Introduction
Overall mortality and morbidity after cardiac arrest
(CA) remain high, despite improvements in resuscitation and
critical care. Neurocognitive disabilities are frequently observed
in survivors from CA. Histological damage including neuronal cell
loss was characterized in multiple experimental global
ischemia-reperfusion insults (1).
Several selectively vulnerable regions have been identified, namely
hippocampus, cerebellar Purkinje neurons, lamina IV cortical layer
and striatum. While early ischemic brain injury is the result of
energy failure, neuro-inflammation could contribute, or even
represent a major cause of delayed neuronal death. Tumor necrosis
factor alpha (TNFα) is a pivotal cytokine that can induce neuronal
apoptosis and/or necroptosis, and increase neuroinflammation
(2-4).
CA also triggers a sepsis-like inflammatory response
with expression of TNFα up-regulated in cerebral ischemia. Systemic
selective anti-TNFα therapies improved early recovery from CA, in
both small and large animal models (4,5). Our
prior studies in multiple models of CA in rats, identified a unique
early cytokine response specifically in striatum, showing a
dramatic >100-fold increase of TNFα vs. other brain regions
including hippocampus, where no increase in TNFα was seen (6-8).
The striatum is a region with selective vulnerable
neuronal death in our model (9).
Surprising is the fact our prior studies showed TNFα localized
immunohistochemically in neurons rather than microglia (6,7).
Thus, unique TNFα production in striatal neurons may mediate the
region-specific neuronal loss in striatum after CA. Thus,
region-specific neuronal therapies may be required to best target
neuronal death after CA.
Among several readily translatable potential
strategies for CA to target TNFα, thalidomide, an inhibitor of TNFα
protein synthesis is readily capable of crossing the blood brain
barrier (BBB) (10). Thalidomide
has been reported to selectively decrease TNFα (11-13)
and shown to be neuroprotective via destruction of TNFα mRNA in
vivo (14) and in vitro
(15). Specifically, thalidomide
and its derivatives, including both lenalidomide and pomalidomide,
termed immunomodulatory imide drugs (IMiDs), are a class of drugs
that target the 3'-untranslated region (3'-UTR) of TNFα mRNA,
inhibiting TNFα production (16).
In this exploratory study, we hypothesized that thalidomide would
attenuate (1) systemic TNFα
levels, (2) neuroinflammation as
reflected by brain tissue TNFα levels, (3) extra-cerebral organ TNFα levels, and
(4) markers of organ injury after
prolonged CA in rats. We have also assessed (5) complex cytokine response to CA to
elucidate the potential downstream effect of thalidomide on other
cytokines. Naïve rats and rats treated with vehicle served as
controls.
Materials and methods
Institutional approval
The study protocol was approved by the Institutional
Animal Care and Use Committee of the University of Pittsburgh
(Protocol no. 13021161; ‘Neuroinflammation after prolonged cardiac
arrest’). We used our previously established model of ventricular
fibrillation (VF) CA (17).
Preparation phase
In brief, adult male Sprague-Dawley rats (350-400 g)
were obtained from a licensed vendor (Hilltop Lab Animals,
Scottdale, PA) and housed under 12/12 h light/dark in a holding
facility for at least two days prior to the experiment. Water was
provided ad libitum until the experiment. Standard chow was removed
12 h prior to experiment. On the day of the experiment, rats were
anesthetized with 4% isoflurane (Baxter) in pure oxygen in a
plexiglass jar, intubated with a 14-gauge cannula (Becton
Dickinson), and mechanically ventilated (Harvard Ventilator 683,
Harvard Rodent Apparatus) with tidal volume 8 ml/kg, PEEP 3 cm
H2O and respiratory rate 30-40/min to maintain
normocapnia. Anesthesia was maintained with 2% isoflurane
(FiO2 of 0.5).
CA and resuscitation phase
Three groups (n=6 per group) were studied: i) Naïve
rats; ii) rats subjected to VFCA without thalidomide (CA); and iii)
rats subjected to VFCA with thalidomide (CA+T). Naïve rats were
deeply anesthetized with isoflurane 4% over 4 min, midline
laparotomy and sternotomy were performed, and rats were perfused
transcardially with 250 ml of ice-cold heparinized normal
saline.
In rats subjected to VFCA, arterial (PE50) and
venous (PE90) femoral catheters were inserted via cut-downs for
blood pressure monitoring and drug administration. For VFCA, 5F
pacing catheter was introduced via the jugular vein to the
conjunction of right atrium and right ventricle.
Electrocardiogram (ECG) and mean arterial pressure
(MAP) were continuously monitored and recorded (Polygraph, Grass
Instruments). Rectal temperature was controlled at 37.0±0.5˚C with
a temperature controlled operating table, overhead heating lamp and
a fan. After surgery, the FiO2 was reduced to 0.3 and
isoflurane was gradually weaned to 0% over 10 min in rats scheduled
for CA.
No-flow was then induced by a 2-minute impulse of 12
V/50 Hz alternating current and ensured by ECG readings and
reduction in MAP <10 mmHg. The pacing catheter was then removed
and jugular vein ligated. After 10 min of VFCA, manual chest
compressions were started at a rate ~360/min along with mechanical
ventilation with 100% oxygen. Epinephrine (Abbott) 0.01 mg/kg was
given with start of compressions. Additional epinephrine 0.005
mg/kg was given at 1 min resuscitation time (RT). Sodium
bicarbonate (Abbott) 1 mEq/kg was also given at start of
resuscitation. At 2 min after the start of resuscitation (2 min
RT), defibrillation was attempted with biphasic 10 J impulse (Zoll
M series defibrillator; Zoll). If unsuccessful, subsequent shocks
were delivered every 30 seconds, with maximum 5 attempts over 4 min
resuscitation effort. Return of spontaneous circulation (ROSC) was
defined as sustained supraventricular rhythm with MAP >50 mmHg.
In rats subjected to thalidomide treatment (CA+T group), 50 mg/kg
thalidomide (Enzo Life Sciences; cat. no. BML-T115-0100) dissolved
in dimethyl sulfoxide (DMSO) was administered i.p. at 5 min after
initiation of resuscitation. This dose was selected based on prior
studies reporting benefits (18-21).
