Tukey's
post hoc test, and P<0.05 was considered to indicate a
statistically significant difference.
Results
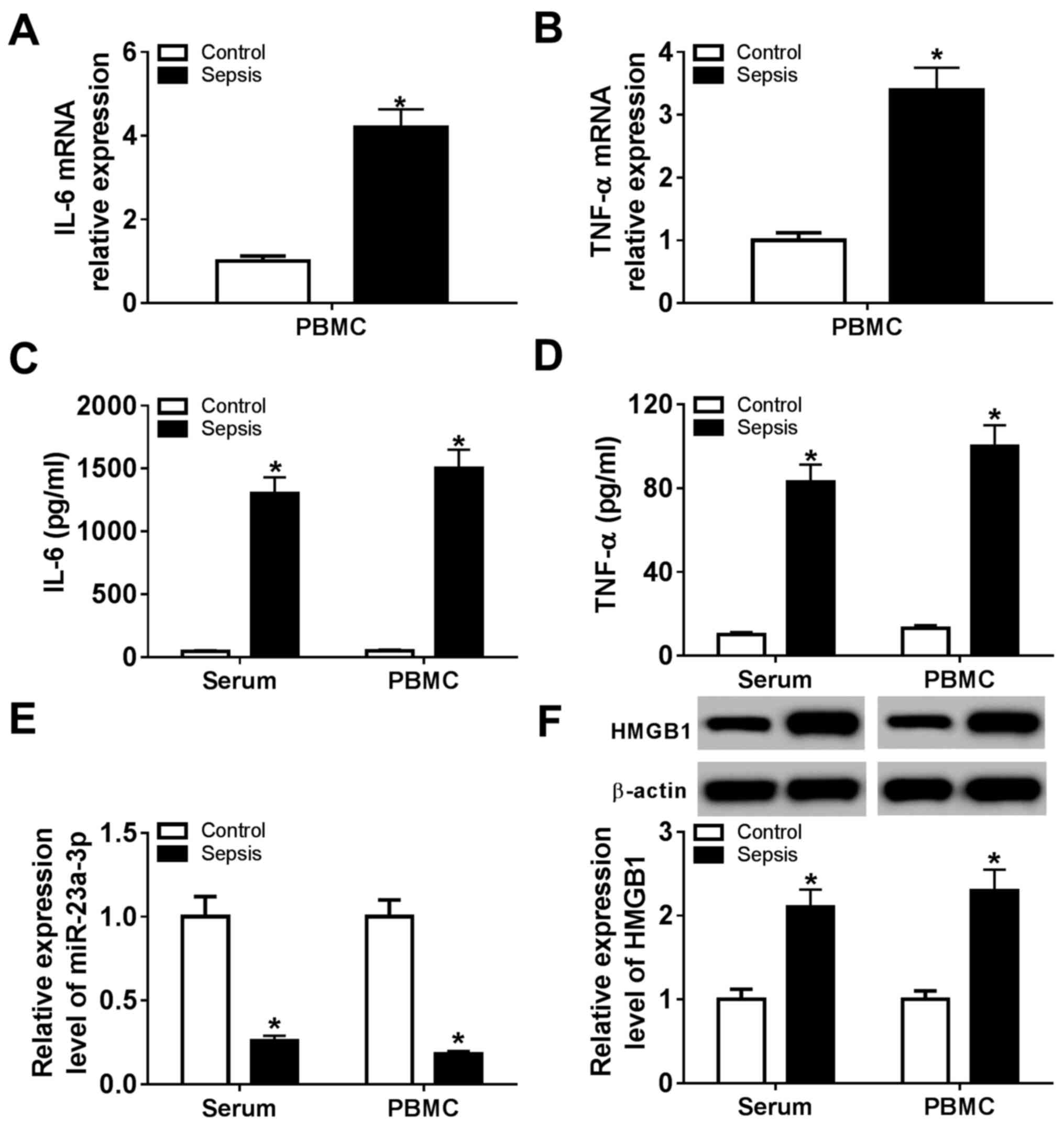
Expression of HMGB1 is increased and
miR-23a-3p is downregulated in patients with sepsis
Whole blood from healthy volunteers (control; n=12)
and patients with sepsis (sepsis; n=20) was collected, and serum
and PBMCs were subsequently isolated. Expression levels of
miR-23a-3p and HMGB1 were measured, as were the levels of
pro-inflammatory cytokines. RT-qPCR analysis demonstrated that mRNA
expression of two critical pro-inflammatory cytokines, IL-6 and
TNF-α, was significantly elevated in sepsis group PBMCs compared
with the control group (Fig. 1A and
B). In addition, ELISA data showed
that the secretion of IL-6 and TNF-α was significantly higher in
serum and PBMCs from the sepsis group compared with the control
group (Fig. 1C and D). In patients with sepsis, expression of
miR-23a-3p was significantly downregulated (~0.22-fold) and the
level of HMGB1 protein was significantly upregulated (~2.15-fold)
in the serum and PBMCs (Fig. 1E and
F) compared with levels in
controls. These results suggested an inhibition of miR-23a-3p, and
a promotion of HMGB1, IL-6 and TNF-α expression in patients with
sepsis, indicating a potential role for miR-23a-3p in the
inflammatory response during sepsis.
