Introduction
Alzheimer's disease (AD) is one of the most common
neurodegenerative diseases, with an estimated global prevalence of
6.2 million (1). AD has multiple
clinical symptoms, including memory loss, confusion regarding time
or place, decline in the ability to make decisions and judgments,
aphasia, apraxia and agnosia, in addition to other symptoms,
including fatigue, sleep disturbance, anxiety, depression and
gastrointestinal dysfunction. Based on the knowledge acquired in
recent years, various genomic factors, such as the presenilin 1
gene, amyloid-β (Aβ) precursor protein (APP) gene and
apolipoprotein E gene, as well as certain epigenetic factors,
contribute to the occurrence and progression of AD (2-4).
A major pathological hallmark of AD is the presence of Aβ plaques.
Aβ is generated from the Aβ precursor protein encoded by the APP
gene, which is widely expressed in the central nervous system. Aβ
peptides cause neurotoxicity in the brain by disrupting synaptic
plasticity and promoting the production of nitric oxide formation.
Furthermore, Aβ induces an influx of calcium ions
(Ca2+), which may then cause neuronal apoptosis. In
fact, mitochondria mainly regulate cellular Ca2+
homeostasis via the expression of mitochondrial Ca2+
uniporter (MCU) to maintain neuronal survival and function
(5,6). Conversely, dysregulation of MCU
induces mitochondrial malfunction, contributing to neuronal
apoptosis (6,7). Furthermore, mitochondrial dysfunction
is another key factor involved in the pathogenesis of AD (8). Fu et al (9) discovered mitochondrial dysfunction in
an AD mouse model. Taken together, these findings demonstrate that
both Aβ and mitochondria have key roles in the pathogenesis of
AD.
Ginkgolides are natural products isolated from
Ginkgo biloba leaves. Ginkgolide K (GK) is a ginkgolide and
a diterpene lactone compound. Multiple pharmacological properties
of GK have been reported in previous studies, including
neuroprotection (10), regulation
of inflammation (11),
antioxidative stress (12) and
potential benefits against ischemic stroke (13). In particular, Ma observed that GK
treatment markedly protected PC12 cells against
H2O2-induced cytotoxicity by ameliorating
oxidative stress and mitochondrial dysfunction (14).
In the present study, the potential neuroprotective
effect of GK on neuronal cell survival in AD pathology was
explored. The results suggested that GK treatment decreased MCU
expression, which contributes to the maintenance of calcium
homeostasis and benefits neuronal cell survival (Fig. S1). Furthermore, GK supplementation
regulated MCU expression in the brains of AD model mice and
improved their cognitive ability.
Materials and methods
Cell culture
The human brain neuroblast cell line SH-SY5Y
(CRL-2266) and the human cell line 293T (CRL-3216) were purchased
from the American Type Culture Collection and were maintained in
Eagle's minimum essential medium (Invitrogen; Thermo Fisher
Scientific, Inc.) or Dulbecco's Modified Eagle's Medium (DMEM,
HyClone; Cytiva), both of which were supplemented with 10%
ultracentrifuged fetal bovine serum (FBS), penicillin (100 U/ml)
and streptomycin (10 mg/ml; all from Invitrogen; Thermo Fisher
Scientific, Inc.) at 37˚C in a humidified atmosphere with 5%
CO2. SH-SY5Y and 293T cells were seeded in a 96-well
plate at 5,000 cells per well and maintained overnight for
attachment. Thereafter, the cells were subjected to the different
treatments for 72 h.
Aβ25-35 was purchased from MilliporeSigma
and diluted in DMSO for use at a concentration of 25 µm (15,16),
while cells were treated with GK at a concentration of 50 µg/ml
(12).
Transfection
SH-SY5Y cells were seeded into the wells of a
12-well plate at 105 cells per well and cultured at 37˚C
with 5% CO2. The cells were then transfected with small
interfering (si)RNA against MCU (5'-GGAAAGGGAGCUUAUUGAA-3') or
negative control siRNA (5'-UUCUCCGAACGUGUCACGU-3') from Sangon
Biotech at a final concentration of 40 nM using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's protocol. The
transfection efficiency was validated by reverse
transcription-quantitative (RT-q)PCR or western blot analysis. At
24 h post-transfection, cells were used for the subsequent
experiments.
Plasmid construction and
transfection
The protein-coding sequences of human MCU cDNAs
(sequence proofed, OriGene Technologies, Inc.) was subcloned into
the pcDNA3.1 expression vector (Thermo Fisher Scientific, Inc.).
The expression vector and 4 µg lentiviral vector (MilliporeSigma)
were then transfected into 293T cells using the calcium phosphate
precipitation method for 48 h following the manufacturer's
protocol. Subsequently, the culture medium of 293T cells was
replaced with DMEM supplemented with 5% FBS, followed by incubation
for 48 h. The viral supernatant was then collected, centrifuged at
250 x g for 5 min at 4˚C and passed through a filter membrane (pore
size, 0.45 µm; EMD Millipore). SH-SY5Y cells were incubated at 37˚C
with the recombinant lentiviral vectors at a multiplicity of
infection of 30 using the FuGENE Transfection Reagent (Roche
Diagnostics) and were used for further experiments after 72 h.
Western blot analysis was performed to verify the interference
efficiency.
Western blot analysis
Total protein was collected from the cells using
RIPA buffer (MilliporeSigma) containing protease inhibitors and the
protein concentrations were determined by the BCA method. Equal
amounts of protein (20 µg/lane) were loaded and separated on
SDS-polyacrylamide gels (8-10%) for electrophoresis. Thereafter,
the protein bands on the gels were transferred to nitrocellulose
membranes (MilliporeSigma). The membranes were then blocked with 5%
bovine serum albumin (diluted in Tris-Cl-buffered saline with 0.1%
Tween-20) for 2 h at room temperature and incubated with primary
antibodies at 1:3,000 dilution overnight at 4˚C. Subsequently, the
membrane was incubated with secondary antibodies (1:3,000 dilution;
anti-mouse IgG or anti-rabbit IgG; cat. nos. ab205719 and ab205718;
Abcam). The blot was then detected using the Western Bright ECL
western blotting detection kit (Bio-Rad Laboratories, Inc.). Equal
sample loading was verified by detection of GAPDH. The primary
antibodies were as follows: Mouse monoclonal anti-GAPDH (cat. no.
sc-32233; Santa Cruz Biotechnology, Inc.), rabbit monoclonal
anti-MCU (cat. no. ab272488; Abcam), rabbit polyclonal anti-Aβ
(cat. no. 51-2700; Invitrogen; Thermo Fisher Scientific, Inc.),
mouse monoclonal anti-tau (cat. no. sc-390476; Santa Cruz
Biotechnology, Inc.) and rabbit monoclonal anti-tau (phospho
Ser214; cat. no. ab170892; Abcam).
