Introduction
Acute myocardial infarction (AMI) is a leading cause
of mortality worldwide. Although the current effective percutaneous
coronary intervention and thrombolytic therapy have saved the lives
of countless patients, ischemia and reperfusion (I/R) injury can
also cause further damage to the heart (1,2).
Following MI, TGFβ1 is markedly upregulated, inducing fibroblast
activation and cell phenotypic conversion from fibroblasts to
myofibroblasts. Myofibroblasts express α-smooth muscle actin
(α-SMA) and secrete large amounts of extracellular matrix (ECM)
proteins (3). Following MI injury
the accumulation of ECM is critical for acute wound healing;
however, in response to myocardial injury, persistent fibroblast
activation and cell phenotypic conversion from fibroblasts to
myofibroblasts result in the excessive production and accumulation
of ECM proteins and excessive fibrosis worsens disease and
accelerates the progression to heart failure (HF) (4).
ECM contains structural components such as collagen
and nonstructural components, including glycoproteins,
proteoglycans and glycosaminoglycans (5). Chondroitin sulfate proteoglycans
(CSPGs) consist of core proteins with covalently attached
chondroitin sulfate (CS) chains (6). CS is a class of sulfated
glycosaminoglycan chains composed of a repeating disaccharide unit
consisting of glucuronic acid (GlcA) and
N-acetylgalactosamine (GalNAc). Biosynthesis of CS is
initiated by addition of the linkage tetrasaccharide sequence to a
specific serine residue in a core protein (7). The sulfation pattern of CS is known
to be important for the specific functions of CS. CSPGs that
promote fibrogenesis are deposited in the fibrosis model induced by
bleomycin (8). A previous study
demonstrated that excessive CSPG deposition is associated with
impaired cardiac function with ischemic HF in animal models and
patients (9). Another study
confirmed that TGFβ induces chondroitin-4-sulfate (C4S)
upregulation in cardiac fibroblasts (CFs) and found that C4S also
accumulates in fibrotic regions of the pathologically remodeled
left ventricle (LV) (5). However,
the mechanisms that induce CSPG expression in fibroblasts following
MI remain to be elucidated.
A previous study confirmed that I/R infarct-derived
CSPGs are important for sustaining denervation of the infarct
(10,11). Another study demonstrated that
CSPGs accumulate in the ECM of the injured central nervous system
(CNS) and are synthesized by reactive astrocytes, limiting axonal
regeneration (12). TGFβ1 signals
through the canonical Smad pathway and several noncanonical
pathways affect cell function (13). A previous study on CNS injury
confirmed that TGFβ-Smad3 mediates astrocyte-secreted 4-sulfation
(14). However, another study
reported that TGFβ mediates CSPG expression through
non-Smad-mediated activation of the PI3K-Akt-mTOR signaling pathway
(15). Notably, a previous study
demonstrated that canonical PI3K/Akt signaling has no effect on
TGFβ1-induced collagen deposition in pulmonary fibroblasts and this
response may be mediated via cooperation between canonical Smad3
and mTORC1 signaling (13).
Therefore, the purpose of the present study was to examine whether
oxygen-glucose deprivation (OGD) and TGFβ1 stimulation induced
myofibroblast transformation and CSPG synthesis in fibroblasts
mediated by cooperation between canonical Smad3 and mTORC1
signaling.
Materials and methods
Primary cell isolation, culture and in
vitro treatment
Adult rat ventricular CFs were isolated as
previously described (16). In
brief, a total of 48 adult male Sprague-Dawley rats (age, 8- to
10-week-old; weight, 150-200 g) were provided by the Laboratory
Animal Centre of the Gansu University of Chinese Medicine. All
experimental subjects were maintained in a specific pathogen-free
grade environment at a temperature of 23±2˚C with a humidity of
50-60%, a 12-h light/dark cycle and free access to food and water.
The rats were anaesthetized via intraperitoneal injection of 3%
sodium pentobarbital (30 mg/kg). Hearts were then excised from
anesthetized rats and rinsed in cold phosphate-buffered saline. The
ventricles were cut into pieces of ~1 mm3 in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.), digested in 0.25% trypsin (Dalian Meilun Biotechnology Co.,
Ltd.) and subsequently digested in DMEM containing 0.1% collagenase
type II (MilliporeSigma). The cells were collected and cultured in
DMEM containing 10% fetal calf serum, 100 U/ml penicillin (Dalian
Meilun Biotechnology Co., Ltd.) and 100 mg/ml streptomycin (Dalian
Meilun Biotechnology Co., Ltd.) at 37˚C and 5% CO2 for 1
h. After pre-plating, the adherent cells were cultured. The purity
of CFs was >95%, as determined by positive staining for
vimentin. Cells at passage 1 were used in the present study. All
experimental procedures were performed in accordance with the Guide
for the Care and Use of Laboratory Animals published by the
National Research Council and were approved by the Animal Care
Committee of the Gansu University of Chinese Medicine (approval
number 2019-212).
Cultured cells subjected to hypoxia and fuel
deprivation provide an efficient in vitro model to examine
the cellular mechanisms mediating ischemic injury (17). Cells were cultured to 70-80%
confluence, at which point the medium was changed to serum- and
glucose-free DMEM (Dalian Meilun Biotechnology Co., Ltd.). Cells
were transferred into a triple gas incubator with hypoxic (5%
CO2, 1% O2 and 94% N2) settings
and incubated under hypoxic conditions and flushed with the same
gas mixture. After exposure to OGD for 12 h, the cells were placed
under normal conditions (DMEM containing serum and glucose) for
reoxygenation for 24 h for RNA detection or 48 h for protein
detection. During reoxygenation, the treated groups were stimulated
with TGFβ1 (10 ng/ml; PeproTech, Inc.). The corresponding control
cells were incubated in DMEM containing serum and glucose under
normoxic conditions for equivalent durations.
Pharmacological inhibitors
The contribution of the PI3K/Akt/mTOR signaling axis
to OGD- and TGFβ1 stimulation-induced myofibroblast transformation
and CSPG synthesis was investigated by evaluating the effect of
highly selective inhibitors of critical components of the
PI3K/Akt/mTOR axis: PI3K (cat. no. ZSTK474; MedChem Express), Akt
(cat. no. MK2206; MedChem Express) and mTOR (cat. no. AZD8055;
MedChem Express). Stock solutions of ZSTK474 (1 mM), MK2206 (1 mM)
and AZD8055 (1 mM) were prepared. All chemicals were prepared in
dimethyl sulfoxide (DMSO; Beijing Solarbio Science & Technology
Co., Ltd.).
All groups except the control group received one of
the following treatments: ZSTK474 (1 µM, 100 nM, 10 nM or 1 nM),
MK2206 (1 µM, 100 nM, 10 nM or 1 nM), AZD8055 (1 µM, 100 nM, 10 nM
or 1 nM) for 4 h before exposure to OGD + TGFβ1. The inhibitor
concentration was always consistent in subsequent processing.
Cell viability by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) assay
CFs were seeded in 96-well plates (density;
1x104 cells/well) in DMEM media and were stimulated
under the conditions indicated for each experiment. Cell viability
was measured using the MTT assay (Beijing Solarbio Science &
Technology Co., Ltd.). Briefly, MTT solution (final concentration,
0.5 mg/ml) was added to each well and the cells were incubated at
37˚C for 4 h. Culture supernatant was then removed and 110 µl DMSO
was added to each well to dissolve formazan crystals and the
absorbance at 490 nm was measured by using an infinite M200PRO
microplate reader (Tecan Group, Ltd.). Data from 3 independent
experiments were used for analysis.
Short interfering (si)RNA
transfection
For siRNA knockdown experiments, rat-specific Smad3
siRNA, Raptor siRNA, Rictor siRNA and negative control siRNA were
purchased from Shanghai GenePharma Co., Ltd. The siRNA was diluted
in serum-free media and rat CFs were either transfected with Smad3
siRNA (10 nM), Raptor siRNA (10 nM), Rictor siRNA (10 nM) or
negative control siRNA (10 nM) using Lipofectamine®
RNAi/MAX Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) at
37˚C for 48 h according to the manufacturer's protocols, after
which the cells were harvested for western blotting analysis to
evaluate the specific silencing effect of siRNA. Following
transfection, the cells were used for subsequent experiments. The
sequences of the Smad3 siRNA were sense 5'-GAUCGAGCUACACCUGAAUTT-3'
and antisense 5'- AUUCAGGUGUAGCUCGAUCTT-3'. The sequences of the
Raptor siRNA were sense 5'-GUGGCAAGUUUGUUUAGAATT-3' and antisense
5'-UUCUAAACAAACUUGCCACTT-3'. The sequences of the Rictor siRNA were
sense 5'-GCAGCCCUGAACUGUUUAATT-3' and antisense
5'-UUAAACAGUUCAGGGCUGCTT-3'. The sequences of the negative control
siRNA were sense 5'-UUCUCCGAACGUGUCACGUTT-3' and antisense
5'-ACGUGACACGUUCGGAGAATT-3'.