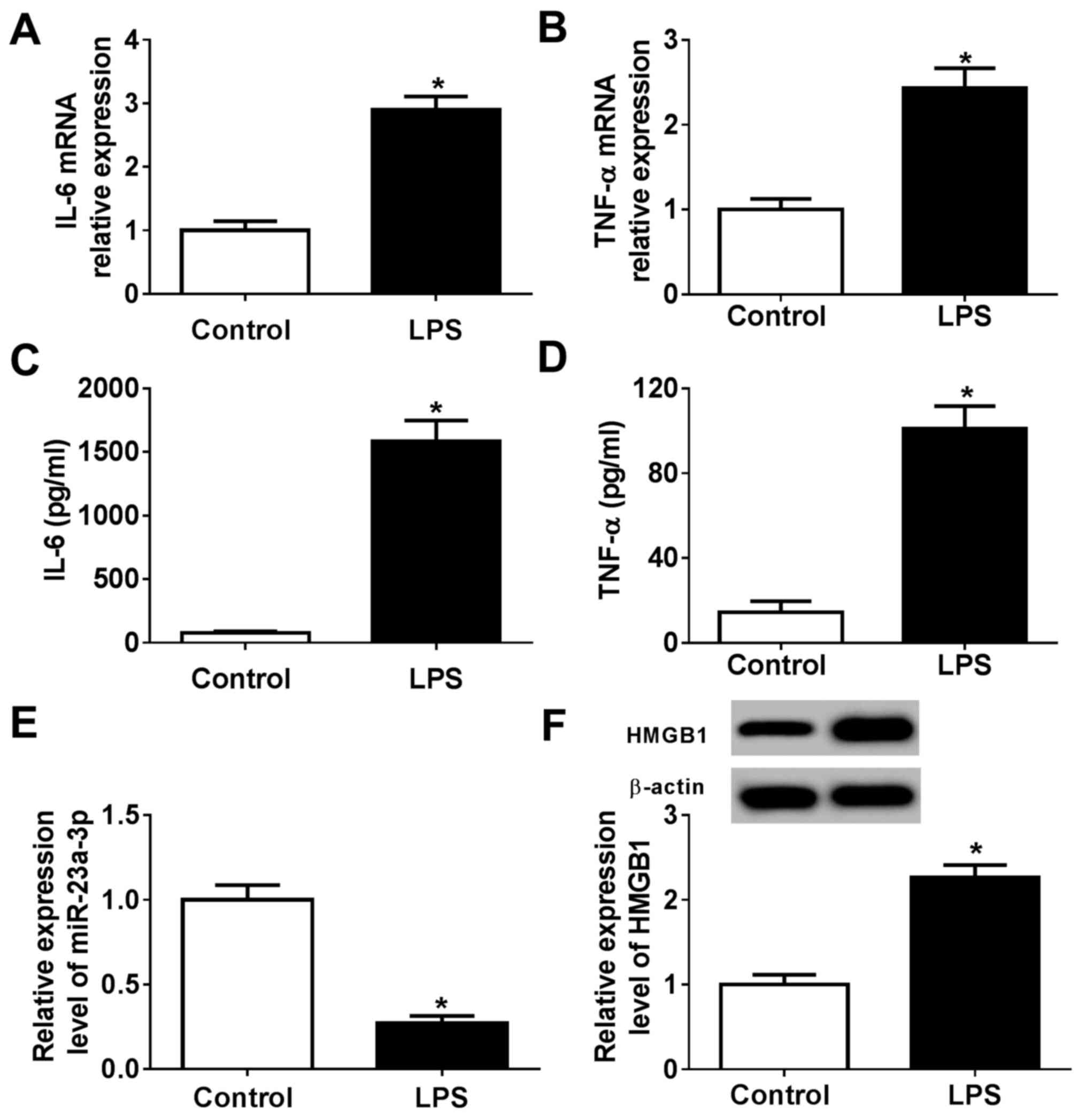
miR-23a-3p and HMGB1 are
differentially expressed following LPS treatment in murine
macrophage cells
To identify the biological function of miR-23a-3p
and HMGB1 in innate immunity, a cell model of sepsis was
constructed using RAW264.7 cells stimulated by LPS. The levels of
pro-inflammatory cytokines released during LPS treatment were
determined compared with control cells without LPS treatment.
RT-qPCR data showed that LPS induced the expression of IL-6 and
TNF-α at the mRNA level compared with the control (Fig. 2A and B), accompanied with increased secretion of
these cytokines into the culture supernatants (Fig. 2C and D). These findings indicated the successful
construction of a LPS-induced inflammation model in murine
macrophages in vitro. RT-qPCR and western blotting
determined that the expression of miR-23a-3p was significantly
reduced, and that of HMGB1 protein was increased, in LPS-stimulated
RAW264.7 cells compared with control cells without LPS treatment
(Fig. 2E and F). These results suggested that LPS
stimulation induced an inflammatory response comparable to sepsis
in RAW264.7 cells, during which miR-23a-3p was downregulated and
HMGB1 was upregulated.