Control rats (CA group) received an identical volume of DMSO at the
corresponding timepoint.
Postoperative care
After ROSC, rats were mechanically ventilated with
FiO2 1.0, Vt 8 ml/kg, PEEP 5 cmH2O and
respiratory rate adjusted to maintain normocapnia. Epinephrine
infusion was titrated to maintain MAP >65 mmHg. Controlled
normothermia (36.5-37.5˚C) was maintained for 3 h. At 3 h RT, serum
samples were obtained, rats were deeply anesthetized with
isoflurane 4%, and perfused transcardially with 250 ml of ice-cold
heparinized normal saline. Rats were then decapitated, hearts and
brains removed and dissected into four regions of interest: cortex,
hippocampus, striatum, and cerebellum. Plasma, heart, and lung
samples were also obtained. Individual tissue samples were then
snap-frozen in liquid nitrogen and stored in -70˚C freezer until
further processing.
TNFα and cytokine measurements
Tissues were then processed for cytokine assessment
for interleukin (IL)-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12,
interferon γ (IFNγ) and granulocyte-macrophage colony stimulating
factor (GMCSF) using Luminex-200 multiplex analyzer (Luminex) using
a rat-specific kit (Millipore). All values were corrected for
protein concentration. The tissue was homogenized in
phosphate-buffered saline (PBS) by using Dounce homogenizer for 20
strokes. The homogenate was then sonicated for 10 seconds for three
times with an interval of 20 seconds, followed by centrifugation at
16,000 x g for 30 min. The supernatant was used for TNFα analysis.
Protein levels in the supernatant was measured using the
bicinchoninic acid (BCA) protein kit (Thermo Fisher Scientific,
Inc.). TNFα concentrations that were read as ‘out of range below’
were nominally assigned a value at one hundredth of the lowest
level on the calibration scale.
Biomarkers of organ-specific
injury
Biomarkers of neuronal injury (neuron-specific
enolase, NSE) and glial injury (S100b) were assessed using kits
(MyBioSource kits catalogue nos. MBS262217 and MBS849461,
respectively; MyBioSource, Inc.) according to manufacturer's
instructions. Myocardial injury was assessed using rat-specific
cardiac troponin T (cTnT) ELISA kit (MyBioSource catalogue no.
MBS730382). Intestinal injury was assess using rat specific ELISA
kit for intestinal fatty acid binding protein (IFABP; MyBioSource
catalogue no. MBS024910).
Statistical analysis
The analyses were performed using IBM SPSS
Statistics 26.0 software (IBM Corp.). The samples size calculations
were based off the results published by us previously. We
hypothesized that thalidomide would reduce TNFα levels in the
striatum by 50%. Using alpha=0.05 and power=80%, number of rats per
group needed was five. Anticipating ~20% mortality in our model, we
randomized 6 rats per group.
Data were tested for normality using the
Kolmogorov-Smirnov test. Hemodynamic and biochemical data are
presented as mean ± standard deviation (SD). Differences between
groups (P-value) were tested using Generalized Estimating Equations
models for treatment (RT5-RT180 in CA vs. CA+T). Cytokine levels
are presented as median and the first and the third quartile.
Differences in plasma cytokines were assessed using one-way
analysis of variance (ANOVA) with post hoc Tukey's test.
Differences in extracerebral organs cytokines at three hours after