RNA isolation and RT-qPCR
Total RNA was extracted from the cells using the
RNeasy kit (Qiagen GmnH) following the manufacturer's protocol and
then reverse-transcribed into cDNA using SuperScript™ III Reverse
Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.). mRNA
expression was measured using an ABI PRISM 7500 Real-Time qPCR
System according to the manufacturer's protocol. Candidate gene
expression was measured using a SYBR Green-based reagent (SYBR
GreenER qPCR SuperMix for iCycler; Invitrogen; Thermo Fisher
Scientific, Inc.) and a real-time qPCR system (ABI PRISM 7500
Real-Time PCR System; Thermo Fisher Scientific, Inc). The cycling
conditions for the reaction were as follows: 10 min at 95˚C for
initial hold; then 40 cycles of 15 sec at 95˚C for denaturation, 30
sec at 60˚C for annealing and a 30 sec extension at 72˚C. The
following primers were used: MCU forward, 5'-ACCGGACGGTACACCAGAG-3'
and reverse, 5'-GATAGGCTTGAGTGTGAACTGAC-3'; and GAPDH forward,
5'-TGTGGGCATCAATGGATTTGG-3' and reverse,
5'-ACACCATGTATTCCGGGTCAAT-3'. All PCR analyses were performed in
triplicate and expression values were quantified with the
corresponding standard curves. The expression of MCU was normalized
to GAPDH expression. Relative quantitation of gene expression was
performed using the 2-ΔΔCq method (17).
Cell viability assay
SH-SY5Y cells with various transfections were seeded
into 96-well plates (5,000 cells/well) and maintained overnight for
attachment. The cells were then treated with Aβ, GK or Aβ+GK
respectively and further cultured for 72 h. Subsequently, cell
viability was measured via the WST-1 assay (Roche Diagnostics)
according to the manufacturer's protocol. The absorbance was read
at 440 nm using a Multimode Plate Reader (Varioskan™ LUX; Thermo
Fisher Scientific, Inc.).
Apoptosis assay
Following cell treatment as specified above, flow
cytometry was applied to evaluate the apoptosis of SH-SY5Y cells
in vitro. After 72 h of treatment, the cells were collected,
washed with PBS and resuspended in 100 µl binding buffer at a
concentration of 106 cells/ml. Subsequently, 5 µl
annexin V-FITC and 10 µl propidium iodide (both purchased from
Beyotime Institute of Biotechnology) were added to the cell
suspension, which was then incubated for 15 min at room temperature
in the dark. Finally, the rate of apoptosis in each cell sample was
examined using a FACScan flow cytometer (BD Biosciences) and the
data were analyzed by FlowJo software (V10.6; BD Biosciences).
Caspase-3/8 activity
The Caspase-3 (cat. no. C1168S) and Caspase-8 (cat.
no. C1152) kits were both purchased from Beyotime Institute of
Biotechnology and used to measure the respective activities of
Caspase-3/8 following the manufacturer's protocols. The cells were
collected after the treatments and total protein was extracted from
the cells using lysis buffer, and then mixed with 85 µl reaction
buffer. Subsequently, 5 µl Leu-Glu-His-Asp-p-nitroanilide was added
to the protein samples, followed by incubation at 37˚C for 2 h. A
multiplate reader (Varioskan™ LUX; Thermo Fisher Scientific, Inc.)
was used to measure the activities of Caspase-3/8 at 450 nm.
Ca2+ uptake assay
The mitochondria were isolated by using a
mitochondria isolation kit for Cultured Cells (cat. no. 89874;
Thermo Fisher Scientific, Inc.) and then dissolved in swelling
buffer provided with the kit. The protein concentration of the
mitochondria solution was determined by the BCA method at 5 mg/ml.
After 30 min of application of CaCl2 (100 µM) to the
solution, the mitochondria were collected by centrifugation (3,000
x g) for 15 min at 4˚C. The pellets were resuspended in swelling
buffer containing 1 µM ruthenium red. After collecting the
mitochondria through centrifugation (3,000 x g) for 15 min at 4˚C,
the pellets were dried and dissolved in 40 µl 0.75 M sulfuric acid
at 95˚C. The solution was then diluted with water and the
Ca2+ concentration of the solution was measured by an
atomic absorbance spectrometer (iCE™ 3300 AAS; Thermo Scientific,
Inc.).
APP/PS1 mice
The procedures and experiments in this study were
approved by the Committee on Ethics of Animal Experiments, Beijing
Geriatric Hospital (no. 2019134). All animal care procedures and
experiments were conducted in accordance with the Animal Research:
Reporting of In Vivo Experiments guidelines (18). The APP/PS1 mice (n=20 in total)
were purchased from The Jackson Laboratory and were used in all
experimental groups (n=10 mice/group; male-to-female ratio, 1:1;
body weight, 21.43±2.42 g). Mice were housed in the Experimental
Animal Facility (five mice per cage) of Beijing Geriatric Hospital
(Beijing, China) under standard laboratory conditions (18-23˚C;
40-60% humidity; 12-h light/dark cycle) with free access to food
and water. Animal health and behavior were monitored every 2
days.
GK was purchased from Shanghai Bohu Biotechnology
and dissolved in DMSO. GK was administered intraperitoneally at 8
mg/kg (10), while the control
mice were treated with DMSO. In total, 20 mice (age, 6 months) were
used in the experiments. The mice received GK treatment or DMSO for
1 month before they were subjected to the water maze test (19). Thereafter, the mice were euthanized
via carbon dioxide inhalation (replacement of 50% of the chamber
volume/min). Death of the mice was confirmed by lack of a
heartbeat, lack of respiration, lack of corneal reflex and presence
of rigor mortis.
During the water maze test, distress was monitored
by observing animal behavior. The water was warmed to room
temperature before the mice are placed in it. In the maze test,
mice were placed gently in the water hindfeet-first to avoid
stress. The mice that were agitated or became unable to swim were
rescued immediately. Overtiring or hypothermia of mice were
prevented by limiting the swimming duration. Mice were dried upon
completion of the task before their return to their home cage. In
addition, the water was changed daily to prevent growth of
pathogenic organisms (20).
Morris water maze test
The spatial learning ability and memory of mice were
assessed using the Morris water maze test by measuring the latency
to find a hidden platform submerged in a pool (21). The training protocol was applied
for five consecutive days, with four trials per day in a water
maze. In each trial, the mice were placed into the water at a
different starting point and were allowed to swim and find the
hidden platform within 90 sec. The mice that failed to find the
platform were guided to the platform manually and kept at the
platform for 10 sec. Thereafter, on day six, the platform was
removed from the pool and the spatial probe test was conducted.