RNA extraction and reverse
transcription-quantitative (RT-q) PCR
Total RNA (5x106 cells) was extracted
using TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. RNA was subsequently
reverse transcribed using PrimeScript RT Master Mix (Takara
Biotechnology Co., Ltd.). The reaction temperature of reverse
transcription was 37˚C for 15 min and 85˚C for 5 sec. RT-qPCR was
performed using SYBR Premix Ex Taq Ⅱ (Takara Biotechnology Co.,
Ltd.). All reaction were performed with the following cycling
parameters: 95˚C for 30 sec, followed by 40 cycles of 95˚C for 5
sec, 60˚C for 34 sec and a final stage of 95˚C for 15 sec, 60˚C for
60 sec and 95˚C for 15 sec. Relative expression of all genes was
calculated by normalizing to the levels of expression of the
internal reference gene β-actin using the 2-ΔΔCq method
(18). Data from 3 independent
experiments were used for analysis of relative gene expression. A
list of the genes and primers used is provided in Table SI.
Sample preparation and western
blotting CFs conditioned media
CF-conditioned medium was placed into a tube
containing complete protease inhibitors (Roche Diagnostics). The
conditioned medium was then centrifuged at 4,000 x g at 4˚C for 20
min to concentrate using an Amicon Ultra-15 NMWL100K centrifugal
filter device (MilliporeSigma). The protein concentration was
measured using a BCA protein assay reagent (Beijing Solarbio
Science & Technology Co., Ltd.). Protein (30 µg) was loaded
onto 8% gels, subjected to SDS-PAGE and then transferred to PVDF
membranes (MilliporeSigma). Following blocking with 5% skimmed milk
at room temperature for 1 h, membranes were incubated with the
following primary antibody: anti-2H6 (Cosmo Bio) at 4˚C overnight.
The secondary antibody was horseradish peroxidase (HRP)-conjugated
anti-mouse IgG (ImmunoWay Biotechnology Company) and was incubated
with the membranes at room temperature for 1 h. Proteins were
visualized using Enhanced Chemiluminescence (ECL) western blotting
Substrate (Dalian Meilun Biotechnology Co., Ltd.) and gel images
were visualized using a Universal Hood Ⅱ (Bio-Rad Laboratories,
Inc.) and quantified using ImageJ 1.51k software (NIH). Catalog
numbers and dilutions of antibodies are provided in Table SII.
Cell lysates
CFs were lysed in RIPA buffer containing 1%
phenylmethylsulfonyl fluoride (Dalian Meilun Biotechnology Co.,
Ltd.) and 1% phosphatase inhibitor cocktail (Beijing Solarbio
Science & Technology Co., Ltd.). Protein concentration was
measured using a BCA protein assay reagent (Beijing Solarbio
Science & Technology Co., Ltd.). Protein (20 µg) was loaded
onto 10% gels for SDS-PAGE and then transferred to PVDF membranes
(MilliporeSigma). Following blocking with 5% skimmed milk at room
temperature for 1 h, membranes were incubated with the following
primary antibodies: anti-α-SMA (Abcam), anti-Akt (Cell Signaling
Technology, Inc.), anti-phospho-Akt(Ser473) (Cell
Signaling Technology, Inc.), anti-PRAS40 (Cell Signaling
Technology, Inc.), anti-phospho-PRAS40(Thr246) (Cell
Signaling Technology, Inc.), anti-p70S6k (Cell Signaling
Technology, Inc.), anti- phospho-p70S6k(Thr389) (Cell
Signaling Technology, Inc.), anti-Smad3 (Cell Signaling Technology,
Inc.), anti-phospho-Smad3(Ser425) (Affinity),
anti-Raptor (Cell Signaling Technology, Inc.), anti-Rictor (Cell
Signaling Technology, Inc.) and anti-β-actin (ImmunoWay
Biotechnology Company) at 4˚C overnight. Secondary antibodies were
HRP-conjugated anti-mouse IgG and HRP-conjugated anti-rabbit IgG
(ImmunoWay Biotechnology Company) at room temperature for 1 h.
Proteins were visualized with ECL western blotting Substrate
(Dalian Meilun Biotechnology Co., Ltd.) and gel images were
visualized using Universal Hood Ⅱ (Bio-Rad Laboratories, Inc.) and
quantified using ImageJ 1.51k software (NIH). Catalog numbers and
dilutions of primary antibodies are provided in Table SII.
Immunofluorescence imaging
Following the aforementioned treatments, cells were
fixed in 4% paraformaldehyde for 15 min at room temperature and
rinsed with phosphate-buffered saline-(0.05%) Tween 20 (PBST).
Cells were then permeabilized using 0.25% Triton X-100 in PBS for
10 min. Next, cells were blocked in 1% bovine serum albumin for 30
min at room temperature. Subsequently, the cells were incubated
with primary antibodies against vimentin (Abcam) and α-SMA (Abcam)
overnight at 4˚C. Cells were then washed with PBST and incubated
with Alexa Fluor 594-conjugated goat anti-rabbit secondary antibody
(ImmunoWay Biotechnology Company) or Alexa Fluor 488-conjugated
goat anti-rabbit/mouse secondary antibody (ImmunoWay Biotechnology
Company) for 1 h at room temperature. Stained slides were then
rinsed and the coverslips were mounted using prolonged gold
anti-fade with DAPI (Beijing Solarbio Science & Technology Co.,
Ltd.). Fluorescent images of labeled cells in three randomly
selected views were captured using an Olympus fluorescence
microscope (Olympus Corporation; magnification, x10). Catalog
numbers and dilutions of primary antibodies are provided in
Table SII.
Statistical analysis
GraphPad Prism 6.0 (GraphPad Software, Inc.) was
used for all statistical analyses. Data are expressed as means ±
standard deviation. Data were analyzed by unpaired Student's
t-tests for studies that had 2 groups and one-way ANOVA with the
Bonferroni post hoc test was used to compare 3 or more groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
OGD and TGFβ1 induce CF to
myofibroblast differentiation and secretion of CSPGs
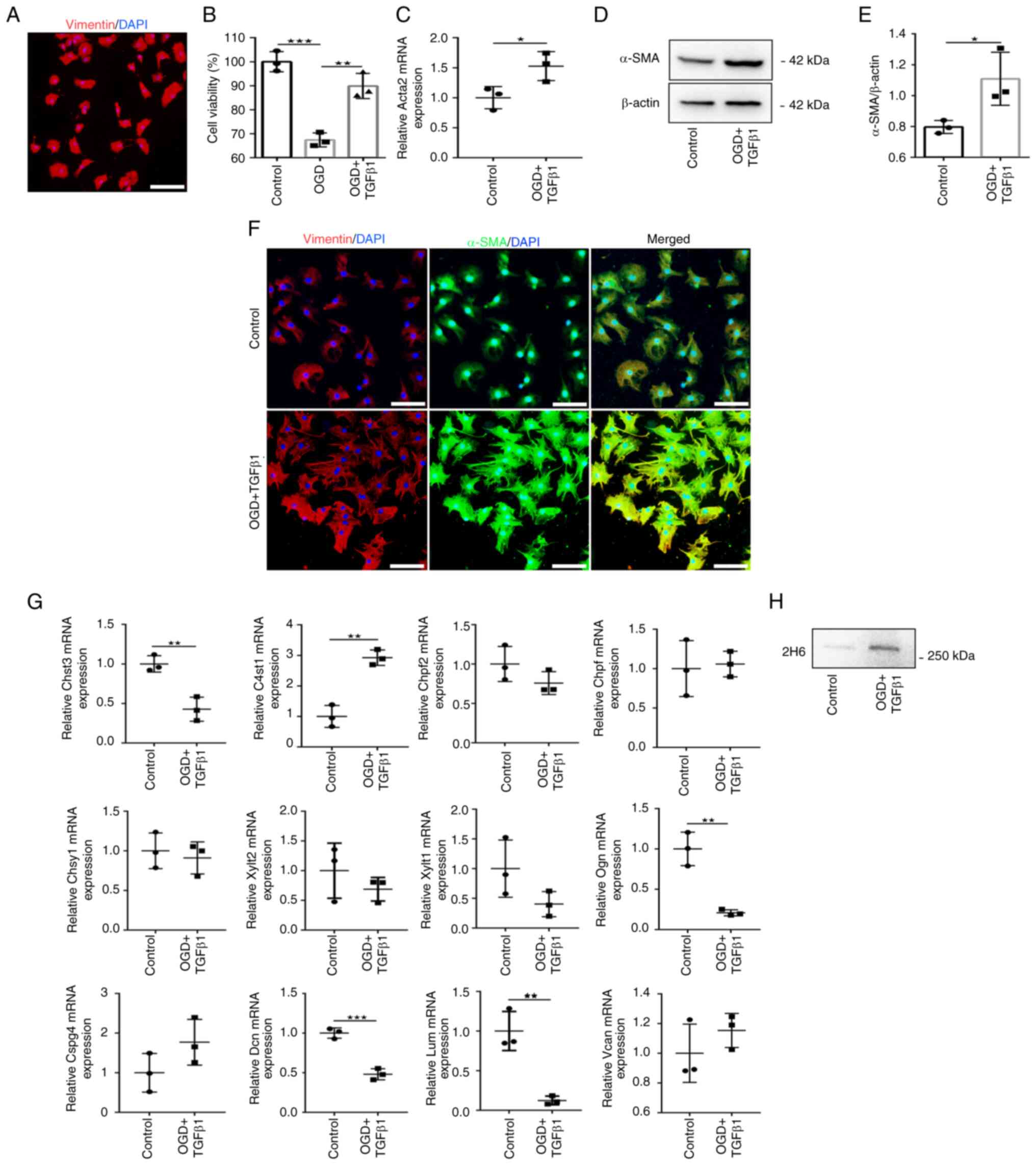
The purity of CFs was >95%, as determined by
positive staining for vimentin (Fig.