CA were assessed using Kruskal-Wallis test followed by Dunn's test
with post hoc Bonferroni test. The differences in biomarkers of
organ-specific damage between groups were evaluated with a one-way
ANOVA with Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Survival
One rat in each group died. One rat in the CA group
died from hemodynamic collapse at RT 10 min, whereas one rat in the
CA+T group did not achieve ROSC. Data from both rats were excluded
from further analyses.
Biochemical and hemodynamic
profiles
As anticipated in our model, CA induced profound
metabolic acidosis with increased lactate that illustrated the
severity of the insult. The metabolic derangements progressively
resolved by the 180 min RT. There were no major differences in
hemodynamic or biochemical profiles between CA and CA+T groups
except pHa (Table I).
 | Table IPhysiologic and biochemical profile
after CA. |
Table I
Physiologic and biochemical profile
after CA.
| Variable | BL | RT5 | RT30 | RT60 | RT120 | RT180 | P-value |
|---|
| HR, bpm | | | | | | | |
|
CA | 324±11 | 272±86 | 324±40 | 364±27 | 374±15 | 390±20 | 0.88 |
|
CA+T | 344±25 | 302±22 | 340±32 | 384±43 | 378±46 | 390±31 | |
| MAP, mmHg | | | | | | | |
|
CA | 86±4 | 146±18 | 75±8 | 70±5 | 80±12 | 85±16 | 0.21 |
|
CA+T | 91±12 | 133±22 | 72±4 | 66±7 | 79±18 | 81±12 | |
| pHa | | | | | | | |
|
CA | 7.39±0.04 | 7.31±0.13 | 7.18±0.04 | 7.43±0.03 | 7.48±0.02 | 7.45±0.01 | <0.0001 |
|
CA+T | 7.40±0.02 | 7.31±0.09 | 7.18±0.04 | 7.35±0.08 | 7.38±0.04 | 7.39±0.03 | |
| paO2,
mmHg | | | | | | | |
|
CA | 189±31 | 343±50 | 354±44 | 366±31 | 369±68 | 370±45 | 0.75 |
|
CA+T | 227±15 | 363±73 | 413±102 | 324±90 | 397±79 | 379±75 | |
| paCO2,
mmHg | | | | | | | |
|
CA | 37±6 | 33±10 | 42±6 | 34±4 | 29±5 | 33±3 | 0.10 |
|
CA+T | 38±5 | 34±7 | 39±4 | 35±7 | 39±6 | 40±6 | |
| BE, mEq/l | | | | | | | |
|
CA | -2.1±1.4 | -9.5±3.8 | -11.9±2.2 | -1.4±2.5 | -1.8±2.7 | -0.8±1.4 | 0.36 |
|
CA+T | -1.1±2.4 | -8.9±2.0 | -13.0±0.7 | -5.3±3.8 | -1.7±4.8 | -0.7±4.8 | |
| Lactate,
mmol/l | | | | | | | |
|
CA | 1.4±0.8 | 9.8±1.6 | 9.4±2.6 | 4.5±1.8 | 3.1±0.9 | 2.5±0.7 | 0.82 |
|
CA+T | 1.4±0.5 | 10.3±1.0 | 8.4±2.7 | 3.9±0.7 | 2.8±0.7 | 3.1±1.0 | |
| Hct, % | | | | | | | |
|
CA | 43±2 | 46±3 | 50±3 | 48±3 | 45±4 | 47±4 | 0.33 |
|
CA+T | 43±3 | 45±2 | 47±5 | 45±4 | 47±1 | 48±3 | |
| Glucose, g/dl | | | | | | | |
|
CA | 166±31 | 105±42 | 121±28 | 142±41 | 145±24 | 161±46 | 0.76 |
|
CA+T | 178±35 | 86±14 | 94±22 | 125±34 | 190±26 | 199±60 | |
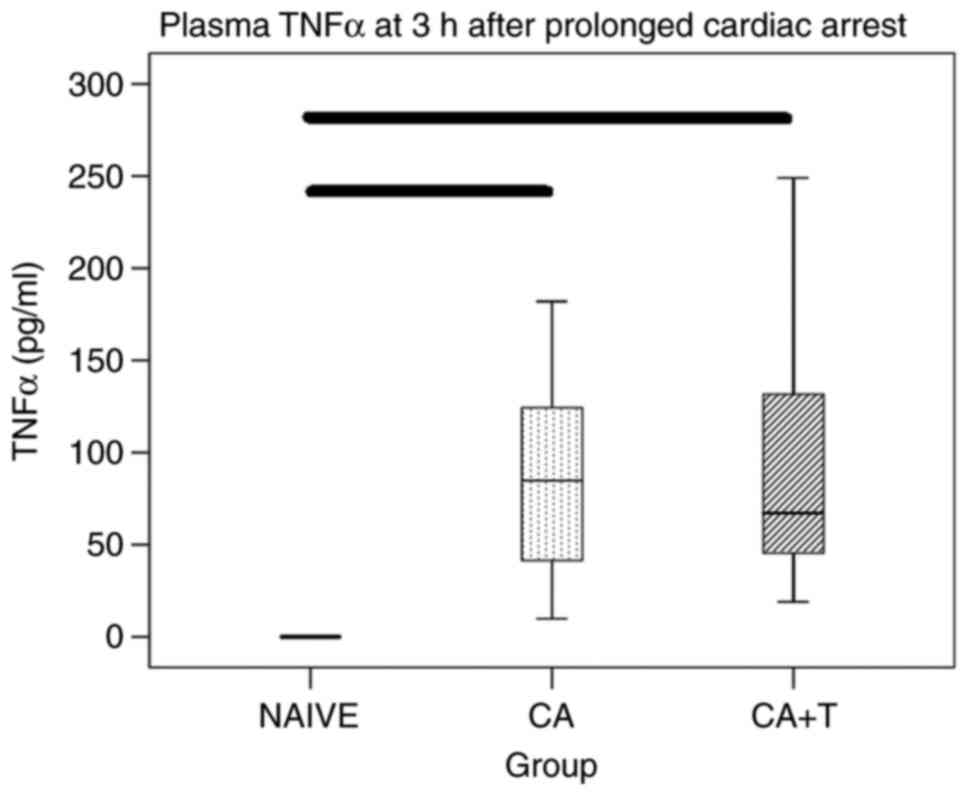
Plasma TNFα
CA resulted in an anticipated marked increase in
plasma TNFα (both naïve vs. CA and naïve vs. CA+T P<0.05).
However, no significant differences were found in plasma TNFα
between CA and CA+T treatment groups (Fig. 1).
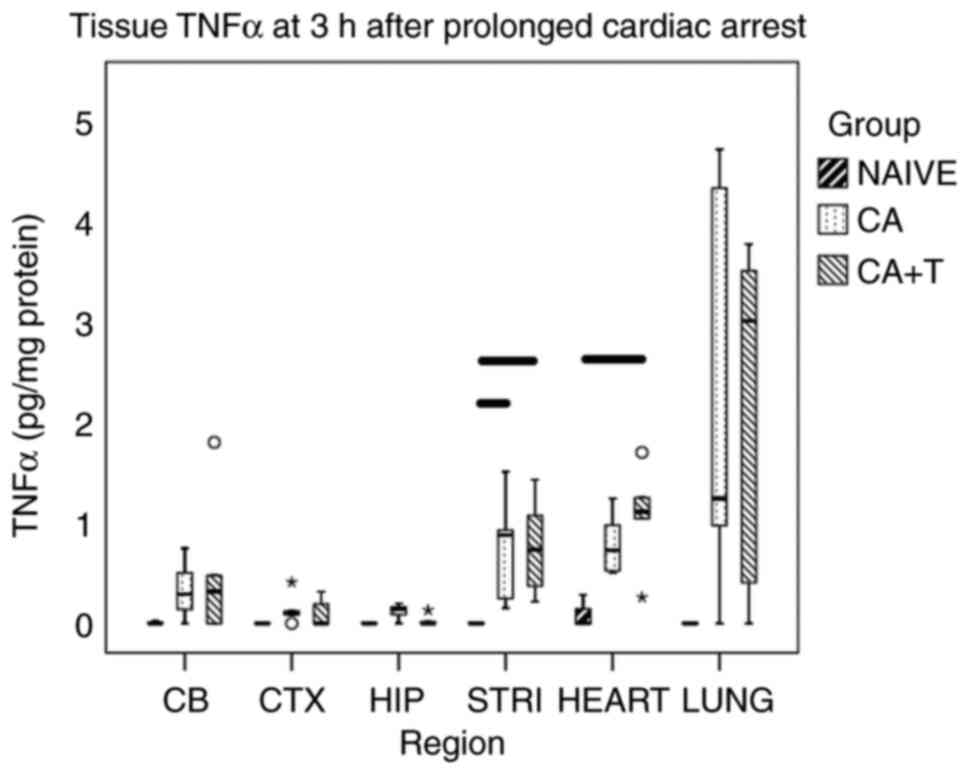
TNFα in individual brain regions
In general, brain cytokines in naïve rats were low
or undetectable. Although the TNFα levels were increased after CA
in most surveilled brain regions, no statistically significant
difference was found between any groups for CA, CA+T and naïve in
the cerebellum (P=0.236), cortex (P=0.297), or in the hippocampus
(P=0.067). As anticipated from our prior studies, TNFα was
significantly increased after CA in the striatum vs. naïve
(P<0.05) but was not found to be significantly different between
CA and CA+T (P=1.0), failing to support our hypothesis (Fig. 2).