Each mouse was placed in the water in a location opposite the
target quadrant, facing the wall of the pool. The time that the
mice spent in the target quadrant was recorded over a period of 90
sec. A tracking camera device (Ethovision 2.0; Noldus) was used to
monitor the behavioral experiments and the recording was analyzed
using video-tracking software (DigBehv Animal Behavior Analysis
Software 1.0; Shanghai Jiliang Software Technology Co., Ltd.).
Immunohistochemistry (IHC)
Brain tissues were fixed with 10% formalin for 24 h
at room temperature and embedded in paraffin. Paraffin-embedded
tissue samples were then cut into 5-µm-thick sections. The sections
on glass slides were deparaffinized with xylene at 55˚C, rehydrated
with a descending alcohol series and then subjected to antigen
retrieval. Next, the sections were blocked with 5% goat serum
(Thermo Fisher Scientific, Inc.) at room temperature for 1 h. The
sections were then stained with antibody (1:200 dilution) against
MCU (cat. no. PA5-120437; Invitrogen; Thermo Fisher Scientific,
Inc.) or Aβ (cat. no. 51-2700; rabbit polyclonal; Invitrogen;
Thermo Fisher Scientific, Inc.) and incubated with a horseradish
peroxidase-labeled dextran polymer coupled with an anti-rabbit
antibody (1:1,000 dilution; Beyotime Institute of Biotechnology) at
room temperature for 1 h. Staining that was clearly distinguishable
from the background was considered positive. The staining results
were visualized using a light microscope (Olympus Corporation). The
expression level of MCU was quantified by the optical density
values in 10 random area fields under a magnification of x400
according to the regular IHC staining grade system (22). The load of Aβ was quantified as the
percentage area of Aβ deposits within the image cubes made with a
multispectral imaging system (23).
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. SPSS 26 software (IBM Corporation) was used to analyze
the data. An unpaired independent Student's t-test or one-way ANOVA
followed by Tukey's post-hoc test were used to compare multiple
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Regulatory effect of MCU on the
viability and apoptosis of SH-SY5Y cells
To investigate the effect of MCU in SH-SY5Y cells,
the expression of MCU was enhanced by transfection of recombinant
lentiviral vectors or by blocking the expression of MCU with siRNA
(Fig. 1A).
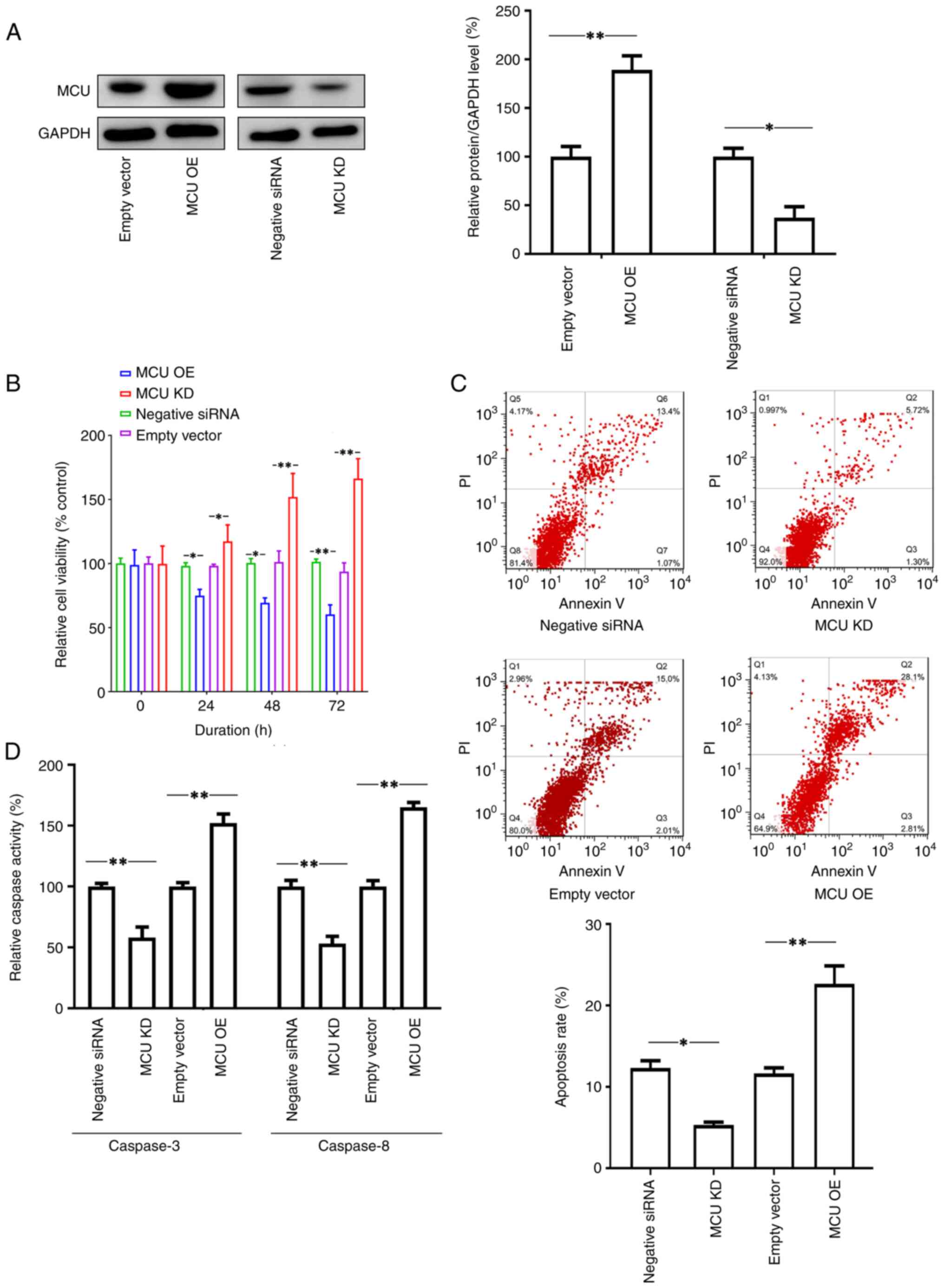 | Figure 1Effects of MCU on the viability and
apoptosis of SH-SY5Y cells. SH-SY5Y cells were cultured in
vitro and then transfected with recombinant lentiviral vectors
to overexpress MCU or knockdown expression MCU by transfection of
MCU siRNA, while cells transfected with empty vector or negative
control siRNA were used as controls. Thereafter, the cells were
cultured for 72 h and cell viability, apoptosis and Caspase-3/8
activities were examined. (A) Transfection of recombinant
lentiviral vectors (MCU OE) increased MCU expression compared to
the transfection of empty vector, while knockdown of MCU by
transfection of siRNA (MCU KD) inhibited MCU expression compared to
the transfection of negative control siRNA. Representative images
of the western blot analysis and quantified results are provided.