1A). As depicted in Fig. 1B,
OGD treatment significantly decreased the cell viability of CF
compared with control cells. TGFβ1 addition during reperfusion
stimulated showed a significant protection in the loss of CF
viability. Treatment of CFs with OGD and TGFβ1 stimulation
(treatment group) induced significant upregulation of the mRNA and
protein expression of the myofibroblast marker α-SMA (encoded by
Acta2) compared with the control group (Fig. 1C-E). In the treatment group,
immunofluorescence staining revealed that the expression of α-SMA
was co-labeled with vimentin and cells displayed visible signs of
hypertrophy (Fig. 1F). These
results demonstrated that OGD and TGFβ1 stimulation induce
fibroblast differentiation into myofibroblasts.
CSPG expression in the control and treatment groups
were next analyzed (Fig. 1G). The
mRNA expression of CSPG protein cores, sulfated glycosaminoglycan
(GAG) chain initiation and elongation enzymes and CS sulfatase
enzymes were profiled. Among these genes, only C4st1, the C4S
sulfotransferase gene, was significantly increased in the treatment
group compared with the control group. The expression of other
genes revealed that Chst3 (also called C6st1, the C6S
sulfotransferase gene), Ogn, Dcn and Lum mRNA levels were
significantly decreased in the treatment group. However, expression
of all other genes was not altered in response to stimulation. It
was confirmed that 2H6 levels, which probe for the major CS
component of C4S, using conditioned media collected from CFs, were
highly expressed in the treatment group compared with the control
group (Fig. 1H). These results
demonstrated that fibroblast differentiation into myofibroblasts
increases the secretion of C4S.
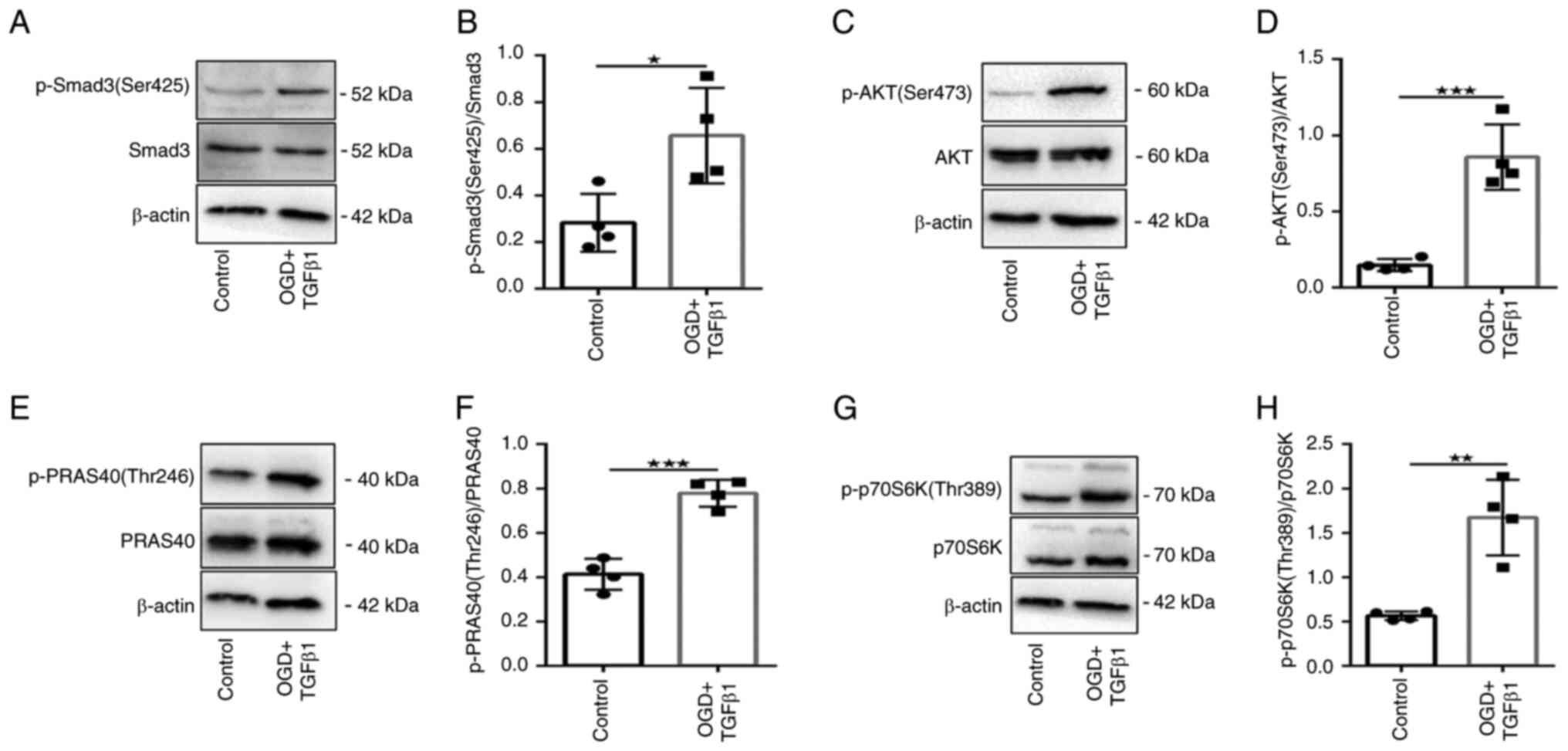
OGD and TGFβ1 induce Smad3 and
PI3K/Akt/mTOR signaling in CFs
As Smad signaling is rapid and relatively
short-lived (13), the
phosphorylation of Smad3 (Ser425) 2 h after TGFβ1 treatment was
detected. The data revealed that Smad3 (Ser425) phosphorylation was
significantly increased in the treatment group compared with the
control group (Fig. 2A and
B).
The phosphorylation of several key targets of the
PI3K/Akt/mTOR signaling pathway were next tested: Akt (Ser473),
PRAS40 (Thr246) and p70S6K (Thr389). Phosphorylation of Akt
(Ser473), PRAS40 (Thr246) and p70S6K (Thr389) was significantly
increased in the treatment group compared with the control group
(Fig. 2C-H). These data confirmed
that CF treatment with OGD and TGFβ1 induces Smad3 and
PI3K/Akt/mTOR signaling activation.