TNFα in extracerebral organs
TNFα levels were numerically increased after CA or
CA+T vs. naïve in lung tissue but this trend did not reach
statistical significance (P=0.077). There were no differences in
lung TNFα between CA and CA+T. In contrast, the myocardial tissue
levels of TNFα were significantly increased after CA+T vs. naïve
(P<0.05) but were not found to be significantly different
between CA and CA+T (Fig. 2).
Biomarkers of organ-specific
injury
Rats subjected to CA had significantly increased
biomarkers of end-organ injury over naïves across all the organs
sampled: NSE (P<0.05), S100b (P<0.05), cardiac troponin T
(P<0.05), and IFABP (P<0.05), without significant differences
between CA and CA+T groups (Table
II).
| Table IIBiomarkers of brain and extracerebral
organ injury after CA. |
Table II
Biomarkers of brain and extracerebral
organ injury after CA.
| Variable | Naïve | CA | CA+T |
|---|
| NSE, ng/ml | 0.01±0.02 |
1.19±0.16a |
1.13±0.22a |
| S100b, pg/ml | 22±1 | 196±26a | 177±37a |
| cTnT, pg/ml | 0.64±0.38 |
1.20±0.06a |
1.17±0.30a |
| IFABP, pg/ml | 239±73 | 368±39a | 393±70a |
Cytokine response in individual brain
regions
Rats subjected to CA showed a significant increase
of IL-6 in the hippocampus and TNFα in the striatum over naïve
rats, with no differences between CA and CA+T groups (Table III).
| Table IIICytokine profile in individual brain
regions at 3 h after CA. |
Table III
Cytokine profile in individual brain
regions at 3 h after CA.
| Variable | CTX, pg/mg
protein | HIP, pg/mg
protein | STRI, pg/mg
protein | CEREB, pg/mg
protein |
|---|
| IL-1a | | | | |
|
N | 0.00
(0.00-1.97) | 0.08
(0.00-1.25) | 0.00
(0.00-0.00) | 0.00
(0.00-0.65) |
|
CA | 1.43
(0.00-3.11) | 2.56
(1.12-4.47) | 2.91
(0.00-4.44) | 0.47
(0.00-2.17) |
|
CA+T | 0.00
(0.00-1.47) | 2.04
(0.10-4.35) | 3.40
(1.47-4.72) | 1.38
(0.00-1.76) |
| IL-1b | | | | |
|
N | 2.90
(2.27-4.42) | 6.45
(3.40-7.96) | 6.90
(2.23-10.62) | 7.87
(7.10-9.02) |
|
CA | 8.02
(4.85-11.77) | 7.93
(7.25-14.23) | 7.12
(6.38-14.44) | 9.72
(4.84-14.44) |
|
CA+T | 6.81
(3.58-9.76) | 8.29
(6.15-10.0) | 10.42
(7.28-11.76) | 12.73
(9.78-13.51) |
| IL-2 | | | | |
|
N | 6.12
(1.41-50.59) | 0.00
(0.00-0.00) | 0.00
(0.00-0.00) | 4.98
(4.08-5.47) |
|
CA | 0.00
(0.00-14.04) | 4.55
(0.00-9.10) | 0.00
(0.00-26.60) | 5.75
(0.00-52.85) |
|
CA+T | 0.00
(0.00-22.33) | 0.00
(0.00-2.82) | 24.28
(2.83-40.66) | 4.08
(0.00-10.12) |
| IL-4 | | | | |
|
N | 0.02
(0.01-0.02) | 0.02
(0.01-0.02) | 0.03
(0.01-0.04) | 0.02
(0.02-0.02) |
|
CA | 0.02
(0.02-0.02) | 0.02
(0.01-0.02) | 0.02
(0.02-0.04) | 0.02
(0.01-0.02) |
|
CA+T | 0.02
(0.02-0.03) | 0.02
(0.01-0.02) | 0.02
(0.02-0.04) | 0.02
(0.02-0.02) |
| IL-6 | | | | |
|
N | 0.00
(0.00-0.00) | 0.00
(0.00-0.34) | 1.01
(0.00-2.05) | 0.41
(0.06-0.90) |
|
CA | 2.88
(1.05-10.92) | 1.52
(0.25-3.05) | 1.88
(0.63-3.47) | 2.29
(0.92-6.82) |
|
CA+T | 2.20
(0.61-3.28) | 1.34
(0.81-2.32) | 2.66
(1.91-4.40) | 3.96
(2.65-4.41) |
| IL-10 | | | | |
|
N | 5.72
(1.06-9.15) | 1.75
(0.00-4.14) | 17.48
(4.27-28.86) | 1.55
(0.00-4.12) |
|
CA | 9.41
(5.96-10.83) | 1.90
(1.19-4.99) | 13.52
(11.28-24.75) | 4.56
(0.00-15.32) |
|
CA+T | 13.76
(7.75-18.65) | 4.83
(2.35-6.94) | 18.51
(8.88-23.71) | 5.45
(1.49-7.99) |
| IL-12 | | | | |
|
N | 0.00
(0.00-2.45) | 1.13
(0.00-2.48) | 0.00
(0.00-0.00) | 1.33
(0.08-2.91) |
|
CA | 1.56
(0.62-3.06) | 1.50
(0.76-3.55) | 0.00
(0.00-0.57) | 0.00
(0.00-1.49) |
|
CA+T | 2.31
(0.90-4.05) | 1.04
(0.39-2.4) | 1.37
(0.58-3.19) | 0.97
(0.00-2.35) |
| IFNγ | | | | |
|
N | 0.48
(0.10-1.12) | 0.33
(0.10-0.48) | 0.33
(0.18-1.32) | 0.42
(0.37-0.50) |
|
CA | 0.38
(0.00-0.57) | 0.26
(0.11-0.37) | 0.31
(0.03-0.78) | 0.24
(0.09-0.54) |
|
CA+T | 0.40
(0.00-0.65) | 0.27
(0.12-0.49) | 0.54
(0.18-1.32) | 0.50
(0.25-0.64) |
| TNFα | | | | |
|
N | 0.00
(0.00-0.00) | 0.00
(0.00-0.00) | 0.00
(0.00-0.00) | 0.00
(0.00-0.16) |
|
CA | 0.10
(0.04-0.27) | 0.14
(0.04-0.18) | 0.88
(0.20-1.22)a | 1.29
(0.07-0.63) |
|
CA+T | 0.00
(0.00-0.25) | 0.00
(0.00-0.08) | 0.74
(0.29-1.25)a | 0.31
(0.00-1.14) |
Cytokine response in plasma and
extracerebral organs
CA resulted in statistically significant increases
of selected cytokines in plasma, namely IL-1β, IL-6, IL-10 and
IL-12. None of these cytokines were affected by thalidomide
administration. Increases of selected cytokines in heart (IL-1α,
IL-1β, IL-6, TNFα) and lung (IL-1α, IL-1β, IL-6, IFNγ) were also
not affected by thalidomide (Table
IV).