(B) Knockdown of MCU promoted cell viability. The cells with
different pretreatments (MCU OE or MCU KD) were cultured in
vitro for 72 h and cell viability was measured by a WST-1
assay. The results indicated that the cells with MCU expression
enhancement had lower cell viability rates compared to the controls
(empty vector transfection), whereas knockdown of MCU increased
cell viability compared to that in cells transfected with negative
siRNA. (C) Apoptotic cells were measured by FACScan after staining
with annexin V and PI. The sum of annexin V-positive cells and
annexin V- + PI-positive cells was used to indicate the total
percentage of apoptotic cells. The expression enhancement of MCU
promoted apoptosis in SH-SY5Y cells, which was significantly higher
than that in the control group. However, blocking the expression of
MCU decreased the rate of apoptosis of the cells. Representative
images and relative quantifications are presented. (D) The data on
apoptotic protein activities (Caspase-3 and Caspase-8) were
consistent with the results of flow cytometry. Expression
enhancement of MCU promoted the activity of Caspase-3 and Caspase-8
in cells. Blocking the expression of MCU resulted in lower activity
of Caspase-3 and Caspase-8 in cells. All results are representative
of three independent experiments performed in triplicate. Values
are expressed as the mean ± standard error of the mean.
*P<0.05; **P<0.01, one-way ANOVA
followed by Tukey's post-hoc test. siRNA, small interfering RNA;
KD, knockdown; OE, overexpression; PI, propidium iodide; Q,
quadrant; MCU, mitochondrial Ca2+ uniporter. |
In the cell viability assay, it was observed that
ectopic expression of MCU by transfection inhibited cell viability,
while knockdown of the expression of MCU by siRNA increased cell
viability (Fig. 1B). Furthermore,
the cells were collected after different treatments, stained with
annexin V and PI and analyzed using flow cytometry to evaluate
apoptosis. The results indicated that MCU expression enhancement by
transfection promoted the percentage of apoptotic SH-SY5Y cells,
whereas blocking the expression of MCU significantly decreased the
apoptotic rate of SH-SY5Y cells (Fig.
1C). In addition, Caspase-3/8 activities were examined in the
cells. Consistently, overexpression of MCU increased the activities
of both Caspase-3 and Caspase-8; by contrast, blocking the
expression of MCU decreased the activities of both Caspase-3 and
Caspase-8 (Fig. 1D).
Effect of GK on the viability and
apoptosis of SH-SY5Y cells
It is known that Aβ is able to induce apoptosis in
neuronal cells and contribute to AD pathology in mammals. In the
present study, Aβ was used to treat SH-SY5Y cells and the results
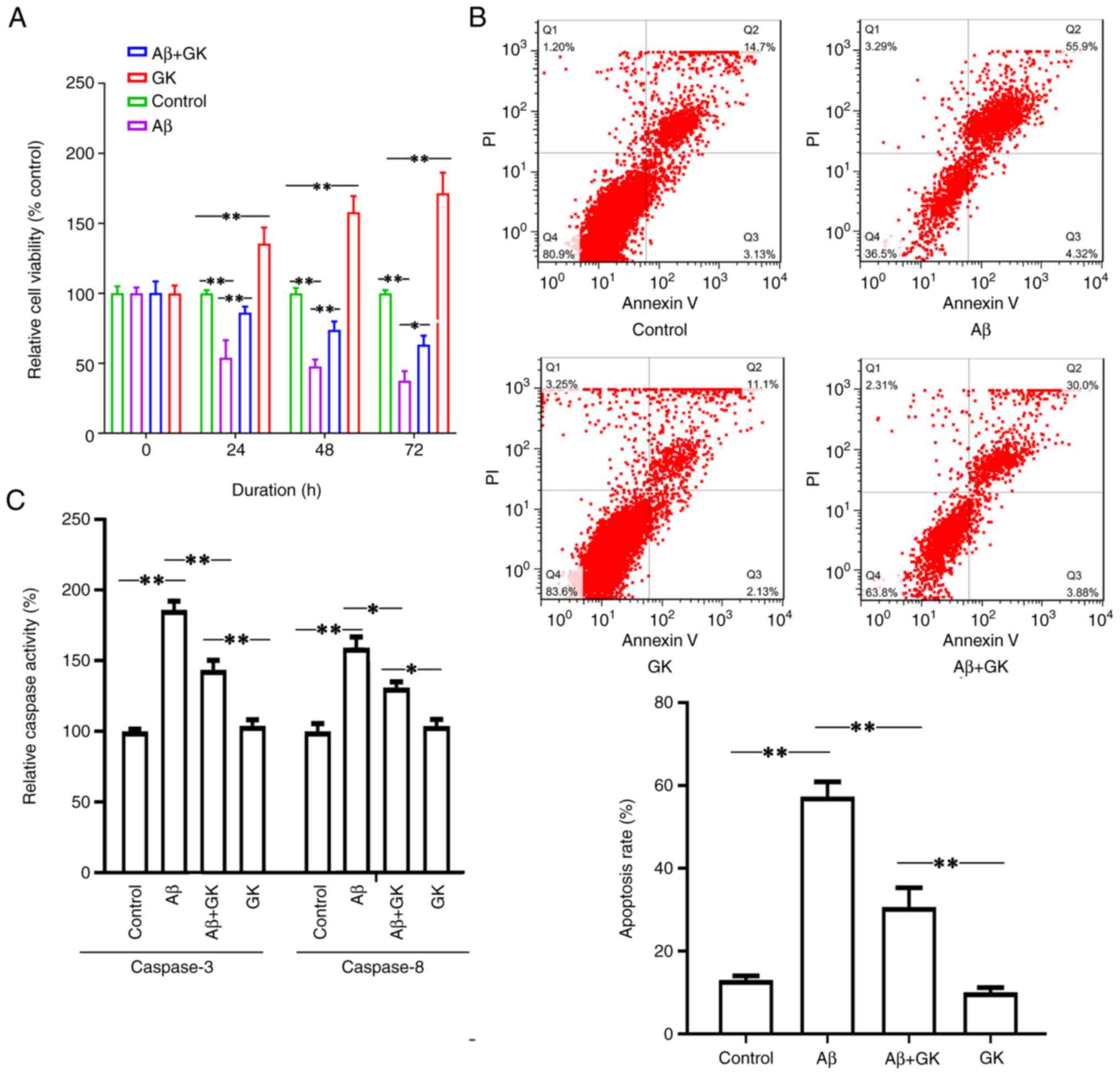
indicated that Aβ treatment inhibited cell viability (Fig. 2A). Furthermore, cotreatment with GK
alleviated the cytotoxicity caused by Aβ. The cells cotreated with
GK had a higher viability rate than the cells without GK treatment
(Fig. 2A). The cell viability
exhibited a significant difference when the cells were treated with
GK alone compared to the control group (treatment with DMSO). In
addition, Aβ treatment increased the cell apoptosis rate, whereas
the administration of GK significantly attenuated Aβ-induced
apoptosis in SH-SY5Y cells with a decrease in the apoptosis rate
and Caspase-3/8 activities (Fig.