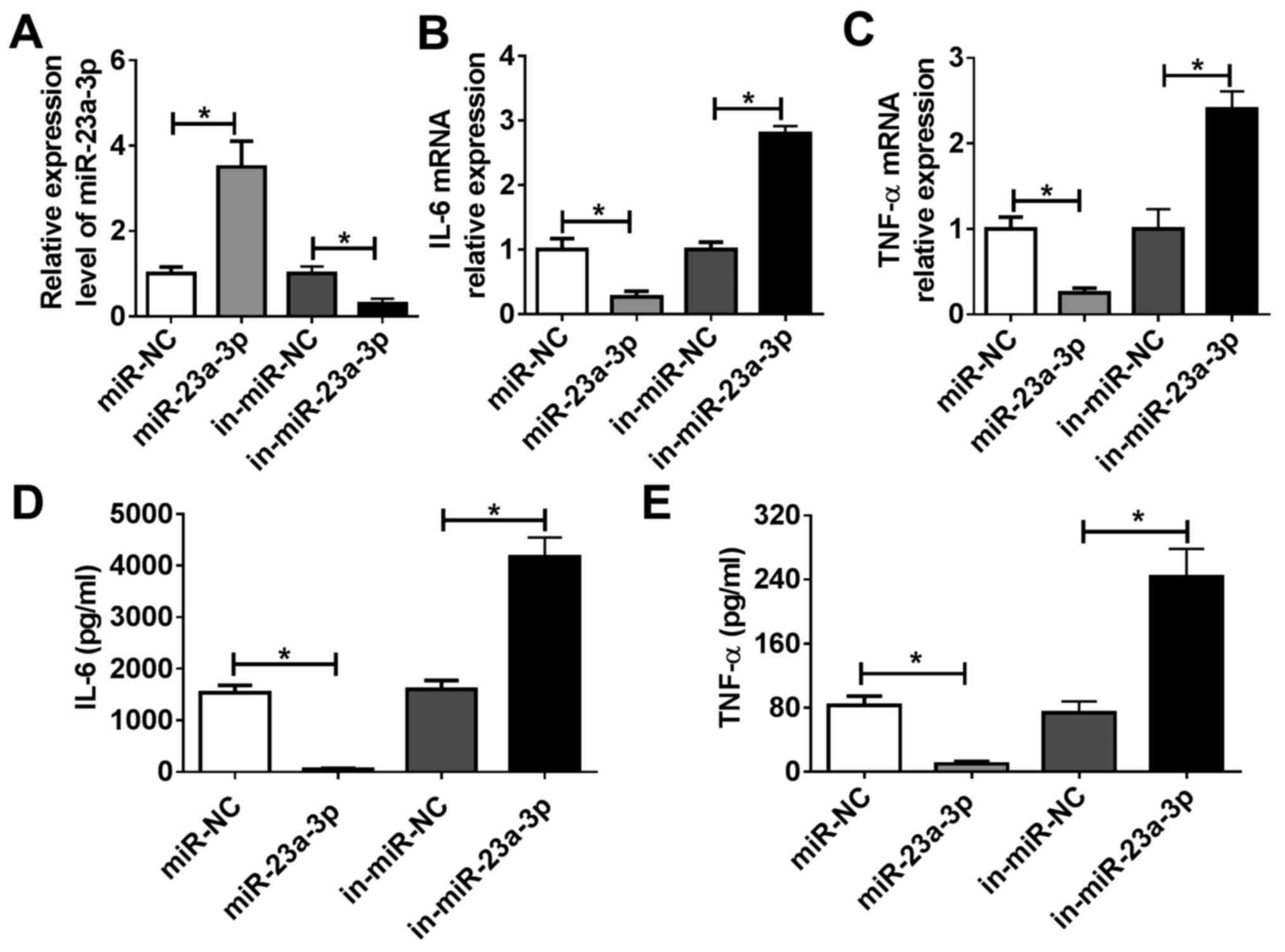
miR-23a-3p modulates LPS-induced
inflammation in murine macrophage cells in vitro
RAW264.7 cells were transfected with miR-23a-3p
mimic to overexpress miR-23a-3p, and they were transfected with
in-miR-23a-3p to silence miR-23a-3p. Efficiency of transfection was
evaluated via RT-qPCR. miR-23a-3p expression was significantly
increased following treatment with miR-23a-3p mimic, whereas it was
inhibited by transfection of in-miR-23a-3p, compared with the
corresponding NCs (Fig. 3A).
RT-qPCR and ELISA were used to analyze LPS-induced TNF-α and IL-6
expression. As indicated by Fig. 3B
and C, overexpression of miR-23a-3p
reduced IL-6 and TNF-α mRNA expression levels in RAW264.7 cells
under LPS stimulation compared with control cells, whereas
silencing of miR-23a-3p had the opposite effect. Moreover, the
release of IL-6 and TNF-α into culture supernatants was decreased
by miR-23a-3p overexpression, but promoted by miR-23a-3p inhibition
(Fig. 3D and E). These data suggested miR-23a-3p may
inhibit LPS-induced inflammation in murine macrophages in
vitro.
miR-23a-3p physically targets HMGB1
via complementary binding
DianaTools in silico data predicted that
there were complementary binding sites of miR-23a-3p in HMGB1 3'UTR
at position 649-669. As presented in Fig. 4A, a HMGB1 MUT was constructed. A
dual luciferase reporter assay showed that miR-23a-3p mimic
transfection induced a significant decrease in the luciferase
activity of HMGB1 WT in RAW264.7 cells, whereas in-miR-23a-3p
transfection caused a significant increase (Fig. 4B and C). These results suggested a direct
targeting relationship between HMGB1 and miR-23a-3p. RT-qPCR and
western blotting data demonstrated that expression of HMGB1 at both
the mRNA and the protein level was significantly inhibited in
RAW264.7 cells transfected with miR-23a-3p, but it was promoted
following in-miR-23a-3p transfection (Fig. 4D and E). These findings indicated that
miR-23a-3p modulated LPS-induced inflammation in murine macrophages
by targeting HMGB1.