| Table IVCytokine profile in extracerebral
organs and plasma at 3 h after CA. |
Table IV
Cytokine profile in extracerebral
organs and plasma at 3 h after CA.
| Variable | Heart, pg/mg
protein | Lung, pg/mg
protein | Plasma, pg/ml |
|---|
| IL-1a | | | |
|
N | 0.00
(0.00-0.00) | 46.49 (44.14-52.17)
(43.95-46.49) | 0.00
(0.00-0.00) |
|
CA | 12.59
(9.97-18.80)a | 1135.50
(699.93-1777.02) | 266.41
(93.34-420.76) |
|
CA+T | 17.39
(12.43-32.68)a | 851.2
(818.11-1231.17)a | 216.04
(105.97-405.33) |
| IL-1b | | | |
|
N | 4.82
(3.23-6.12) | 45.21
(43.90-47.95) | 2.68
(0.07-11.99) |
|
CA | 54.57
(34.17-75.14)a | 1,421.24
(559.38-1,887.66)a | 255.81
(141.62-619.25) |
|
CA+T | 59.50
(37.90-79.31)a | 1,135.49
(784.93-1,534.19)a | 330.60
(229.21-863.37)a |
| IL-2 | | | |
|
N | 118.71
(86.42-142.46) | 0.00
(0.00-0.00) | 179.61
(80.27-1495.17) |
|
CA | 84.71
(65.65-120.83) | 13.38
(1.95-18.84) | 1033.3
(661.27-1,903.21) |
|
CA+T | 148.08
(49.9-194.49) | 24.15
(0.00-64.48) | 1,053.10
(672.88-2,526.03) |
| IL-4 | | | |
|
N | 0.28
(0.12-0.25) | 0.02
(0.02-0.04) | 0.17
(0.14-1.02) |
|
CA | 0.07
(0.04-0.25) | 0.03
(0.02-0.04) | 0.38
(0.32-0.87) |
|
CA+T | 0.19
(0.02-0.25) | 0.04
(0.03-0.05) | 0.41
(0.26-0.76) |
| IL-6 | | | |
|
N | 0.00
(0.00-1.85) | 0.00
(0.00-0.00) | 2.31
(0.00-7.29) |
|
CA | 61.26
(26.02-117.38) | 131.05
(20.75-165.54)a | 6,316.17
(2,533.21-8462.35)a |
|
CA+T | 129.84
(73.25-333.64)a | 119.86
(86.35-169.09)a | 6,756.38
(2,561.31-7775.65)a |
| IL-10 | | | |
|
N | 145.01
(96.93-194.58) | 10.74
(1.91-16.33) | 0.00
(0.00-0.00) |
|
CA | 75.55
(48.46-157.16) | 17.02
(4.06-24.36) | 925.86
(453.14-1390.16)a |
|
CA+T | 133.14
(27.23-147.75) | 16.22
(13.02-16.84) | 868.66
(597.01-1336.45)a |
| IL-12 | | | |
|
N | 8.24
(6.54-12.78) | 21.69
(18.22-30.02) | 1,479.62
(1,142.40-1704.93) |
|
CA | 9.45
(6.81-15.84) | 32.29
(27.04-67.66) | 4,754.44
(2,533.69-9116.59)a |
|
CA+T | 10.26
(7.93-13.91) | 31.53
(17.88-63.99) | 4,049.25
(2,830.96-7046.72)a |
| IFNγ | | | |
|
N | 3.04
(2.26-3.55) | 0.37
(0.25-0.48) | 1.89
(0.35-7.90) |
|
CA | 1.35
(0.90-2.05) | 0.63
(0.50-2.40) | 17.39
(7.74-190.14) |
|
CA+T | 3.01
(0.55-3.35) | 0.78
(0.70-1.50)a | 30.86
(12.58-105.17) |
| TNFα | | | |
|
N | 0.00
(0.00-0.21) | 0.00
(0.00-0.00) | 0.00
(0.00-0.00) |
|
CA | 0.73
(0.52-1.05) | 1.24
(0.49-4.50) | 84.74
(25.68-153.13)a |
|
CA+T | 1.11
(0.65-1.48)a | 3.01
(0.20-3.64) | 67.14
(32.24-190.30)a |
Discussion
We previously reported that this model of 10 min of
VFCA results in extensive neuronal death and dramatic increases in
cytokines. In the current study, we confirmed our findings of early
marked increases in TNFα levels in the striatum, and also
demonstrated that CA resulted in an early marked TNFα response both
systemically, and in other target organs. Contrasting our
hypothesis, however, thalidomide in a dose reported previously to
improve outcome of in vivo models of brain ischemia did not
decrease TNFα levels in the striatum, a brain region that showed
the most pronounced response to ischemia in our model of
experimental CA. Unfortunately, TNFα levels in plasma, other brain
regions or extracerebral organs were also not attenuated by
thalidomide. Selected pro- and anti-inflammatory cytokines
increased in brain, plasma, heart or lung after CA were not
affected by thalidomide. The reason for the failure of this
approach to attenuate the increase in striatal and/or other levels
of TNFα in our model and/or exhibit neuroprotective effects is
unclear.