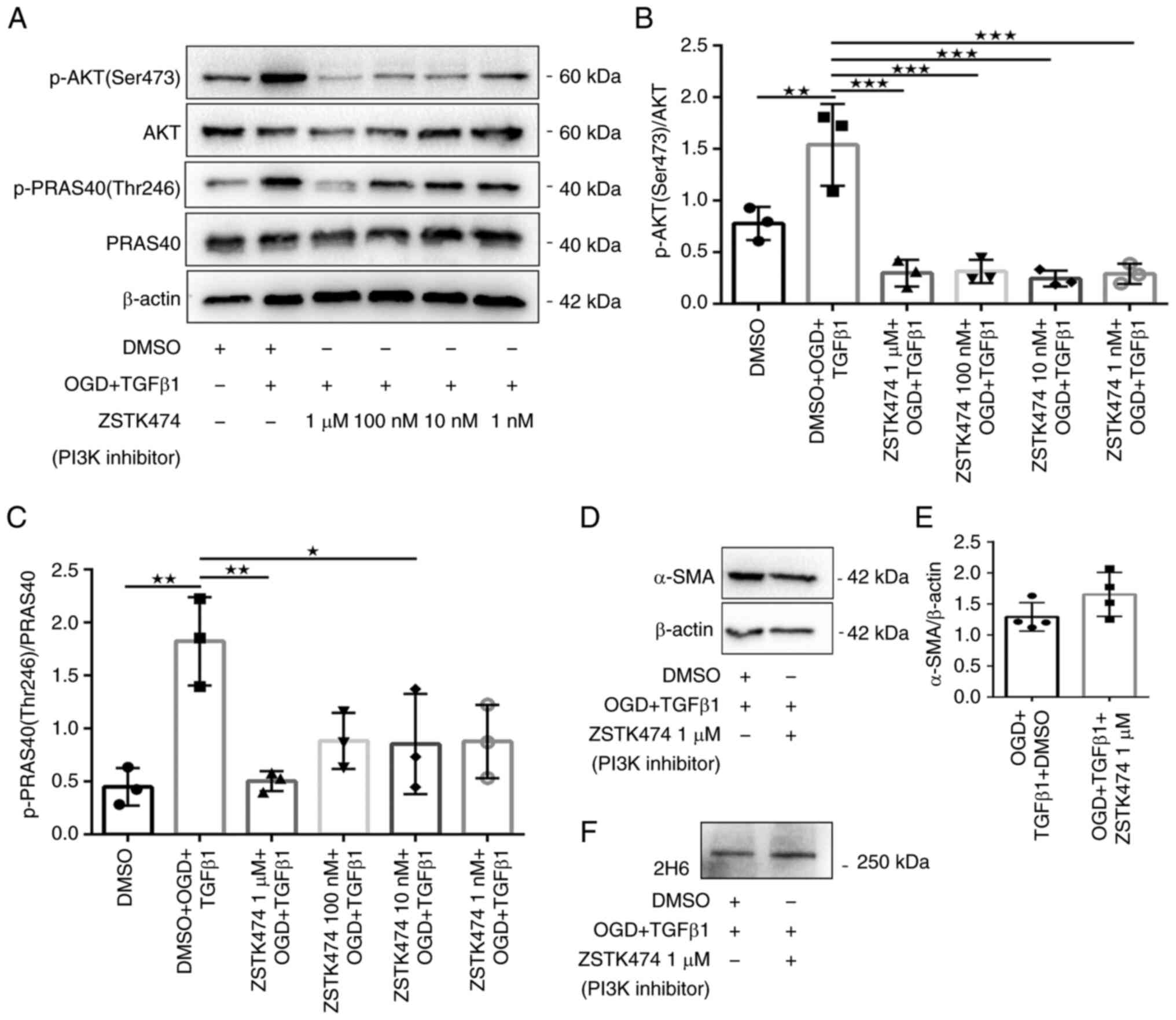
The effect of PI3K/Akt/mTOR pathway
inhibition on induced myofibroblast transformation and secreted
C4S
The role of the PI3K/Akt/mTOR signaling axis in OGD-
and TGFβ1-induced myofibroblast transformation and C4S secretion
were next investigated. Therefore, the effect of highly selective
inhibitors of critical components of the PI3K/Akt/mTOR axis were
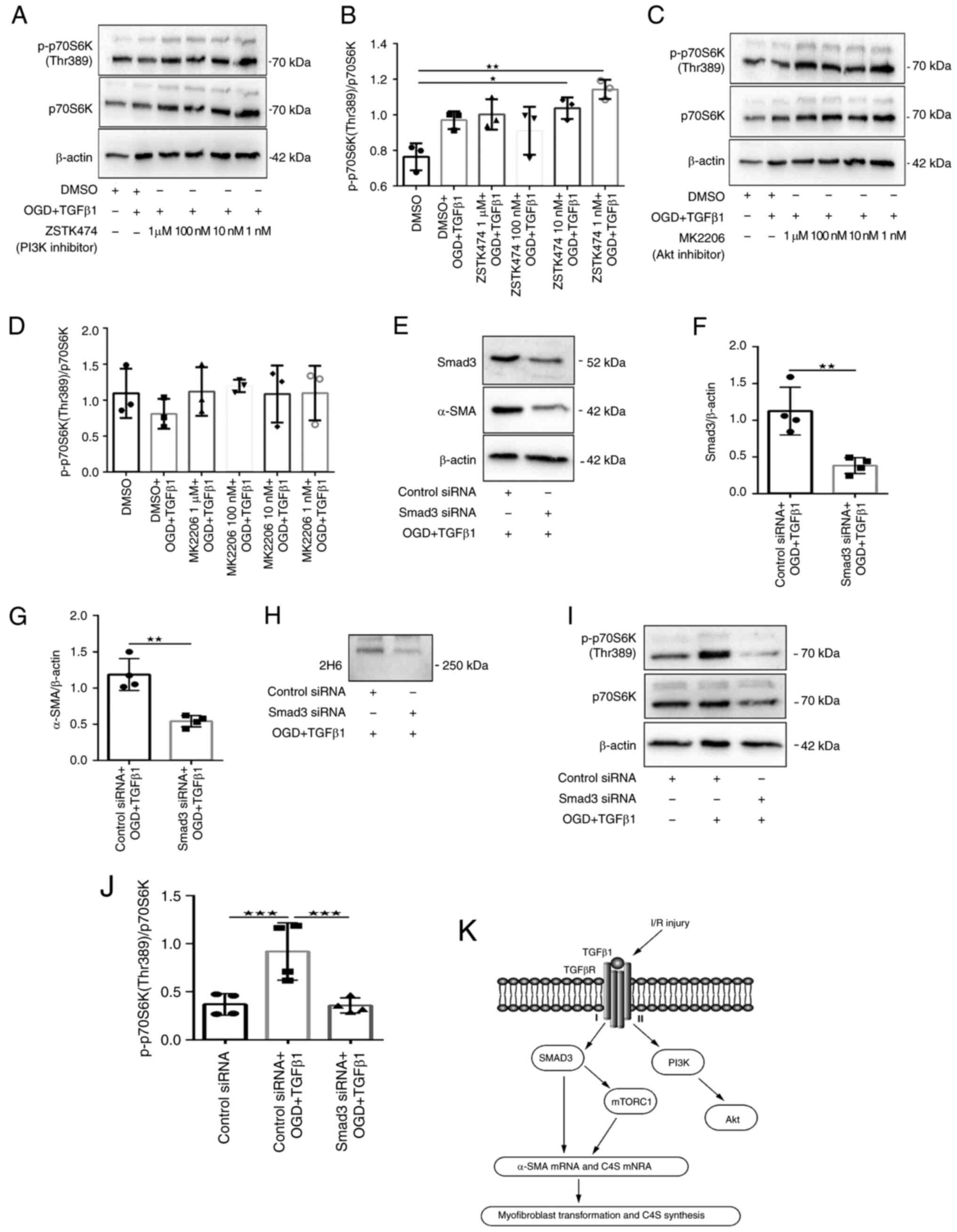
examined: PI3K (ZSTK474), Akt (MK2206) and mTOR (AZD8055). ZSTK474
reduced Akt Ser473 phosphorylation and inhibited the
phosphorylation of PRAS40 Thr246, a downstream substrate of Akt
(Fig. 3A-C). By contrast,
treatment of fibroblasts with 1 µM ZSTK474 had no effect on the
protein expression of α-SMA or 2H6 compared with DMSO-treated
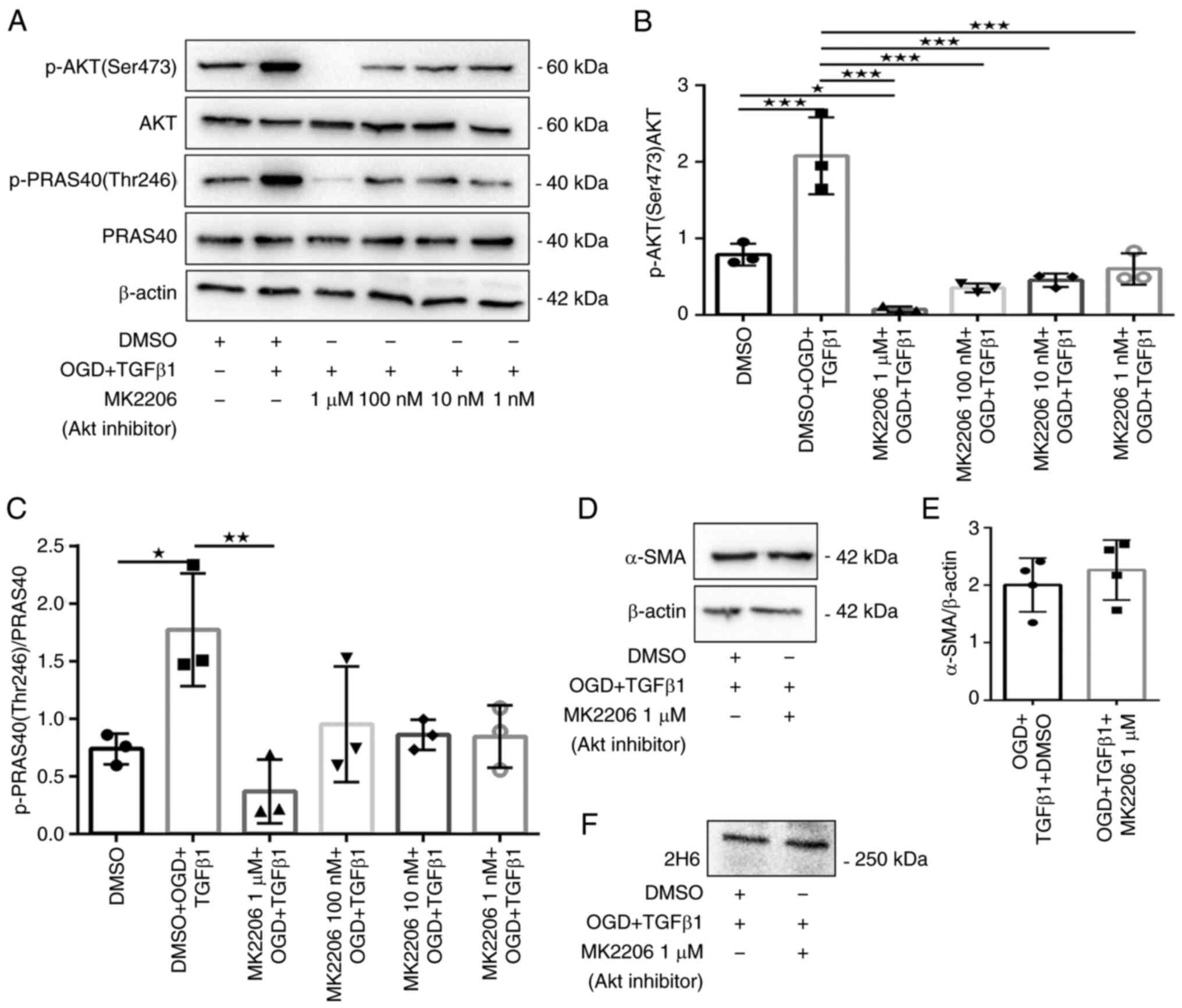
fibroblasts that received OGD and TGFβ1 stimulation (Fig. 3D-F). Similarly, fibroblasts treated
with MK2206 exhibited robust attenuation of Akt Ser473 and PRAS40
Thr246 phosphorylation (Fig.