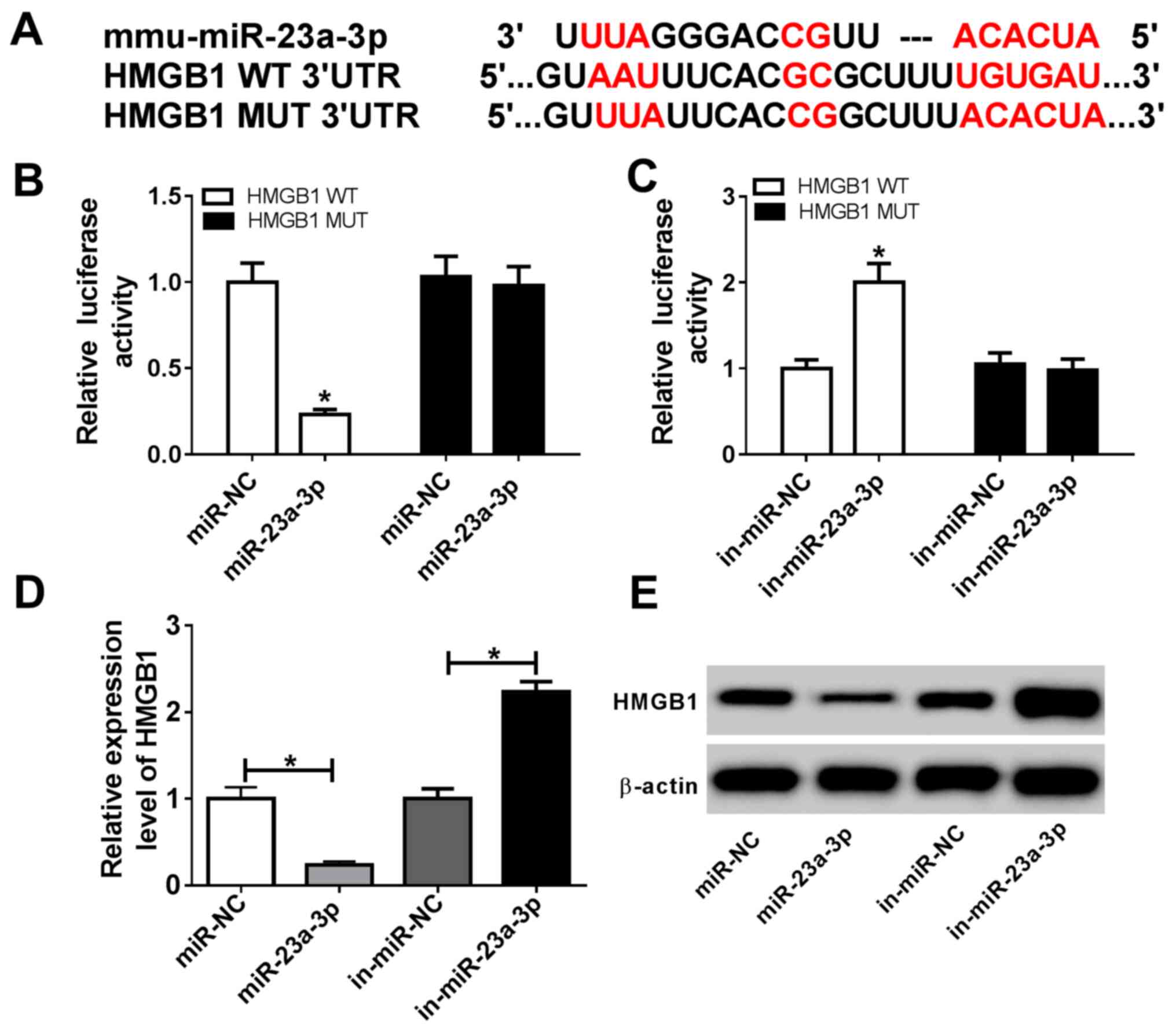 | Figure 4miR-23a-3p targets the 3'UTR of HMGB1
in murine macrophage cells. (A) DianaTools revealed ae binding site
between miR-23a-3p and HMGB1 WT 3'UTR. Dual-luciferase reporter
assays investigating the luciferase activity of vectors carrying
HMGB1 WT 3'UTR or HMGB1 MUT 3'UTR in RAW264.7 cells transfected
with (B) miR-23a-3p or (C) in-miR-23a-3p. (D) Reverse
transcription-quantitative PCR and (E) western blot analyses of
HMGB1 expression in RAW264.7 cells transfected with miR-23a-3p,
miR-NC, in-miR-23a-3p or in-miR-NC. *P<0.05 vs.