We used a dose previously reported to be effective
in an experimental incomplete brain ischemia in mice. However, only
pre-treatment with that dose was effective; post-treatment was not
(20). To evaluate the potential
of this approach in a clinically relevant translational CA model,
we used a post-treatment paradigm.
Early brain cytokine response to global brain
ischemia has been documented by us (6,7) and
others (22). Microglia are deemed
to be a major source of brain TNFα in the later phases after the
insult or in neuro-inflammatory diseases in which thalidomide was
shown to be effective, e.g., Alzheimer's disease (23). However, we previously reported that
in the early phase post resuscitation, TNFα is produced by neurons
(6,7). This observation has been supported by
others (24,25). It is conceivable that neuronal
origin of TNFα in this early phase of reperfusion could contribute
to the lack of a definitive effect of thalidomide that primarily
targets glia cells in brain (16).
Also, thalidomide selectively targets microglia-mediated
neuroinflammatory response rather than astrocytes (26). We reported that the cytokine
production at the early stages of post-CA syndrome is not mediated
by microglia but rather neurons and astrocytes (7).
Most studies focused on neuronal loss in
hippocampus, a selectively vulnerable region with extensive
neuronal degeneration after cerebral ischemia. In recent studies
focused on the neuro-inflammatory response to CA, however, we noted
dramatic regional dependence of the cytokine response in brain
after CA (6,8). These studies suggest the potential
need for a paradigm shift in the approach to the development of
neuroprotective therapies in CA-namely, region specific therapies
tailored to individual brain regions. Dopaminergic neurons in the
striatum may represent an alternative, selectively vulnerable
region in the prolonged CA setting. The early surge of TNFα in the
striatum as described by us earlier was selected as a primary
target structure in our current study.
TNFα has also been documented to be increased after
ischemia-reperfusion also in plasma and extracerebral organs.
In rats subjected to a shorter, 6 min VFCA,
increased levels of TNFα, IL-6, and IL-10 were observed in the
jejunum from 6 h until 7 d but not in serum in rats (27). Similarly, Wender et al
(28) did not observe increased
serum TNFα levels past 24 h in a rat CA model. In contrast, Yang
et al (29) reported that
serum IL-6 and TNFα levels were both increased at 6 h after 6 min
VFCA in rats.
In a porcine model of VFCA, systemic TNFα was
detected early (15 min after reperfusion), and peak plasma levels
coincided with myocardial depression (30) and with poor survival (31). Zhu et al (32) recently reported early increases of
both serum TNFα and IL-6 levels that continued to rise over 6
h.
In an experimental model of intestinal
ischemia-reperfusion injury, thalidomide was effective to decrease
both systemic and tissue (intestine, lung) TNFα levels, effectively
ameliorating biomarkers of injury, edema and resulting histologic
damage. However, pre-treatment with a large dose (400 mg/kg p.o.)
was used (33). Not all studies
reported decrease of systemic TNFα with thalidomide treatment.
After a lipopolysaccharide challenge resulting in massive cytokine
response, neither plasma nor hepatic TNFα levels were decreased
with thalidomide (34,35).
Our study has several limitations. Although
thalidomide has been shown to cross the BBB, most studies
documenting the salutary effects of thalidomide were performed in
models with markedly disrupted BBB, (e.g., traumatic brain injury,
stroke (20) or
lipopolysaccharide-induced chronic neuroinflammation). Mohammed
et al (36) reported a
salutary effect of thalidomide was elicited by direct injection of
the drug into the hippocampus, obviating the need for transport
across the BBB. In this light, it is possible that newly
synthetized derivatives of thalidomide, e.g.,
3,6'-dithiothalidomide or pomalidomide, that penetrate BBB more
easily, could have been more effective in ameliorating neuronal
injury (37-39).
We also did not perform a dose response in our
model. Instead, we selected for our study the highest (and the only
effective) dose used in other studies (18,19,21).
Importantly the same dose given to naïve rats did not elicit
notable adverse effects (19). In
that regard, pomalidomide has been shown to produce a TNFα
inhibitory effect that is 50,000 times greater than thalidomide,
and thus might have more potential in a CA scenario, where a rapid
inhibitor effect is needed in the setting of an intact BBB
(16). Also, the time-course of
TNFα response in traumatic brain injury and models of chronic
neuroinflammation seems to be delayed (40), providing a more favorable scenario
for thalidomide or its derivatives to exert the effects on the
injured brain even with oral administration (16). However, the lack of effect of
thalidomide on increases in TNFα levels in plasma and extracerebral
organs argue against brain penetration as the explanation for
failure to effect target engagement and/or reduce secondary
injury.
We explored TNFα levels only at a singular timepoint
(3 h RT). We chose this timepoint based on results from our prior
study using VFCA model in which we have explored both early (3 and
6 h RT) and late periods (14 days). Earlier timepoints showed more
robust cytokine response (6).
Moreover, TNFα-as the major target for thalidomide effects-peaks
early, perhaps within the first hour after reperfusion (22). We have also tested a single, high
dose of thalidomide shown previously to be beneficial and safe.
However, we cannot rule out that alternative dosing regimen or
assessments at other time-points might yield different results.
Biomarkers of end-organ injury were also assessed at an early
timepoint. Despite this limitation, we were able to document
significant increases in CA groups over naïve controls.
We studied healthy young male rats only. More
pronounced systemic cytokine response was observed in aged rats
subjected to asphyxial CA (41). A
significant difference in plasma cytokine response to CA between
sexes have been observed by others (30).
Finally, we observed a trend toward reduced TNFα
levels in the hippocampus in rats treated with thalidomide
(P=0.067). We cannot rule out a possible effect in that brain
region that would need to be explored with a larger sample size to
appropriately test that hypothesis given the modest increase in
TNFα seen in that brain region vs striatum in our model.