4A-C), but fibroblasts treated with 1 µM MK2206 exhibited no
effects on protein expression of α-SMA or 2H6 (Fig. 4D-F). Fluorescence
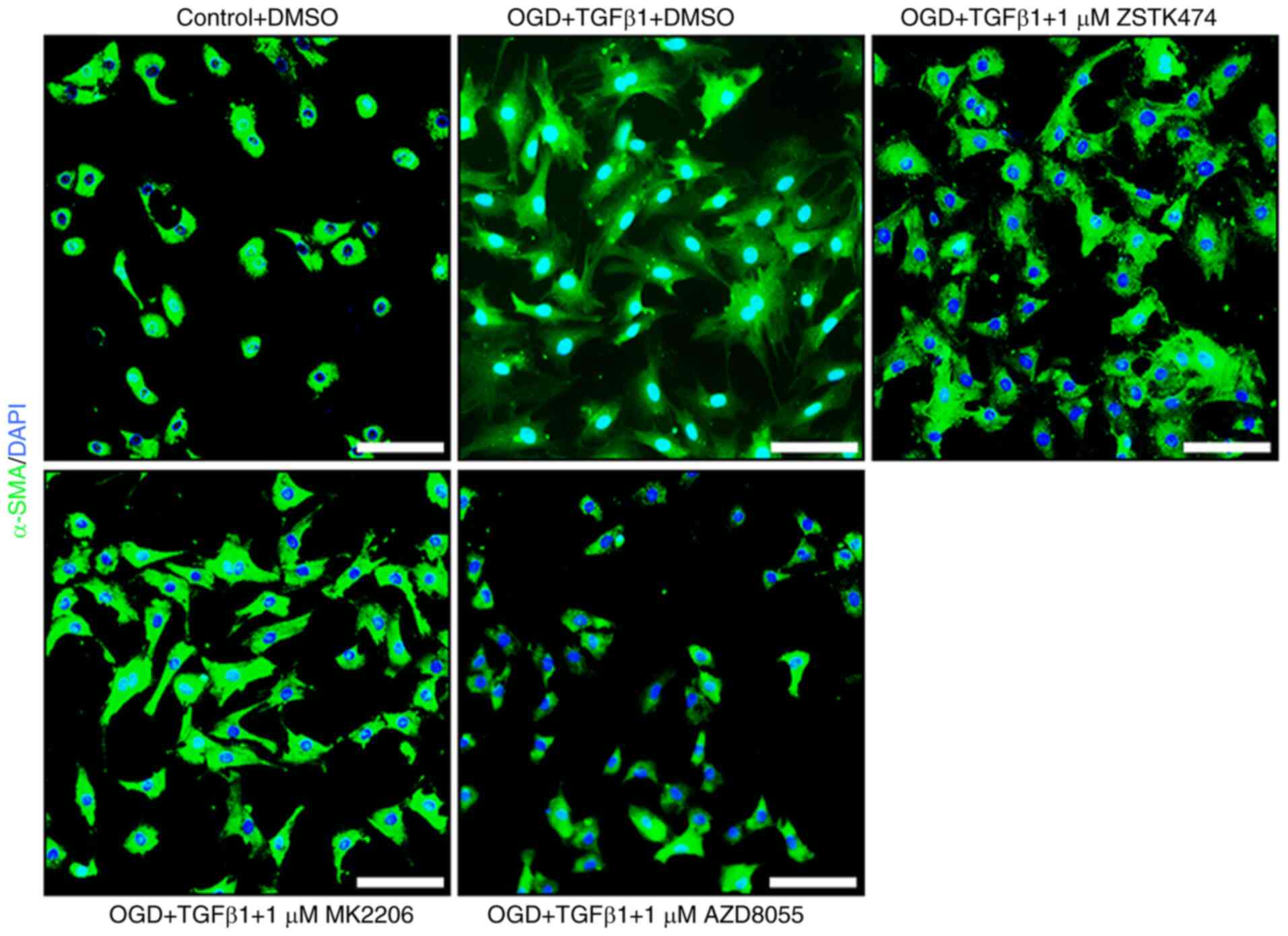
immunohistochemistry for α-SMA was then performed and fibroblasts
retained a reactive phenotype in the presence of 1 µM ZSTK474 or
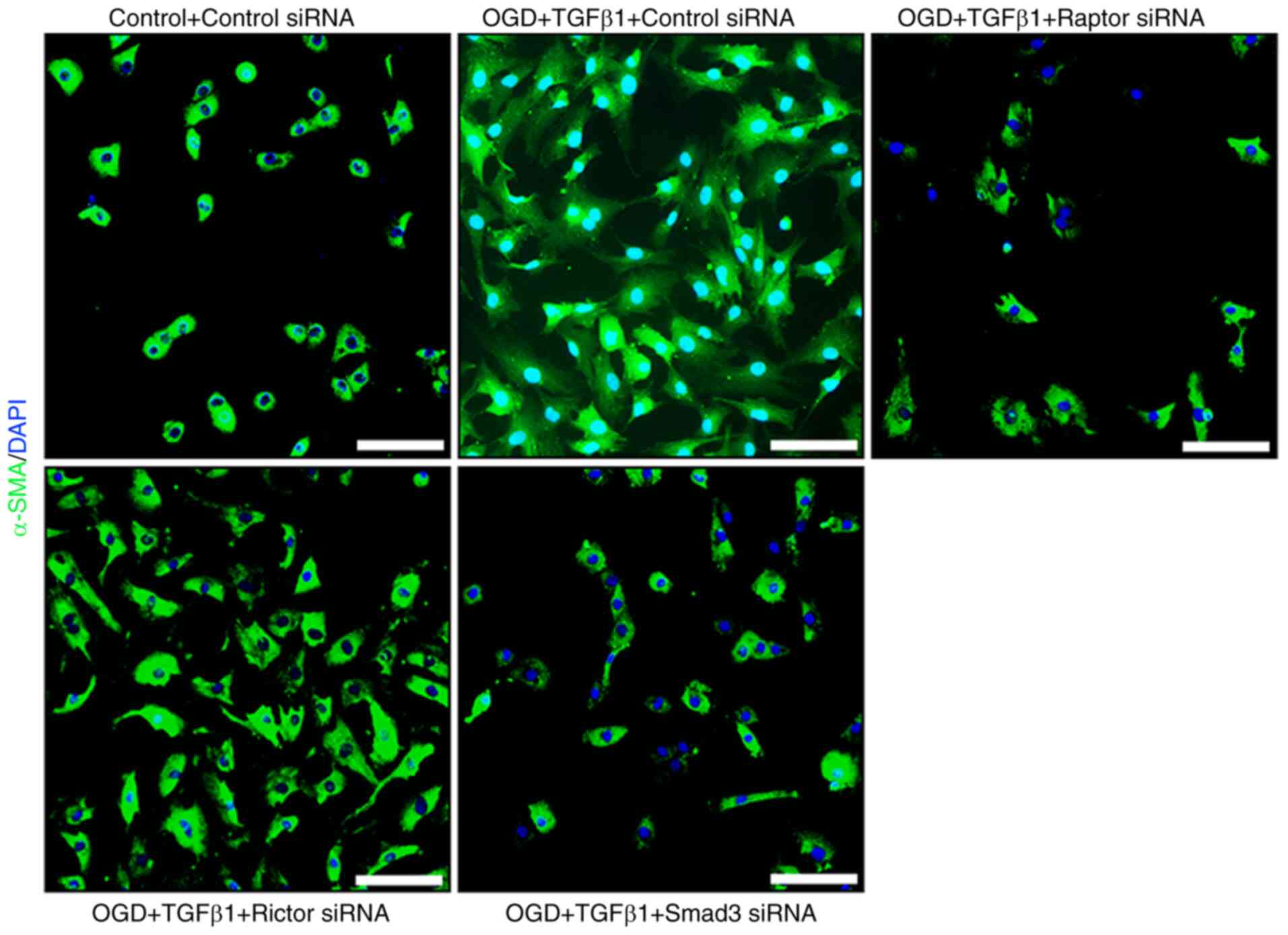
MK2206 (Fig. 5). These results
suggested that canonical PI3K/Akt signaling is redundant for
myofibroblast transformation and secretion of C4S.