2B and C).
The present results indicated that GK failed to
promote cell viability and inhibit apoptosis when MCU expression
was knocked down. When MCU expression was knocked down, there were
no significant differences in cell viability (Fig. 3A), apoptosis rate (Fig. 3B) or Caspase-3/8 activities
(Fig. 3C) between the Aβ+GK
treatment group and the GK treatment group.
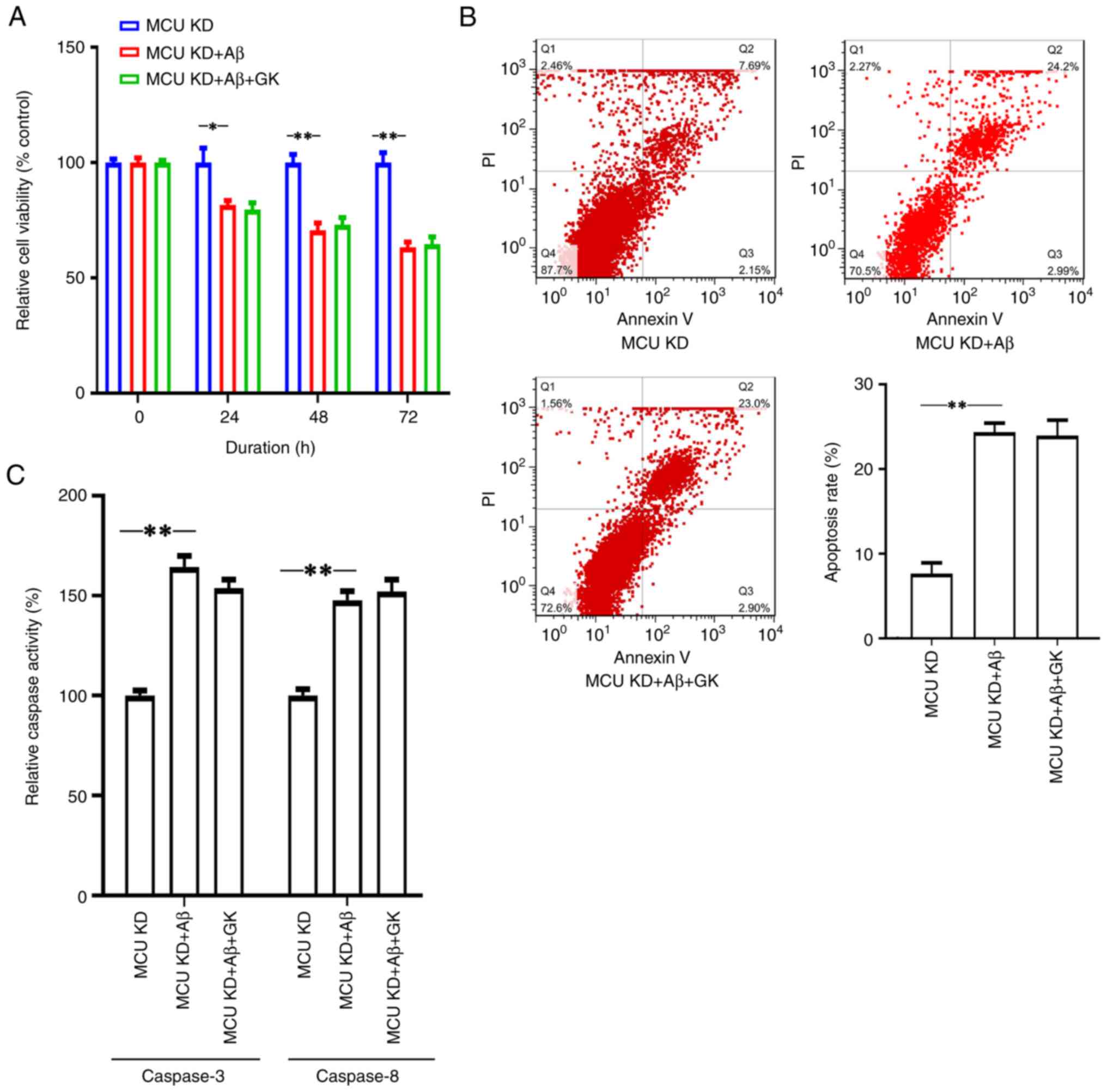 | Figure 3Knocking down the expression of MCU
alleviates the promotion effect of GK on SH-SY5Y cells in the
presence of Aβ. SH-SY5Y cells were cultured in vitro and the
expression of MCU was knocked down by siRNA transfection. The cells
were then treated with Aβ (25 µM) or GK (50 µg/ml)+Aβ (25 µM) for
72 h, while cells treated with DMSO were used as controls. Cell
viability was measured using WST-1. Apoptotic cells were measured
by FACScan after staining with annexin V and PI. The sum of annexin
V-positive cells and annexin V- + PI-positive cells was used to
indicate the total percentage of apoptotic cells. Treatment with Aβ
(A) decreased cell viability, (B) promoted cell apoptosis and (C)
increased the activities of Caspase-3 and Caspase-8 compared to the
control (DMSO). However, cotreatment with GK failed to alleviate
(A) the inhibitory effect on cell viability or (B and C) the
promotion effect on cell apoptosis by Aβ in SH-SY5Y cells with MCU
knocked down in terms of (B) the apoptotic rate measured by flow
cytometry and (C) caspase activity. All experiments were performed
in triplicate. Values are expressed as the mean ± standard error of
the mean. *P<0.05; **P<0.01 by one-way