In conclusion, this exploratory study suggests that
TNFα is increased early after CA systemically, in the brain and in
extracerebral organs. Thalidomide, used early after reperfusion at
high-dose previously showed to confer benefits, failed to decrease
TNFα levels or other increased cytokines assessed at this timepoint
in our experimental CA model. Biomarkers of end-organ injury were
markedly increased after CA without any effect of thalidomide.
Early systemic and organ-specific cytokine response after CA
remains a valuable therapeutic target for future interventions in
both acute and longitudinal studies.
Acknowledgements
The authors would like to thank Mr. Manuel S.
Lombardero (Senior Statistician, Department of Epidemiology, School
of Public Health, University of Pittsburgh, Pittsburgh, PA, USA)
for his invaluable help with the statistical analyses and reviewing
of the manuscript.
Funding
Funding: This work was supported by the Laerdal Foundation
(TD).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AAP, PMK and TD designed the study, analyzed and
interpreted the data, and wrote the manuscript. JPS performed the
experiments. KJF performed the Luminex assays and ELISAs. JPS and
KJF confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Institutional
Animal Care and Use Committee of the University of Pittsburgh
(protocol no. 13021161; ‘Neuroinflammation after prolonged cardiac
arrest’), in compliance with ARRIVE guidelines and the AVMA
euthanasia guidelines 2020.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Janata A, Drabek T, Magnet IA, Stezoski
JP, Janesko-Feldman K, Popp E, Garman RH, Tisherman SA and Kochanek
PM: Extracorporeal versus conventional cardiopulmonary
resuscitation after ventricular fibrillation cardiac arrest in
rats: A feasibility trial. Crit Care Med. 41:e211–e222.
2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Badiola N, Malagelada C, Llecha N, Hidalgo
J, Comella JX, Sabriá J and Rodríguez-Alvarez J: Activation of
caspase-8 by tumour necrosis factor receptor 1 is necessary for
caspase-3 activation and apoptosis in oxygen-glucose deprived
cultured cortical cells. Neurobiol Dis. 35:438–447. 2009.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Liu Q, Qiu J, Liang M, Golinski J, van
Leyen K, Jung JE, You Z, Lo EH, Degterev A and Whalen MJ: Akt and
mTOR mediate programmed necrosis in neurons. Cell Death Dis.
5(e1084)2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang W, Xie L, Zou X, Hu W, Tian X, Zhao G
and Chen M: Pomelo peel oil suppresses TNF-α-induced necroptosis
and cerebral ischaemia-reperfusion injury in a rat model of cardiac
arrest. Pharm Biol. 59:401–409. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Niemann JT, Youngquist ST, Shah AP, Thomas
JL and Rosborough JP: TNF-α blockade improves early
post-resuscitation survival and hemodynamics in a swine model of
ischemic ventricular fibrillation. Resuscitation. 84:103–107.
2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Janata A, Magnet IA, Uray T, Stezoski JP,
Janesko-Feldman K, Tisherman SA, Kochanek PM and Drabek T: Regional
TNFα mapping in the brain reveals the striatum as a
neuroinflammatory target after ventricular fibrillation cardiac
arrest in rats. Resuscitation. 85:694–701. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Drabek T, Wilson CD, Janata A, Stezoski
JP, Janesko-Feldman K, Garman RH, Tisherman SA and Kochanek PM:
Unique brain region-dependent cytokine signatures after prolonged
hypothermic cardiac arrest in rats. Ther Hypothermia Temp Manag.
5:26–39. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Uray T, Dezfulian C, Palmer AA, Miner KM,
Leak RK, Stezoski JP, Janesko-Feldman K, Kochanek PM and Drabek T:
Cardiac arrest induced by asphyxia versus ventricular fibrillation
elicits comparable early changes in cytokine levels in the rat
brain, heart, and serum. J Am Heart Assoc.
10(e018657)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Janata A, Magnet IA, Schreiber KL, Wilson
CD, Stezoski JP, Janesko-Feldman K, Kochanek PM and Drabek T:
Minocycline fails to improve neurologic and histologic outcome
after ventricular fibrillation cardiac arrest in rats. World J Crit
Care Med. 8:106–119. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Huang YJ, Liao JF and Tsai TH: Concurrent
determination of thalidomide in rat blood, brain and bile using
multiple microdialysis coupled to liquid chromatography. Biomed
Chromatogr. 19:488–493. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Sampaio EP, Sarno EN, Galilly R, Cohn ZA
and Kaplan G: Thalidomide selectively inhibits tumor necrosis
factor alpha production by stimulated human monocytes. J Exp Med.
173:699–703. 1991.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Klausner JD, Freedman VH and Kaplan G:
Thalidomide as an anti-TNF-alpha inhibitor: Implications for
clinical use. Clin Immunol Immunopathol. 81:219–223.
1996.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Alam HB: Translational barriers and
opportunities for emergency preservation and resuscitation in
severe injuries. Br J Surg. 99 (Suppl 1):S29–S39. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Majumder S, Sreedhara SR, Banerjee S and
Chatterjee S: TNF α signaling beholds thalidomide saga: A review of
mechanistic role of TNF-α signaling under thalidomide. Curr Top Med
Chem. 12:1456–1467. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang L, Qu Y, Tang J, Chen D, Fu X, Mao M
and Mu D: PI3K/Akt signaling pathway is required for
neuroprotection of thalidomide on hypoxic-ischemic cortical neurons
in vitro. Brain Res. 1357:157–165. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jung YJ, Tweedie D, Scerba MT and Greig
NH: Neuroinflammation as a factor of neurodegenerative disease:
Thalidomide analogs as treatments. Front Cell Dev Biol.
7(313)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Nora GJ, Harun R, Fine DF, Hutchison D,
Grobart AC, Stezoski JP, Munoz MJ, Kochanek PM, Leak RK, Drabek T
and Wagner AK: Ventricular fibrillation cardiac arrest produces a
chronic striatal hyperdopaminergic state that is worsened by
methylphenidate treatment. J Neurochem. 142:305–322.
2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Andrade P, Visser-Vandewalle V, Del
Rosario JS, Daemen MA, Buurman WA, Steinbusch HW and Hoogland G:
The thalidomide analgesic effect is associated with differential
TNF-α receptor expression in the dorsal horn of the spinal cord as
studied in a rat model of neuropathic pain. Brain Res. 1450:24–32.