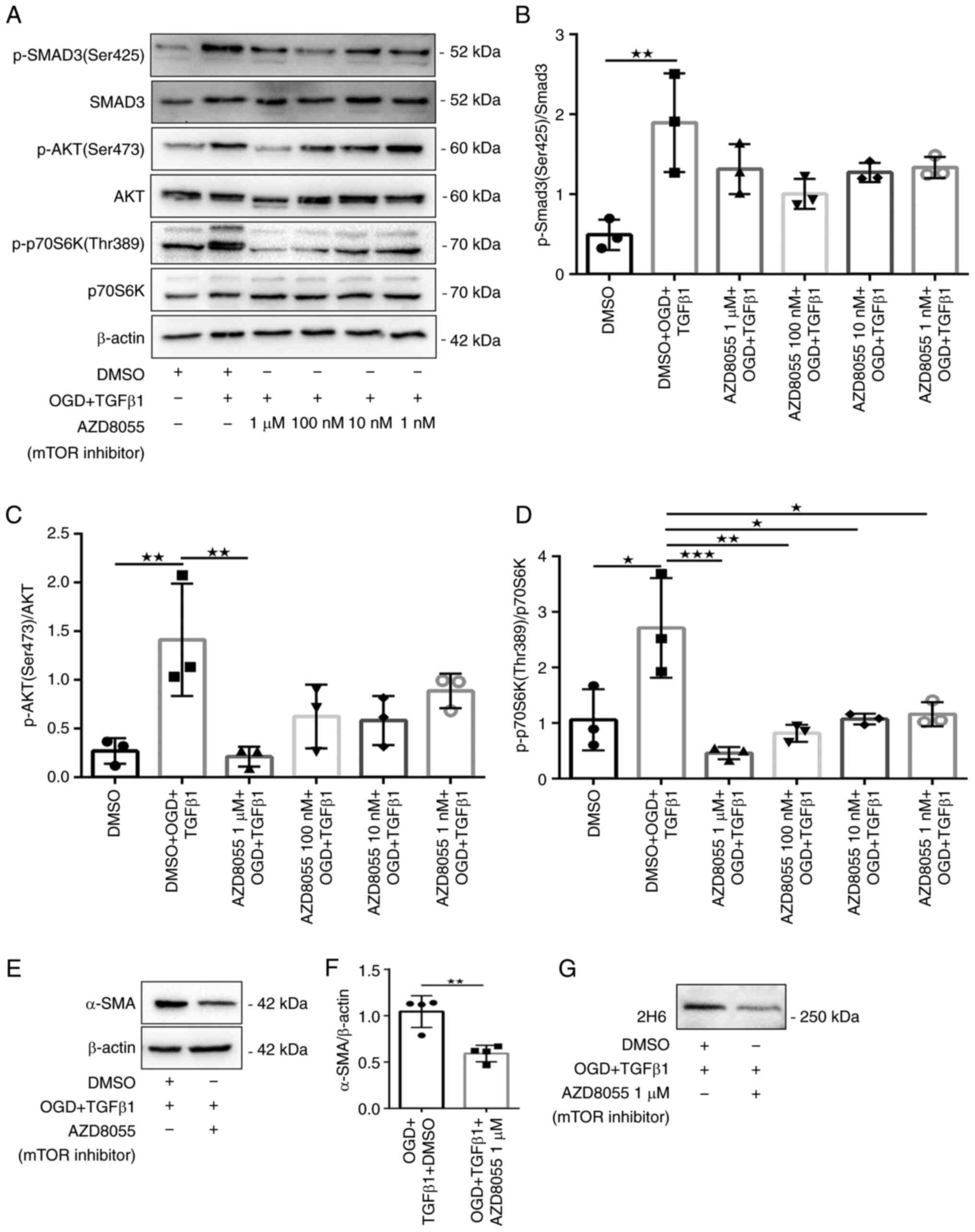
It was further tested whether the mTOR inhibitor
AZD8055, which targets both mTORC1 and mTORC2, was required for
OGD- and TGFβ1-induced myofibroblast transformation and secretion
of C4S. AZD8055 significantly attenuated the phosphorylation of
p70S6K (Thr389) and Akt (Ser473) (Fig.
6A, C and D). Fibroblasts treated with 1 µM AZD8055
also robustly inhibited the protein expression of α-SMA and 2H6
(Fig. 6E-G). They also exhibited
reduced cellular hypertrophy and immunoreactivity for α-SMA
compared with DMSO-treated fibroblasts that received OGD and TGFβ1
stimulation (Fig. 5). These data
suggested that mTOR signaling is necessary for myofibroblast
transformation and secretion of C4S.
Knockdown of mTORC1 or mTORC2
differentially regulates myofibroblast transformation and secretion
of C4S
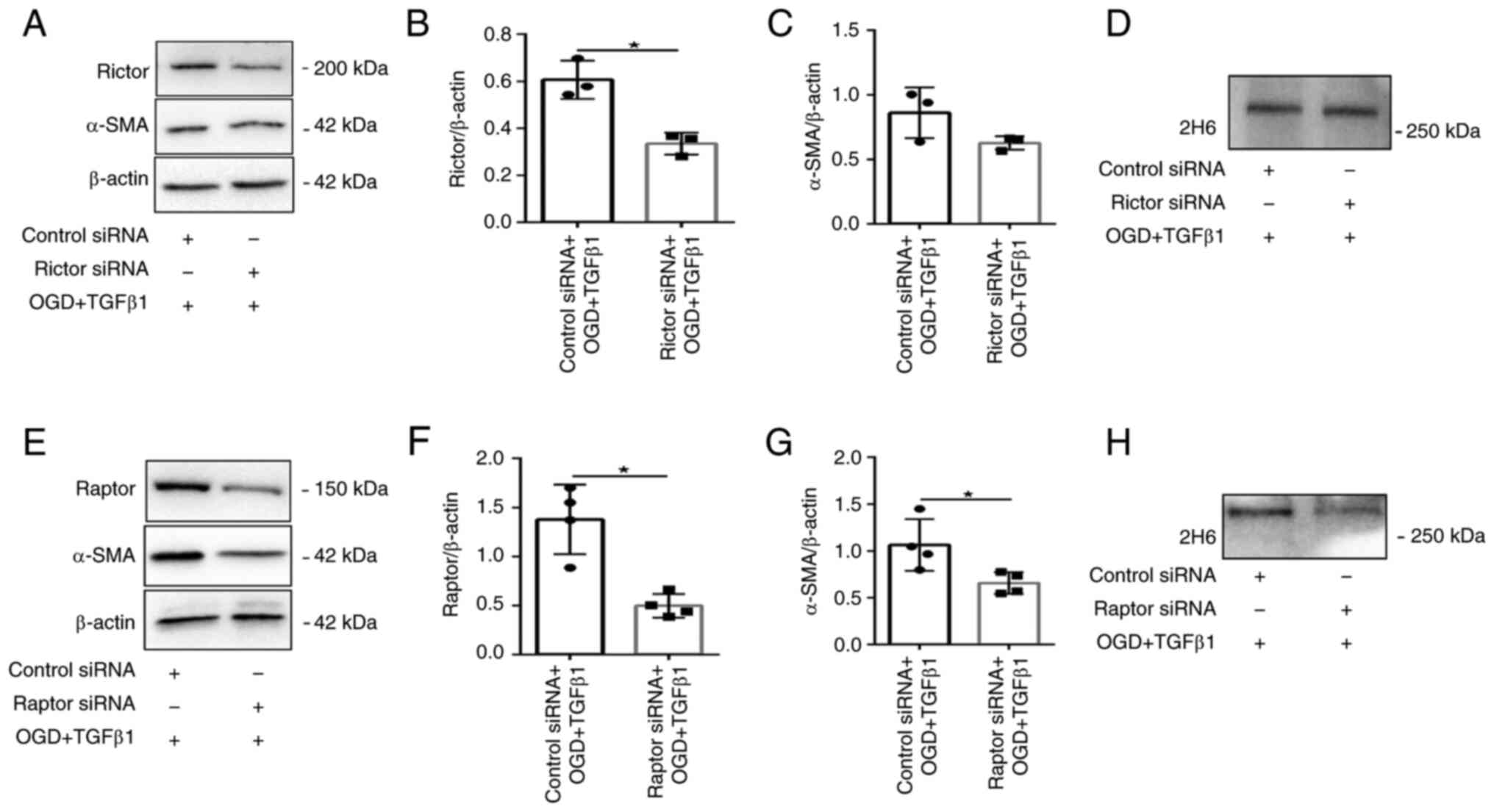
siRNA was used to silence the expression of Raptor
or Rictor in fibroblasts. Western blot analysis demonstrated a
significant knockdown of Rictor (mTORC2) or Raptor (mTORC1) in the
treatment group (Fig. 7A-B and
E-F). OGD- and TGFβ1-induced
expression of α-SMA and 2H6 protein levels was maintained in Rictor
siRNA knockdown cells (Fig. 7A,
C and D). It also exerted no clear effect on
cellular hypertrophy or α-SMA immunoreactivity (Fig. 8). By contrast, Raptor siRNA
knockdown resulted in significant inhibition of OGD- and
TGFβ1-induced protein expression of α-SMA and 2H6 (Fig. 7E, G and H).