ANOVA followed by Tukey's post-hoc test. MCU, mitochondrial
Ca2+ uniporter; PI, propidium iodide; Q, quadrant; GK,
ginkgolide K; Aβ, amyloid β; siRNA, small interfering RNA; KD,
knockdown; OE, overexpression. |
GK regulates Ca2+ levels
through MCU expression in mitochondria
The potential interaction between GK and MCU was
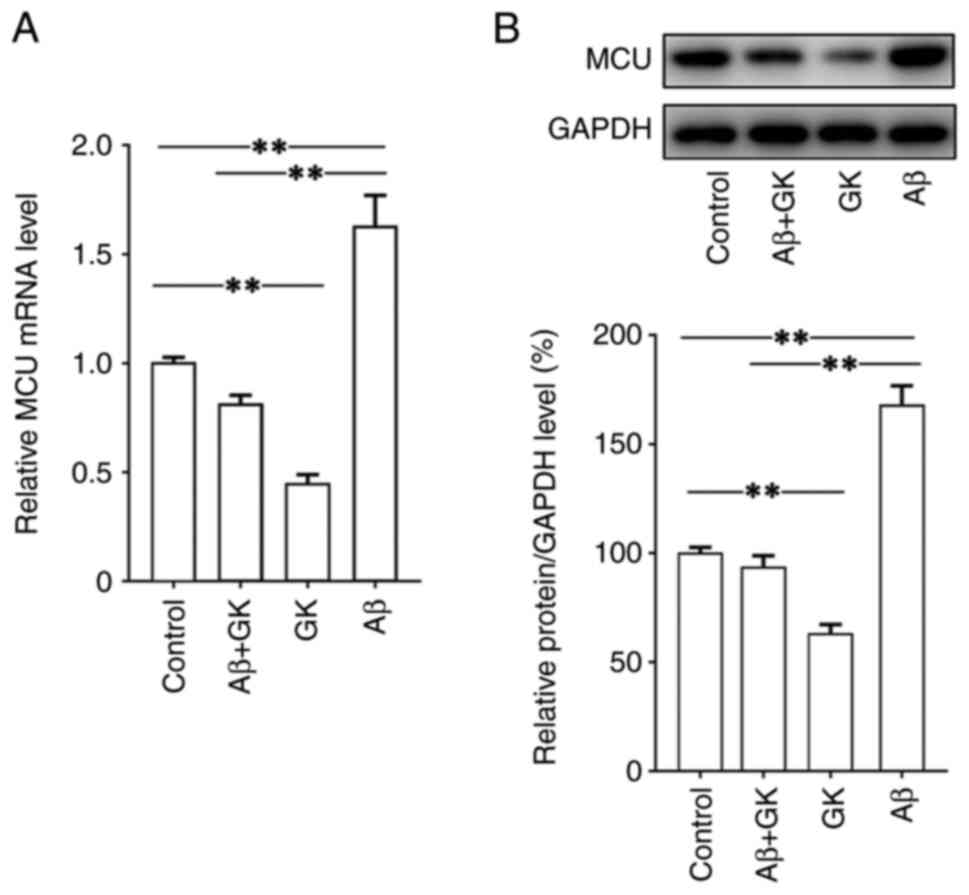
then investigated. The present results indicated that Aβ
significantly increased the expression of MCU, while GK decreased
the expression of MCU at both the mRNA and protein levels (Fig. 4). Furthermore, in SH-SY5Y cells,
the promoting effect of Aβ on MCU expression was clearly inhibited
by cotreatment with GK. In addition, treatment with GK did not
affect the expression levels on Aβ, tau protein and
phosphorylated-tau protein (Fig.
S2).
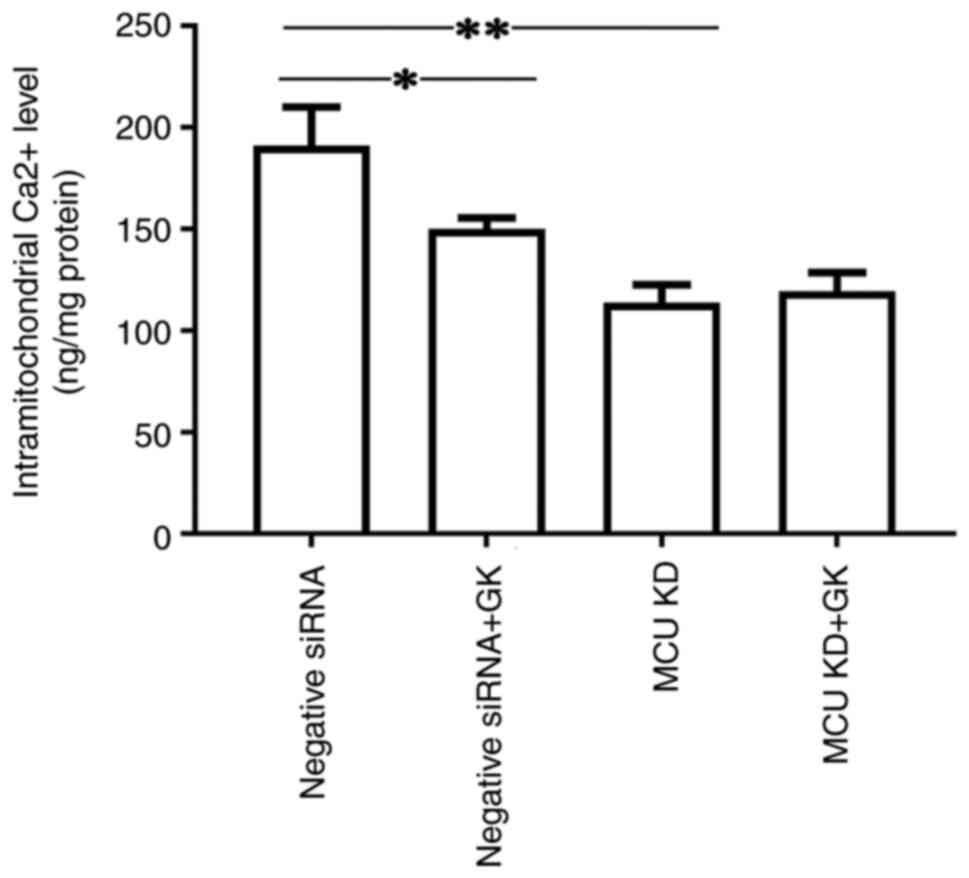
To investigate the effect of MCU on the
Ca2+ levels in mitochondria, MCU expression was knocked
down in cells using siRNA (Fig.
1). It was observed that a deficit in MCU expression decreased
the level of Ca2+ in the mitochondria of SH-SY5Y cells
(Fig. 5). Furthermore, treatment
with GK reduced the levels of Ca2+ in the mitochondria
of SH-SY5Y cells. The inhibitory effect of GK on the levels of
Ca2+ in the mitochondria of SH-SY5Y cells was alleviated
by blocking MCU (Fig. 5). No
significant difference was observed in Ca2+ levels in
mitochondria between the MCU knockdown cells with and those without
GK treatment.
Thus, the present results suggested that GK was able
to regulate MCU expression and then reduce the levels of
Ca2+ in the mitochondria of the cells.