miR-NC, in-miR-NC or as indicated. 3'UTR, 3'untranslated region;
HMGB1, high mobility group box 1; in-miR, miR inhibitor;
miR-23a-3p, microRNA-23a-3p; MUT, mutant; NC, negative control; WT,
wild type. |
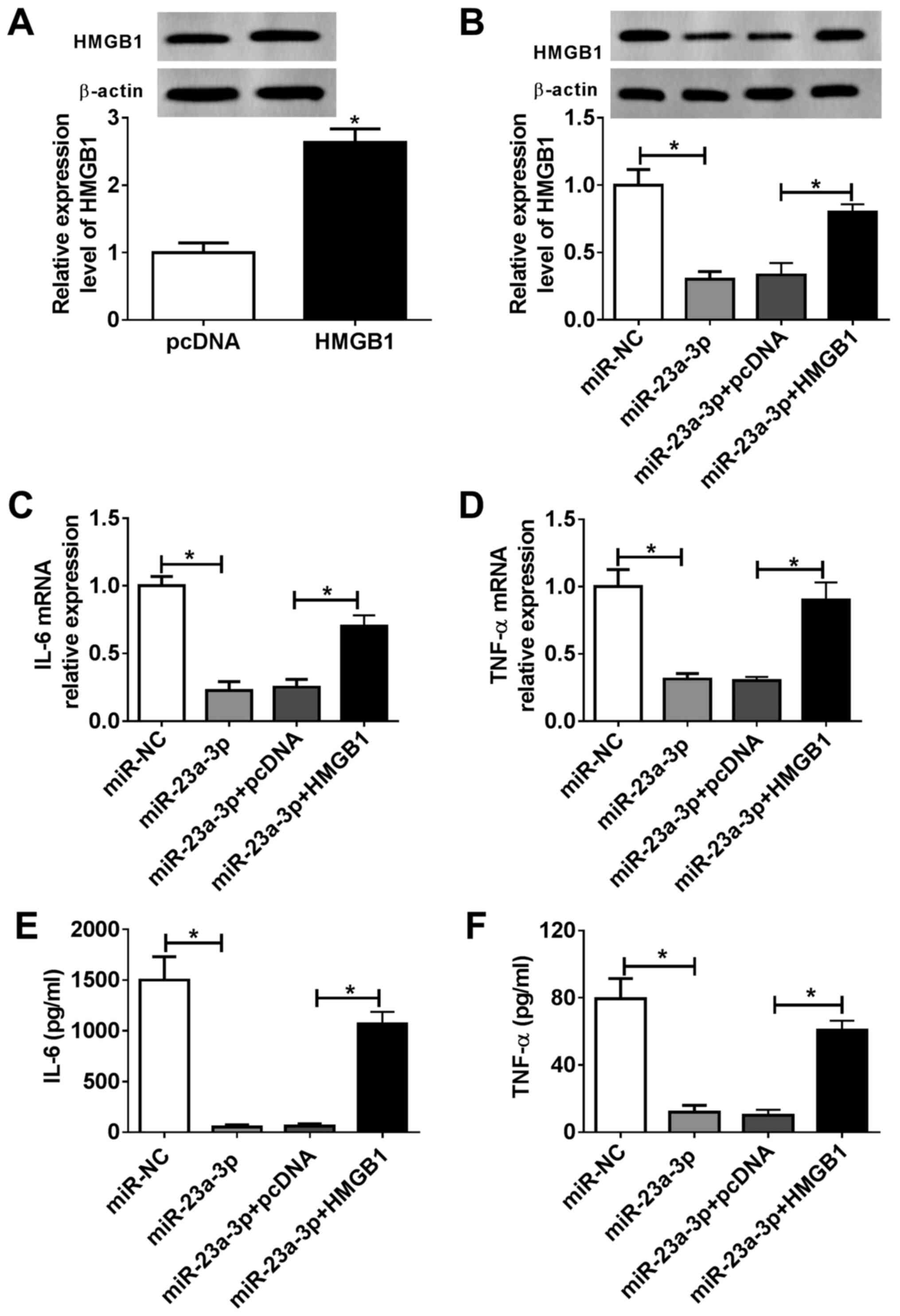
Overexpression of HMGB1 attenuates the
effects of miR-23a-3p overexpression in LPS-induced inflammation in
vitro
RAW264.7 cells were divided into four transfection
groups: miR-NC, miR-23a-3p, miR-23a-3p + pcDNA and miR-23a-3p +
pcDNA-HMGB1. The expression of HMGB1 in RAW264.7 cells was
evaluated via western blotting, and HMGB1 protein expression was
significantly increased following pcDNA-HMGB1 transfection compared
with the empty vector control (Fig.
5A). Furthermore, miR-23a-3p transfection inhibited HMGB1 in
RAW264.7 cells, and this inhibition was attenuated following
pcDNA-HMGB1 transfection (Fig. 5B).
As presented in Fig. 5C and
D, overexpression of miR-23a-3p
reduced IL-6 and TNF-α mRNA expression levels in RAW264.7 cells
under LPS stimulation compared with the NC, but overexpression of
HMGB1 reversed this effect. Moreover, the release of IL-6 and TNF-α
into the culture supernatant was suppressed by miR-23a-3p, which
was subsequently reversed by HMGB1 restoration (Fig. 5E and F). These results suggested that the
suppressive effect of miR-23a-3p overexpression on LPS-induced
inflammation in murine macrophages was partially mediated via
downregulation of HMGB1.
Discussion
Several preclinical studies in lethal sepsis have
suggested that HMGB1 may be a promising target for improving
therapeutic outcomes (34,35). In the present study, an increase of
HMGB1 in patients with sepsis and LPS-challenged macrophages was
observed alongside decreased expression of miR-23a-3p. In addition,
overexpression of miR-23a-3p reduced, whereas silencing of
miR-23a-3p elevated, pro-inflammatory cytokine expression (IL-6 and
TNF-α) in RAW264.7 cells under LPS stimulation, and the inhibitory
effect of miR-23a-3p overexpression on LPS-induced inflammation was
attenuated following HMGB1 upregulation. Of note, HMGB1 was
targeted by miR-23a-3p. These findings indicated a novel mechanism
of HMGB1 in sepsis regulated by miR-23a-3p.