Cells displayed reduced α-SMA immunoreactivity and cellular
hypertrophy (Fig. 8). These data
provided evidence that mTORC1 is a key signaling node involved in
myofibroblast transformation and secretion of C4S.
The PI3K pathway is considered to be the primary
pathway that activates mTORC1. However, neither PI3K nor Akt
inhibition affected the downstream signaling of mTORC1 (Fig. 9A-D). This may suggest another
potential PI3K/Akt-independent pathway regulating mTORC1 activation
in response to ODG and TGFβ1 stimulation. A previous study reported
that inhibition of Erk1/2 activity did not have a significant
effect on TGFβ-mediated CSPG expression (15). Another study confirmed that
canonical Smad signaling preceded the activation of mTORC1
signaling stimulated by TGFβ1(13). Therefore, the role of Smad3 in OGD-
and TGFβ1-induced myofibroblast transformation and secreted C4S
were next investigated.
Knockdown of Smad3 regulates
myofibroblast transformation and secretion of C4S
siRNA was used to silence the expression of Smad3 in
fibroblasts. Western blot analysis demonstrated a significant
knockdown of Smad3 in the treatment group (Fig. 9E-F). OGD- and TGFβ1-induced
expression of α-SMA and 2H6 protein levels was significantly
reduced in fibroblasts with Smad3 siRNA knockdown (Fig. 9E, G and H).
Cells displayed reduced α-SMA immunoreactivity and cellular
hypertrophy (Fig. 8). These data
provided evidence that Smad3 signaling regulates myofibroblast
transformation and secretion of C4S.
Myofibroblast transformation and C4S
expression may be mediated by cooperation between canonical Smad3
and mTORC1 signaling
Whether Smad is necessary to activate mTORC1 was
next examined. The data showed that mTORC1-dependent
phosphorylation of p70S6K (Thr389) was significantly reduced in
fibroblasts with Smad3 siRNA knockdown, suggesting that activation
of mTORC1 is Smad3 dependent (Fig.
9I-J). It was also confirmed that the mTOR inhibitor AZD8055
had no significant downregulation of phosphorylation of Smad3
(Ser425) (Fig. 6A-B). These
results may indicate that Smad signaling is temporally upstream of
mTOR. It was also confirmed that Smad3 knockdown was associated
with a significant reduction in the protein expression of α-SMA and
2H6. These data may suggest that OGD and TGFβ1 stimulation induce
myofibroblast transformation and secretion of C4S, which may be
mediated by cooperation between canonical Smad3 and mTORC1
signaling.
Discussion
The present study revealed that OGD and TGFβ1
stimulation induced fibroblast differentiation into myofibroblasts
and C4S synthesis in vitro. The data demonstrated that
mTORC1 was critical for OGD and TGFβ1 stimulation inducing
myofibroblast transformation and C4S expression, whereas the
upstream canonical PI3K/Akt axis was dispensable. This response may
be mediated via cooperation between canonical Smad3 and mTORC1
signaling.
It is well known that TGFβ1 exerts cardioprotective
effects and anti-apoptotic activity in CF under simulated I/R
(19). The data from the present
study are in accordance with the previous observations (19,20);
it was also demonstrated that TGFβ1 partly prevented OGD induced
CFs death. Following MI, TGFβ1 was also markedly upregulated and
the induced fibroblasts underwent myofibroblast
transdifferentiation and ECM protein synthesis (3). It was also found that CFs exposed to
OGD and TGFβ1 stimulation displayed visible signs of hypertrophy
and robust upregulation of α-SMA mRNA and protein levels compared
with the control group. This result suggested fibroblast activation
and differentiation into myofibroblasts. The cardiac ECM contains
collagens, glycosaminoglycans, glycoproteins and proteoglycans
(21). A previous study showed
that I/R infarct-derived CSPGs act through protein tyrosine
phosphatase receptor σ on sympathetic neurons, leading to
persistent postinfarction sympathetic denervation (10,11).
Another study demonstrated that excessive CSPG deposition is
associated with impaired cardiac function with ischemic HF in
animal models and patients (9).
The present study investigated the mRNA expression of CSPG protein
cores, GAG chain initiation and elongation enzymes and CS sulfatase
enzymes. Notably, compared with the control group, only C4st1, the
C4S sulfotransferase gene that was increased the 4-sulfation of GAG
chains, was highly expressed in the treatment group. It was also
confirmed that 2H6, which probes for the major CS component of C4S,
was highly expressed in the treatment group compared with the
control group. A previous study evaluated TGFβ-induced C4S
upregulation in CFs and found that C4S also accumulates in fibrotic
regions of the pathologically remodeled LV. It also showed that the
relative CS composition remains unchanged between healthy and
diseased individuals and C4S was the predominant CS component
(5). Another study on CNS injury
demonstrates that the GAG chains of CSPGs, especially those with
4-sulfated sugars within the glial scar, serve a major role in
inhibiting axonal regeneration (22).
Little is known regarding the molecular mechanisms
underlying CSPG expression by CFs following I/R. A previous study
confirms TGFβ-Smad3-mediated induction of 4-sulfation as a critical
determinant of astrocyte-secreted CSPGs (14). However, another study reported that
TGFβ mediated CSPG expression through non-Smad-mediated activation
of the PI3K-Akt-mTOR signaling pathway and showed that Smad and
MEK1/2 signaling were not required for TGFβ-induced CSPG expression
(15). As these two studies
reached opposing conclusions, this phenomenon aroused the authors'
interest.
The present study found that compared with the
control group, Smad3 (Ser425) phosphorylation was strongly
upregulated in the treatment group. Previous studies have indicated
that Smad3 is a critical mediator of myofibroblast differentiation
in vitro and is the dominant effector of cardiac fibrosis
in vivo, but Smad2 is not necessary for this response
(23,24). Therefore, Smad3 was knocked down
using siRNA in the treatment group and under these conditions,
expression of α-SMA and 2H6 was significantly reduced. Smad3
knockdown also reduced α-SMA immunoreactivity and cellular
hypertrophy. These data led to the conclusion that knocking down
Smad3 to inhibit the reactive phenotype of CFs also reduced C4S
synthesis.
In addition to the canonical Smad-mediated signaling
pathway, TGFβ can activate numerous Smad-independent signaling
pathways. The PI3K-Akt-mTOR signaling pathway regulates diverse
biological processes, such as metabolism, cell cycle progression,
proliferation, growth, autophagy and protein synthesis. The
PI3K-Akt-mTOR signaling pathway has previously been implicated in
the transformation of cardiac fibroblasts to a myofibroblast
phenotype and ECM synthesis (25,26).
In the present study, phosphorylated Akt (Ser473), PRAS40 (Thr246)
and p70S6K (Thr389) were also clearly upregulated in the treatment
group compared with the control group. These data confirmed that CF
treatment with OGD and TGFβ1 induced PI3K/Akt/mTOR signaling
activation. A previous study showed that canonical PI3K/Akt
signaling has no effect on TGFβ1-induced collagen deposition
(13). Thus, the present study
further investigated the effect of the PI3K-Akt-mTOR signaling
pathway on inducing α-SMA and C4S expression in the treatment
group. The present study confirmed that CFs treated with class 1
PI3K or Akt inhibitors (ZSTK474 or MK2206) in the treatment group
markedly attenuated Akt signaling but had no effect on the protein
expression of α-SMA or 2H6. Fibroblasts retained a reactive
phenotype in the presence of 1 µM ZSTK474 or MK2206. This result
contrasted with a previous study reporting a role for PI3K in
TGFβ1-mediated CSPG synthesis. This may be due to the earlier study
using the first-generation PI3K inhibitor LY294002, which can
inhibit a variety of other PI3K-related proteins, including mTOR
(13). The present study used
ZSTK474, which exhibits excellent selectivity for all class 1 PI3K
isomers over mTOR.
The present study further demonstrated that an mTOR
inhibitor (AZD8055) attenuated the phosphorylation of p70S6K
(Thr389) and Akt (Ser473) and significantly reduced the protein
expression of α-SMA and 2H6 in the treatment group. mTOR inhibition
also reduced cellular hypertrophy and immunoreactivity. This
suggested that mTOR serves a key role in myofibroblast
transformation and the expression of C4S. The present study further
investigated the relative contributions of mTORC1 and mTORC2 by
knocking down either Raptor or Rictor using siRNA. The data
suggested that mTORC1 mediated the expression of α-SMA and C4S in
CFs induced by OGD and TGFβ1 stimulation.