Administration of GK decreases the
levels of MCU and Aβ deposits and improves cognitive ability in
APP/PS1 mice
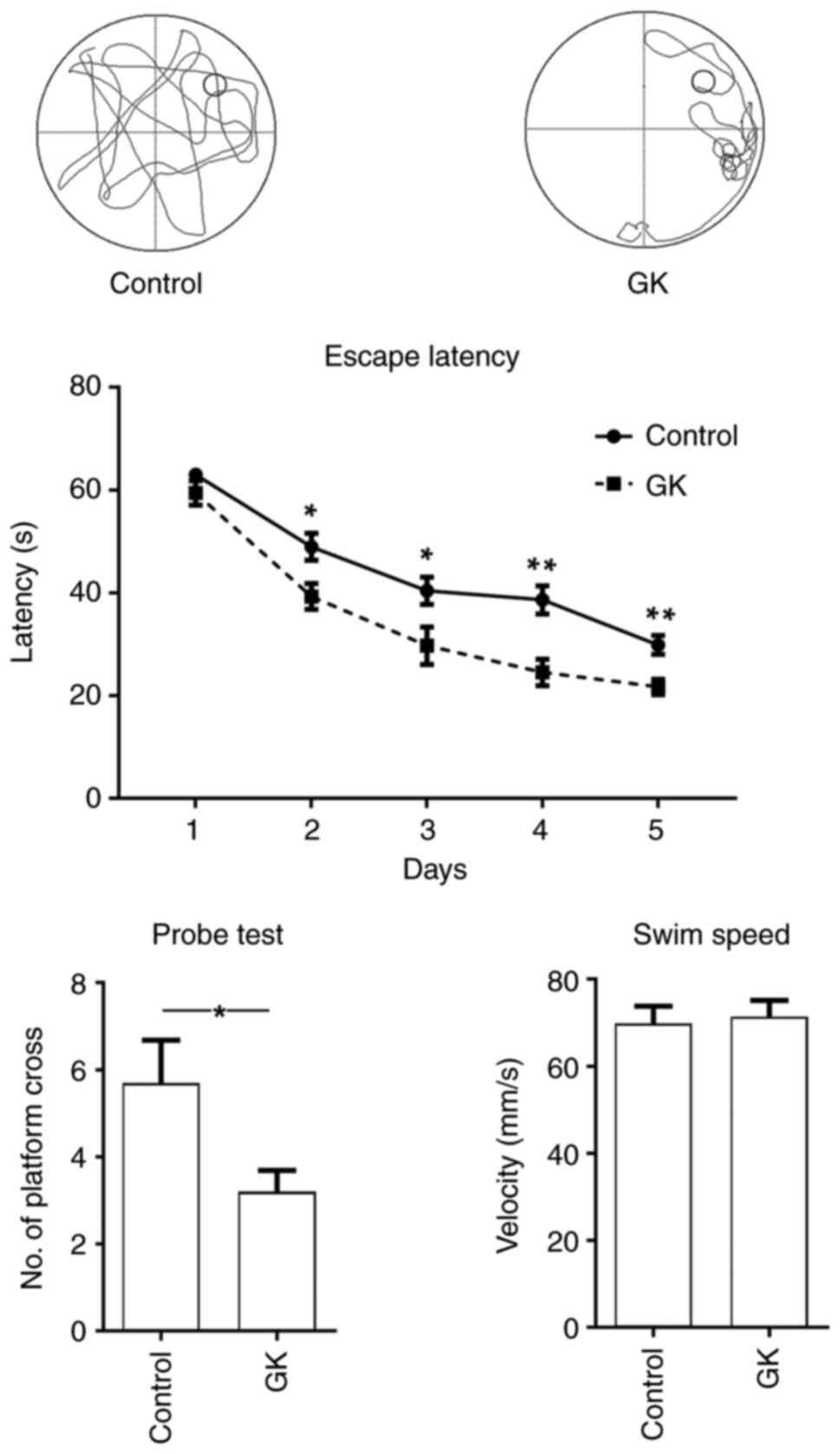
APP/PS1 mice (age, 6 months) received GK for 1
month. Thereafter, the Morris water maze test was used to assess
spatial learning and cognitive ability. In the mice with GK
supplementation, the latency to find the hidden platform was
significantly decreased compared with that of the control mice, and
the performance of the mice with GK supplementation was
significantly improved in terms of the numbers of platform
crossings (Fig. 6). Furthermore,
mouse cortex tissues were collected and IHC staining for MCU in
brain sections revealed strong MCU expression in the cortex. IHC
analysis also indicated that GK supplementation decreased the
expression level of MCU protein (Fig.
7A).
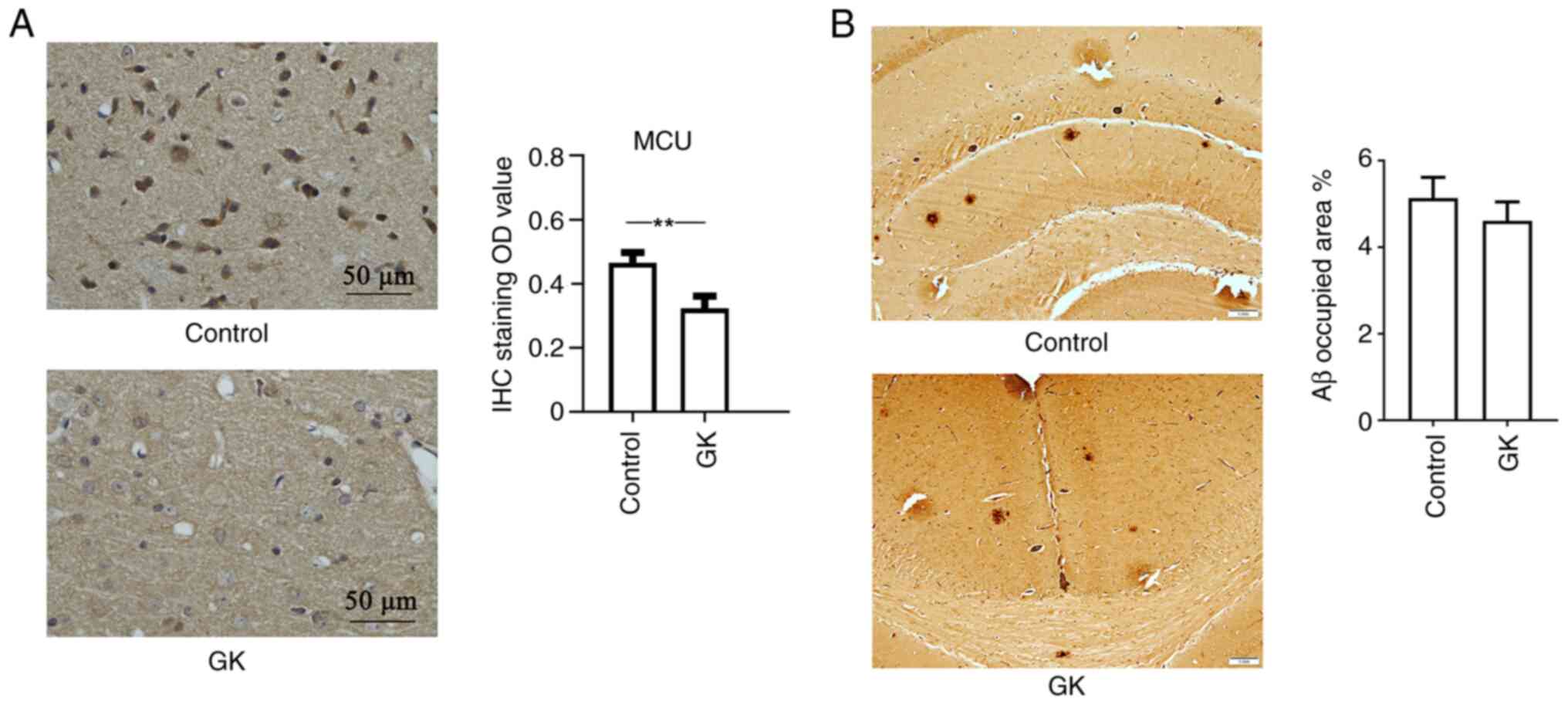 | Figure 7MCU expression and Aβ deposits in
APP/PS1 mice with GK supplementation. APP/PS1 mice (age, 6 months
old; n=10 mice/group; male-to-female ratio, 1:1) were administered
8 mg/kg GK intraperitoneally for 1 month, while mice treated with
DMSO were used as controls. After the treatments, the mouse brains
were collected and cut into sections. The sections were then
stained with antibody against MCU. The staining results were
visualized using a light microscope. (A) IHC staining of the cortex
of mice indicated clear positive expression of MCU in neuronal
cells. Quantitative data indicated that GK supplementation
significantly inhibited the expression of MCU in the cerebral
cortex of mice. The expression levels of MCU were semiquantified
according to the standard IHC staining grade system (scale bar, 50
µm). (B) IHC staining of brain tissues was performed using
antibodies against Aβ. Aβ load was estimated by stereology and
presented as % positive staining of the image area. Qualitative
assessment of Aβ deposition did not indicate any significant
difference in the number of Aβ plaques between mice with and those
without GK administration (scale bar, 1 mm). Values are expressed
as the mean ± standard error of the mean. **P<0.01,
by 2-tailed unpaired Student's t-test. MCU, mitochondrial
Ca2+ uniporter; GK, ginkgolide K; IHC,
immunohistochemistry; Aβ, amyloid β. |
In addition, IHC staining against Aβ identified
extracellular Aβ-positive deposits and intracellular granules in
the neuronal cell body in the brain cortex. The deposits were dense
and spherical. Qualitative assessment of Aβ deposition did not
indicate any obvious difference in the number of Aβ plaques in mice
with/without GK administration (Fig.
7B).
Discussion
In the present study, the effect of GK on AD
pathology was examined and the underlying mechanisms were
investigated. It was revealed that GK was able to promote cell
viability and prevent apoptosis induced by Aβ by regulating the
expression of MCU in vitro. In line with this, GK treatment
decreased the expression of MCU in the mouse brain and alleviated
the impairment in the cognitive ability of APP/PS1 mice.
In the brain, the amyloid precursor protein is
cleaved by β-site amyloid precursor protein-cleaving enzyme 1 to
generate Aβ. Increased Aβ activity has consistently been detected
in the brain tissue of patients with AD and has a key role in the
occurrence and progression of AD (24). It was observed that treatment with
Aβ resulted in neuronal cell apoptosis. Aβ may exert its neurotoxic
effects via multiple pathways. Aβ contributes to the generation of
lipid peroxides and carbonyls, which then induce damage to neuronal
cells (25). Furthermore, it has