HMGB1 is a member of the HMGB family, which
contributes to the regulation of gene expression (14,32).
However, in response to LPS stimulation, HMGB1 is translocated from
the nuclei to the cytoplasm of macrophages (14). Moreover, superfluous HMGB1 has been
demonstrated to be secreted by macrophages and monocytes, thus
participating in the occurrence of sepsis (36,37).
In sepsis studies, Wang et al (9) indicated that HMGB1 and LPS in harmful
concentrations were synergistically toxic or lethal. Additionally,
it was reported that HMGB1 has a role in LPS-induced cytotoxicity
for reasons that have not been fully elucidated (16,38).
Deng et al (39) discovered
that hepatocyte-released HMGB1 could bind and target LPS into the
lysosomes of macrophages and endothelial cells. Therefore, HMGB1
enabled LPS to reach cytosolic caspase-11, thus forming multiple
critical inflammatory mediators (40,41).
In the present study, miR-23a-3p mimic-mediated upregulation of
HMGB1 attenuated the expression of IL-6 and TNF-α in LPS-stimulated
murine macrophages. Overexpression of HMGB1 promoted the production
of IL-6 and TNF-α. Collectively, HMGB1 could be important for LPS
to express its cytotoxicity.
A growing number of studies have indicated that
multiple miRNAs are involved in the inflammatory response of
macrophages by regulating HMGB1 expression. For example, miR-212-3p
has been claimed to inhibit inflammatory responses in LPS-treated
RAW264.7 cells by targeting HMGB1(32). Zhou et al (29) demonstrated that HMGB1 was regulated
by a handful of miRNAs, and that miR-205-5b expression was
negatively associated with HMGB1 expression. Peroxisome
proliferator-activated receptor γ suppresses inflammatory gene
expression and pro-inflammatory transcription-factor signaling
pathways in various cell types, and its agonist troglitazone
mediates HMGB1 inhibition, which is associated with the
upregulation of miR-142-3p in inflammatory responses in
vitro and in vivo (42).
In the present study, it was observed that miR-23a-3p was
downregulated and inversely expressed with HMGB1 in LPS-induced
RAW264.7 cells; overexpression of miR-23a-3p reduced the expression
of IL-6, TNF-α and HMGB1 both at the mRNA and protein level by
targeting HMGB1. These data indicated the protective effect of
miR-23a-3p in sepsis, and suggested that miR-23a-3p may act as a
novel negative regulator of macrophage inflammation. Unfortunately,
the role of miR-23a-3p in inflammation-related signal pathways,
such as the MAPK (32),
JAK/STAT1(43) and NF-κB (44) pathways, was not further investigated
in the present study. Animal experiments with miR-23a-3p should be
performed for further investigation of the expression of IL-6,
TNF-α and HMGB1 in serum and organs, including the liver, lung and
kidney (29). In addition,
miR-23a-3p together miRNAs were significantly decreased in both
sepsis-induced acute kidney injury (AKI) and other AKI groups, and
potential target genes of miR-23a-3p were predicted, including
IL-6(27). In the present study, it
was suggested that IL-6 could be directly and indirectly regulated
by miR-23a-3p in sepsis cell models. However, the target
relationship between miR-23a-3p and IL-6 in LPS-induced RAW264.7
cells still requires further investigation.
miR-23a-3p functions are extensive. Accumulating
evidence indicates that miR-23a-3p is involved in multiple
diseases, including cancer, ischemia injury and inflammation. For
example, in renal cell carcinoma (RCC), miR-23a-3p targeted
proline-rich nuclear receptor coactivator 2 to act as an oncogene
by enhancing cell proliferation and cell mobility, and inhibiting
apoptosis in ACHN and 786-O cells, thus being a potential
prognostic biomarker for RCC (45).
miR-23a-3p, together with 8 other miRNAs, was observed to exhibit
increased expression in brain tissue, leukocytes and blood plasma
48 h after onset of photochemically-induced focal cerebral ischemia
(24). Moreover, it was also
reported that oxidative stress injury was alleviated in a mouse
model of focal cerebral ischemia-reperfusion (46). Several studies indicated the
downregulation of miR-23a-3p under inflammation. For example,
inflammation response affected the miRNA profile of the male
reproductive tract; five miRNAs, including miR-23a-3p, let-7f-5p,
miR-200c-3p, miR-23b-3p and miR-98-5p, exhibited >2-fold
downregulation after intraperitoneal injection of LPS in rats for 3
h (26). Serum miR-23a-3p was lower
in sepsis-induced human AKI, as well as miR-4456, miR-142-5p,
miR-22-3p and miR-191-5p (27).
Furthermore, 20 circulating inflammation-associated miRNAs were
downregulated in sepsis compared with SIRS, and miR-23a-3p was one
of the top 6 most differentially expressed miRNAs in severe sepsis
(1). However, there remained a lack
of detailed information upon the dysregulation of miR-23a-3p and
its molecular regulatory mechanism. Therefore, the expression of
miR-23a-3p in sepsis was investigated. The results showed
miR-23a-3p was downregulated to ~0.22-fold in patients with sepsis
and an LPS-induced cell model of sepsis. Functionally, upregulation
of miR-23a-3p resulted in the inhibitory influence on inflammation
with decreased expression of IL-6 and TNF-α via targeting
HMGB1.