2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Choi JI, Kim WM, Yoon MH and Lee HG:
Antiallodynic effect of thalidomide and morphine on rat spinal
nerve ligation-induced neuropathic pain. Korean J Pain. 23:172–178.
2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hyakkoku K, Nakajima Y, Izuta H, Shimazawa
M, Yamamoto T, Shibata N and Hara H: Thalidomide protects against
ischemic neuronal damage induced by focal cerebral ischemia in
mice. Neuroscience. 159:760–769. 2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ribeiro RA, Vale ML, Ferreira SH and Cunha
FQ: Analgesic effect of thalidomide on inflammatory pain. Eur J
Pharmacol. 391:97–103. 2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Saito K, Suyama K, Nishida K, Sei Y and
Basile AS: Early increases in TNF-alpha, IL-6 and IL-1 beta levels
following transient cerebral ischemia in gerbil brain. Neurosci
Lett. 206:149–152. 1996.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ryu JK and McLarnon JG: Thalidomide
inhibition of perturbed vasculature and glial-derived tumor
necrosis factor-alpha in an animal model of inflamed Alzheimer's
disease brain. Neurobiol Dis. 29:254–266. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu T, Clark RK, McDonnell PC, Young PR,
White RF, Barone FC and Feuerstein GZ: Tumor necrosis factor-alpha
expression in ischemic neurons. Stroke. 25:1481–1488.
1994.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pettigrew LC, Kindy MS, Scheff S, Springer
JE, Kryscio RJ, Li Y and Grass DS: Focal cerebral ischemia in the
TNFalpha-transgenic rat. J Neuroinflammation. 5(47)2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ryu JK, Jantaratnotai N and McLarnon JG:
Thalidomide inhibition of vascular remodeling and inflammatory
reactivity in the quinolinic acid-injected rat striatum.
Neuroscience. 163:601–608. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Schroeder DC, Maul AC, Mahabir E, Koxholt
I, Yan X, Padosch SA, Herff H, Bultmann-Mellin I, Sterner-Kock A,
Annecke T, et al: Evaluation of small intestinal damage in a rat
model of 6 Minutes cardiac arrest. BMC Anesthesiol.
18(61)2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wender M, Michalowska-Wender G and Szczech
J: Minimal changes of TNF-alpha and MCP-1 expression in blood serum
of rats subjected to experimental cardiac arrest. Folia
Neuropathol. 43:109–111. 2005.PubMed/NCBI
|
|
29
|
Yang M, Hua T, Yang Z, Chen L, Zou Y,
Huang X and Li J: The protective effect of rhBNP on
postresuscitation myocardial dysfunction in a rat cardiac arrest
model. Biomed Res Int. 2020(6969053)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Niemann JT, Rosborough JP, Youngquist S,
Shah AP, Lewis RJ, Phan QT and Filler SG: Cardiac function and the
proinflammatory cytokine response after recovery from cardiac
arrest in swine. J Interferon Cytokine Res. 29:749–758.
2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Youngquist ST, Shah AP, Rosborough JP and
Niemann JT: High serum tumor necrosis factor levels in the early
post-cardiac arrest period are associated with poor short-term
survival in a swine model of ventricular fibrillation. J Interferon
Cytokine Res. 36:575–579. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhu F, Zhong X, Zhou Y, Hou Z, Hu H, Liang
L, Chen J, Chen Q, Ji X and Shang D: Protective effects of
nicorandil against cerebral injury in a swine cardiac arrest model.
Exp Ther Med. 16:37–44. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Camara-Lemarroy CR, Guzman-de la Garza FJ,
Alarcon-Galvan G, Cordero-Perez P, Munoz-Espinosa LE and
Fernandez-Garza NE: Effects of thalidomide and pentoxyphylline over
local and remote organ injury after intestinal
ischemia/reperfusion. Transplant Proc. 42:1624–1626.
2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Fernandez-Martinez E, Morales-Rios MS,
Perez-Alvarez V and Muriel P: Immunomodulatory effects of
thalidomide analogs on LPS-induced plasma and hepatic cytokines in
the rat. Biochem Pharmacol. 68:1321–1329. 2004.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ilhan N, Susam S, Gul HF, Bardas R and
Ilhan N: Which one is more effective for the treatment of rat
sepsis model: Thalidomide or etanercept? Bratisl Lek Listy.
118:283–287. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Mohammed RA, El-Yamany MF, Abdel-Rahman
AA, Nassar NN and Al-Shorbagy MY: Role of pERK1/2-NFĸB signaling in
the neuroprotective effect of thalidomide against cerebral ischemia
reperfusion injury in rats. Eur J Pharmacol.
895(173872)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lin CT, Lecca D, Yang LY, Luo W, Scerba
MT, Tweedie D, Huang PS, Jung YJ, Kim DS, Yang CH, et al:
3,6'-dithiopomalidomide reduces neural loss, inflammation,
behavioral deficits in brain injury and microglial activation.
Elife. 9(e54726)2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Batsaikhan B and Wang JY, Scerba MT,
Tweedie D, Greig NH, Miller JP, Hoffer BJ, Lin CT and Wang JY:
Post-injury neuroprotective effects of the thalidomide Analog
3,6'-Dithiothalidomide on traumatic brain injury. Int J Mol Sci.
20(502)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Tsai YR, Tweedie D, Navas-Enamorado I,
Scerba MT, Chang CF, Lai JH, Wu JC, Chen YH, Kang SJ, Hoffer BJ, et
al: Pomalidomide reduces ischemic brain injury in rodents. Cell
Transplant. 28:439–450. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Baratz R, Tweedie D, Wang JY, Rubovitch V,
Luo W, Hoffer BJ, Greig NH and Pick CG: Transiently lowering tumor
necrosis factor-α synthesis ameliorates neuronal cell loss and
cognitive impairments induced by minimal traumatic brain injury in
mice. J Neuroinflammation. 12(45)2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Secher N, Ostergaard L, Tonnesen E, Hansen
FB and Granfeldt A: Impact of age on cardiovascular function,
inflammation, and oxidative stress in experimental asphyxial
cardiac arrest. Acta Anaesthesiol Scand. 62:49–62. 2018.PubMed/NCBI View Article : Google Scholar
|