Notably, the results suggested that neither PI3K nor
Akt inhibition affected mTORC1 downstream signaling expression in
the treatment group. A previous study showed that in the lung
fibroblast response to TGFβ1 stimulation, maximal phosphorylation
of mTORC1 substrates is consistently observed at least 10 h before
maximal phosphorylation of Akt (13). This may suggest another potential
PI3K/Akt-independent pathway regulating mTORC1 activation. The
present study confirmed that Smad3 is necessary for the activation
of mTORC1(13). It also examined
whether this reflection affected the expression of α-SMA and C4S in
the treatment group and reached a similar conclusion: Targeted
knockdown of Smad3 with siRNA in the treatment group attenuated
mTORC1-dependent phosphorylation of p70S6K and significantly
reduced the protein expression of α-SMA and 2H6. In addition, it
reduced cellular hypertrophy. The present study also demonstrated
that AZD8055 had no effect on the phosphorylation of Smad3
(Ser425). This indicated that Smad3 preceded the activation of
mTORC1 and that the activation of mTORC1 was Smad3-dependent. These
data may indicate that OGD and TGFβ1 stimulation induced
myofibroblast transformation and that C4S expression is mediated by
cooperation between canonical Smad3 and mTORC1 signaling.
There are some limitations to the present study.
First, due to limited funding, only a few PI3K/Akt/mTOR pathway
targets were selected to verify the hypothesis and no rescue
experiments for gene overexpression or follow-up experiments were
performed. Second, evidence from in vivo experiments was
lack and others should further verify the findings using in
vivo experiments.
In conclusion, the present study presented evidence
that mTORC1 was critical for promoting ODG- and TGFβ1-induced
expression of α-SMA and C4S, whereas the upstream canonical
PI3K/Akt axis was dispensable. This response may be mediated by
cooperation between canonical Smad3 and mTORC1 signaling. Although
digesting CSPG GAG chains with the bacterial enzyme chondroitinase
ABC (ChABC) or rhASB has been shown to promote axonal extension in
the CNS (22,27), ChABC has failed to reach clinical
trials. Therefore, the present study tried to link the synthesis of
C4S with the transdifferentiation of cardiac fibroblasts. These
observations suggested that inhibiting myofibroblast
differentiation may reduce the expression of C4S. This may provide
additional insight into the regeneration of sympathetic nerves and
the reduction of fibrosis after MI at the cellular level.
Supplementary Material
Primer list for reverse
transcription-quantitative PCR.
Antibodies list.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CL and ZZ contributed to the conception and design
of the present study. CL performed the experiments, collected the
data and performed statistical analysis with the help of YP, YZ,
WK, YL and YH. CL drafted the manuscript, which was corrected and
revised by ZZ. ZZ and YP confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were performed in
accordance with the Guide for the Care and Use of Laboratory
Animals published by the National Research Council and were
approved by the Animal Care Committee of the Gansu University of
Chinese Medicine (approval number 2019-212).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wu Y, Liu H and Wang X: Cardioprotection
of pharmacological postconditioning on myocardial
ischemia/reperfusion injury. Life Sci. 264(118628)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lai TC, Lee TL, Chang YC, Chen YC, Lin SR,
Lin SW, Pu CM, Tsai JS and Chen YL: MicroRNA-221/222 mediates
ADSC-Exosome-Induced cardioprotection against ischemia/reperfusion
by targeting PUMA and ETS-1. Front Cell Dev Biol.
8(569150)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chen W and Frangogiannis NG: Fibroblasts
in post-infarction inflammation and cardiac repair. Biochim Biophys
Acta. 1833:945–953. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Molkentin JD, Bugg D, Ghearing N, Dorn LE,
Kim P, Sargent MA, Gunaje J, Otsu K and Davis J:
Fibroblast-specific genetic manipulation of p38 mitogen-activated
protein kinase in vivo reveals its central regulatory role in
fibrosis. Circulation. 136:549–561. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhao RR, Ackers-Johnson M, Stenzig J, Chen
C, Ding T, Zhou Y, Wang P, Ng SL, Li PY, Teo G, et al: Targeting
chondroitin sulfate glycosaminoglycans to treat cardiac fibrosis in
pathological remodeling. Circulation. 137:2497–2513.
2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chelini G, Pantazopoulos H, Durning P and
Berretta S: The tetrapartite synapse: A key concept in the
pathophysiology of schizophrenia. Eur Psychiatry. 50:60–69.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hayes AJ and Melrose J: Neural tissue
homeostasis and repair is regulated via CS and DS proteoglycan
motifs. Front Cell Dev Biol. 9(696640)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kai Y, Tomoda K, Yoneyama H, Kitabatake M,
Nakamura A, Ito T, Yoshikawa M and Kimura H: Silencing of
carbohydrate sulfotransferase 15 hinders murine pulmonary fibrosis
development. Mol Ther Nucleic Acids. 6:163–172. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Barallobre-Barreiro J, Radovits T, Fava M,
Mayr U, Lin WY, Ermolaeva E, Martínez-López D, Lindberg EL,
Duregotti E, Daróczi L, et al: Extracellular matrix in heart
failure: Role of ADAMTS5 in proteoglycan remodeling. Circulation.
144:2021–2034. 2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gardner RT and Habecker BA:
Infarct-derived chondroitin sulfate proteoglycans prevent
sympathetic reinnervation after cardiac ischemia-reperfusion
injury. J Neurosci. 33:7175–7183. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Gardner RT, Wang L, Lang BT, Cregg JM,
Dunbar CL, Woodward WR, Silver J, Ripplinger CM and Habecker BA:
Targeting protein tyrosine phosphatase sigma after myocardial
infarction restores cardiac sympathetic innervation and prevents
arrhythmias. Nat Commun. 6(6235)2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Miyata S and Kitagawa H: Chondroitin
sulfate and neuronal disorders. Front Biosci (Landmark Ed).
21:1330–1340. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Woodcock HV, Eley JD, Guillotin D, Platé
M, Nanthakumar CB, Martufi M, Peace S, Joberty G, Poeckel D, Good
RB, et al: The mTORC1/4E-BP1 axis represents a critical signaling
node during fibrogenesis. Nat Commun. 10(6)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Susarla BT, Laing ED, Yu P, Katagiri Y,
Geller HM and Symes AJ: Smad proteins differentially regulate
transforming growth-β-mediated induction of chondroitin sulfate
proteoglycans. J Neurochem. 119:868–878. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jahan N and Hannila SS: Transforming
growth factor β-induced expression of chondroitin sulfate
proteoglycans is mediated through non-Smad signaling pathways. Exp
Neurol. 263:372–384. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tian K, Liu Z, Wang J, Xu S, You T and Liu
P: Sirtuin-6 inhibits cardiac fibroblasts differentiation into
myofibroblasts via inactivation of nuclear factor kappaB signaling.
Transl Res. 165:374–386. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ryou MG and Mallet RT: An in vitro
oxygen-glucose deprivation model for studying ischemia-reperfusion
injury of neuronal cells. Methods Mol Biol. 1717:229–235.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Olivares-Silva F, Espitia-Corredor J,
Letelier A, Vivar R, Parra-Flores P, Olmedo I, Montenegro J,
Pardo-Jiménez V and Díaz-Araya G: TGF-β1 decreases CHOP expression
and prevents cardiac fibroblast apoptosis induced by endoplasmic
reticulum stress. Toxicology In vitro. 70(105041)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vivar R, Humeres C, Ayala P, Olmedo I,
Catalán M, García L, Lavandero S and Díaz-Araya G: TGF-β1 prevents
simulated ischemia/reperfusion-induced cardiac fibroblast apoptosis
by activation of both canonical and non-canonical signaling
pathways. Biochim Biophys Acta. 1832:754–762. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Frangogiannis NG: Cardiac fibrosis: Cell
biological mechanisms, molecular pathways and therapeutic
opportunities. Mol Aspects Med. 65:70–99. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Pearson CS, Mencio CP, Barber AC, Martin
KR and Geller HM: Identification of a critical sulfation in
chondroitin that inhibits axonal regeneration. ELife.
7(e37139)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Khalil H, Kanisicak O, Prasad V, Correll
RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al:
Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac
fibrosis. J Clin Invest. 127:3770–3783. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Huang S, Chen B, Su Y, Alex L, Humeres C,
Shinde AV, Conway SJ and Frangogiannis NG: Distinct roles of
myofibroblast-specific Smad2 and Smad3 signaling in repair and
remodeling of the infarcted heart. J Mol Cell Cardiol. 132:84–97.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang J, Fan G, Zhao H, Wang Z, Li F,
Zhang P, Zhang J, Wang X and Wang W: Targeted inhibition of focal
adhesion kinase attenuates cardiac fibrosis and preserves heart
function in adverse cardiac remodeling. Sci Rep.
7(43146)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bradley JM, Spaletra P, Li Z, Sharp TE
III, Goodchild TT, Corral LG, Fung L, Chan KW, Sullivan RW,
Swindlehurst CA and Lefer DJ: A novel fibroblast activation
inhibitor attenuates left ventricular remodeling and preserves
cardiac function in heart failure. Am J Physiol Heart Circ Physiol.
315:H563–H570. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kwok JC, Heller JP, Zhao RR and Fawcett
JW: Targeting inhibitory chondroitin sulphate proteoglycans to
promote plasticity after injury. Methods Mol Biol. 1162:127–138.
2014.PubMed/NCBI View Article : Google Scholar
|