The present study provided novel insight into
regulation of HMGB1 by miR-23a-3p, and the miR-23a-3p/HMGB1 axis
may represent a clinically relevant potential pharmacological
target for effective therapeutic intervention in sepsis.
Nevertheless, it should also be noted that targeted therapies,
including those involving monoclonal antibodies or antagonists,
could be limited, due to redundancy in the inflammatory response
(47).
Collectively, it was demonstrated that miR-23a-3p
negatively regulated LPS-induced inflammatory cytokine secretion in
murine macrophages in vitro. Additionally, a novel mechanism
for HMGB1 in sepsis was uncovered that was mediated by miR-23a-3p.
Functional experiments suggested an inhibitory effect of miR-23a-3p
on inflammatory cytokine expression via direct downregulation of
HMGB1.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QS and BW analyzed and interpreted the patient data,
performed all experiments and wrote the first draft. ML designed
the experiments, agreed to be accountable for all aspects of the
work, revised this manuscript critically and gave the final
approval of the version to be published. All authors read and
approved the final manuscript. QS and BW confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Ethical approval was granted by the Ethics Committee
of Jingzhou Central Hospital of Hubei. Participants provided their
written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Caserta S, Kern F, Cohen J, Drage S,
Newbury SF and Llewelyn MJ: Circulating plasma microRNAs can
differentiate human sepsis and systemic inflammatory response
syndrome (SIRS). Sci Rep. 6(28006)2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tracey KJ: The inflammatory reflex.
Nature. 420:853–859. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fleischmann C, Scherag A, Adhikari NK,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K:
International Forum of Acute Care Trialists. Assessment of global
incidence and mortality of Hospital-treated sepsis. Am J Respir
Crit Care Mede. 193:259–272. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Angus DC, Linde-Zwirble WT, Lidicker J,
Clermont G, Carcillo J and Pinsky MR: Epidemiology of severe sepsis
in the United States: Analysis of incidence, outcome, and
associated costs of care. Crit Care Med. 29:1303–1310.
2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cho W, Koo JY, Park Y, Oh K, Lee S, Song
JS, Bae MA, Lim D, Lee DS and Park SB: Treatment of sepsis
pathogenesis with high mobility group box protein 1-regulating
anti-inflammatory agents. J Med Chem. 60:170–179. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Adib-Conquy M and Cavaillon JM: Stress
molecules in sepsis and systemic inflammatory response syndrome.
FEBS Lett. 581:3723–3733. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lyle NH, Pena OM, Boyd JH and Hancock RE:
Barriers to the effective treatment of sepsis: Antimicrobial
agents, sepsis definitions, and host-directed therapies. Ann N Y
Acad Sci. 1323:101–114. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Silman NJ: Rapid diagnosis of sepsis using
biomarker signatures. Crit Care. 17(1020)2013.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gonelevue S, Bandyopadhyay A, Bhagat S,
Alam MI and Khan GA: Sterile inflammatory role of high mobility
group Box 1 protein: Biological functions and involvement in
disease. J Vasc Res. 55:244–254. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao
L, Huang J, Yu Y, Fan XG, Yan Z, et al: HMGB1 in health and
disease. Mol Aspects Med. 40:1–116. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Magna M and Pisetsky DS: The role of HMGB1
in the pathogenesis of inflammatory and autoimmune diseases. Mol
Med. 20:138–146. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ulloa L and Tracey KJ: The ʻCytokine
Profileʼ: A code for sepsis. Trends Mol Med. 11:56–63.
2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang H, Yang H and Tracey KJ:
Extracellular role of HMGB1 in inflammation and sepsis. J Intern
Med. 255:320–331. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gibot S, Massin F, Cravoisy A, Barraud D,
Nace L, Levy B and Bollaert PE: High-mobility group box 1 protein
plasma concentrations during septic shock. Intensive Care Med.
33:1347–1353. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Andersson U and Tracey KJ: HMGB1 is a
therapeutic target for sterile inflammation and infection. Annu Rev
Immunol. 29:139–162. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kingsley SMK and Bhat BV: Role of
microRNAs in sepsis. Inflamm Res. 66:553–569. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bulun SE and Nezhat C: Aromatase,
microRNA, and inflammation: A complex relationship. Fertil Steril.
106:552–553. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Essandoh K and Fan GC: Role of
extracellular and intracellular microRNAs in sepsis. Biochim
Biophys Acta. 1842:2155–2162. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yao Y, Sun F and Lei M: miR-25 inhibits
sepsis-induced cardiomyocyte apoptosis by targetting PTEN. Biosci
Rep. 38(BSR20171511)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ma Y, Liu Y, Hou H, Yao Y and Meng H:
MiR-150 predicts survival in patients with sepsis and inhibits
LPS-induced inflammatory factors and apoptosis by targeting NF-κB1
in human umbilical vein endothelial cells. Biochem Biophys Res
Commun. 500:828–837. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Benz F, Roy S, Trautwein C, Roderburg C
and Luedde T: Circulating MicroRNAs as biomarkers for sepsis. Int J
Mol Sci. 17(78)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chhabra R, Dubey R and Saini N:
Cooperative and individualistic functions of the microRNAs in the
miR-23a~27a~24-2 cluster and its implication in human diseases. Mol
Cancer. 9(232)2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gusar VA, Timofeeva AV, Zhanin IS, Shram
SI and Pinelis VG: Estimation of Time-dependent microRNA expression
patterns in brain tissue, leukocytes, and blood plasma of rats
under photochemically induced focal cerebral ischemia. Mol Biol
(Mosk). 51:683–695. 2017.PubMed/NCBI View Article : Google Scholar : (In Russian).
|
|
25
|
Lozano-Bartolomé J, Llauradó G,
Portero-Otin M, Altuna-Coy A, Rojo-Martinez G, Vendrell J, Jorba R,
Rodríguez-Gallego E and Chacón MR: Altered expression of
miR-181a-5p and miR-23a-3p is associated with obesity and
TNFα-induced insulin resistance. J Clin Endocrinol Metab.
103:1447–1458. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Parker MI and Palladino MA: MicroRNAs
downregulated following immune activation of rat testis. Am J
Reprod Immunol: 77, 2017 doi: 10.1111/aji.12673.
|
|
27
|
Ge QM, Huang CM, Zhu XY, Bian F and Pan
SM: Differentially expressed miRNAs in sepsis-induced acute kidney
injury target oxidative stress and mitochondrial dysfunction
pathways. PLoS One. 12(e0173292)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chen X, Liu Y, Gao Y, Shou S and Chai Y:
The roles of macrophage polarization in the host immune response to
sepsis. Int Immunopharmacoly. 96(107791)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhou W, Wang J, Li Z, Li J and Sang M:
MicroRNA-2055b inhibits HMGB1 expression in LPS-induced sepsis. Int
J Mol Med. 38:312–318. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chen W, Ma X, Zhang P, Li Q, Liang X and
Liu J: miR-212-3p inhibits LPS-induced inflammatory response
through targeting HMGB1 in murine macrophages. Exp Cell Res.
350:318–326. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Paraskevopoulou MD, Georgakilas G,
Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C,
Dalamagas T and Hatzigeorgiou AG: DIANA-microT web server v5.0:
Service integration into miRNA functional analysis workflows.
Nucleic Acids Res. 41:W169–W173. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Stevens NE, Chapman MJ, Fraser CK, Kuchel
TR, Hayball JD and Diener KR: Therapeutic targeting of HMGB1 during
experimental sepsis modulates the inflammatory cytokine profile to
one associated with improved clinical outcomes. Sci Rep.
7(5850)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yang H, Ochani M, Li J, Qiang X, Tanovic
M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, et al:
Reversing established sepsis with antagonists of endogenous
high-mobility group box 1. Proc Natl Acad Sci USA. 101:296–301.
2004.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Charoensup J, Sermswan RW, Paeyao A,
Promakhejohn S, Punasee S, Chularari C, Krabkraikaew S,
Lertanekawattana S and Wongratanacheewin S: High HMGB1 level is
associated with poor outcome of septicemic melioidosis. Int J
Infect Dis. 28:111–116. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Guo ZS, Liu Z, Bartlett DL, Tang D and
Lotze MT: Life after death: Targeting high mobility group box 1 in
emergent cancer therapies. Am J Cancer Res. 3:1–20. 2013.PubMed/NCBI
|
|
38
|
Andersson U, Yang H and Harris H:
Extracellular HMGB1 as a therapeutic target in inflammatory
diseases. Expert Opin Ther Targets. 22:263–277. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Deng M, Tang Y, Li W, Wang X, Zhang R,
Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al: The endotoxin
delivery protein HMGB1 mediates Caspase-11-dependent lethality in
sepsis. Immunity. 49:740–753.e7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kim HM and Kim YM: HMGB1: LPS delivery
vehicle for caspase-11-mediated pyroptosis. Immunity. 49:582–584.
2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wu D, Pan P, Su X, Zhang L, Qin Q, Tan H,
Huang L and Li Y: Interferon regulatory factor-1 mediates alveolar
macrophage pyroptosis during LPS-induced acute lung injury in mice.
Shock. 46:329–338. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yuan Z, Luo G, Li X, Chen J, Wu J and Peng
Y: PPARγ inhibits HMGB1 expression through upregulation of
miR-142-3p in vitro and in vivo. Cell Signal. 28:158–164.
2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Park EJ, Kim YM, Kim HJ and Chang KC:
Degradation of histone deacetylase 4 via the TLR4/JAK/STAT1
signaling pathway promotes the acetylation of high mobility group
box 1 (HMGB1) in lipopolysaccharide-activated macrophages. FEBS
Open Bio. 8:1119–1126. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Lee W, Ku SK and Bae JS: Zingerone reduces
HMGB1-mediated septic responses and improves survival in septic
mice. Toxicol Appl Pharmacol. 329:202–211. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Quan J, Pan X, Li Y, Hu Y, Tao L, Li Z,
Zhao L, Wang J, Li H, Lai Y, et al: miR-23a-3p acts as an oncogene
and potential prognostic biomarker by targeting PNRC2 in RCC.
Biomed Pharmacother. 110:656–666. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhao H, Tao Z, Wang R, Liu P, Yan F, Li J,
Zhang C, Ji X and Luo Y: MicroRNA-23a-3p attenuates oxidative
stress injury in a mouse model of focal cerebral
ischemia-reperfusion. Brain Res. 1592:65–72. 2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Minnich DJ and Moldawer LL: Anti-cytokine
and anti-inflammatory therapies for the treatment of severe sepsis:
Progress and pitfalls. Proc Nutr Soc. 63:437–441. 2004.PubMed/NCBI View Article : Google Scholar
|