been indicated that the toxic properties of Aβ are mediated by
several other mechanisms, including inflammation, synaptic
dysfunction and excitotoxicity (26).
Another potential mechanism of the effect of Aβ on
AD pathology may be through Ca2+ regulation of neuronal
cells. Aβ may cause the formation of Ca2+-permeable
pores in artificial membranes (27) and then regulate Ca2+
entry into the cytoplasm of brain cells (28). Furthermore, Ca2+
homeostasis in mitochondria maintains regular neuronal function
(8). In the present study, it was
observed that increased Ca2+ levels in mitochondria
resulted in a higher apoptosis rate of neuronal cells. In fact, the
loss of Ca2+ homeostasis in the mitochondria of neuronal
cells of patients with AD has been observed by previous studies
(29,30). Mitochondrial dysfunction is another
factor in AD pathogenesis that is involved in cell survival and
synaptic plasticity (31). Perez
et al (32) reported
mitochondrial Ca2+ dysregulation in the fibroblasts of
patients with AD. Calvo-Rodriguez et al (33) observed elevated Ca2+
levels in neuronal mitochondria after significant Aβ plaque
deposition in an AD mouse model. In the present study, it was
observed that treatment with Aβ resulted in apoptosis of neuronal
cells, as well as increased Ca2+ levels in mitochondria,
which may explain the potential interaction between Aβ deposits and
mitochondrial Ca2+ dyshomeostasis in AD pathology. Aβ is
able to increase mitochondrial Ca2+ levels and result in
neuronal death in an AD mouse model, which has been observed in
multiple studies (33-35).
Aβ is also able to promote excessive Ca2+ release from
the endoplasmic reticulum to mitochondria and induce mitochondrial
Ca2+ overload, triggering neuronal cell death (36). By contrast, decreasing the
mitochondrial Ca2+ levels with the MCU blocker RU360
alleviated the impact on cognitive ability in an AD rat model
(37).
The potential protective effect of GK in neuronal
cells has been previously reported. Liu et al (12) indicated that GK protected SH-SY5Y
cells against oxygen/glucose deprivation stress. The present study
reported that treatment with GK promoted viability and prevented
apoptosis of neuronal cells. Furthermore, GK exerted a prosurvival
effect by regulating the function of mitochondria. Zhou et
al (10) suggested that GK
inhibits mitochondrial fission and membrane permeability, while Ma
et al (14) suggested that
GK contributes to maintaining the function of mitochondria.
Similarly, in the present study, it was observed that GK inhibited
the expression of MCU in neuronal cells. MCU is one of the most
prominent calcium uniporters on the inner membrane of mitochondria.
In cells, efficient mitochondrial uniporter-mediated
Ca2+ uptake is required for MCU action. However,
dysfunction of MCU may lead to loss of Ca2+ homeostasis,
promoting the influx of Ca2+ in the mitochondria. The
present results indicated that blocking the expression of MCU
decreased Ca2+ in the mitochondria. In AD pathology,
increased activity of MCU may contribute to Ca2+ influx
and excessive Ca2+ in mitochondria would then inhibit
ATP production and trigger neuronal cell apoptosis and synapse
dysfunction (38). The results of
the present study suggested data that overexpression of MCU led to
an increase in apoptosis and a decrease in viability of neuronal
cells, whereas blocking the expression of MCU promoted the survival
of neuronal cells with decreased apoptosis and increased viability.
Furthermore, it was observed that Aβ treatment promoted the
expression of MCU in vitro, which is consistent with
previous studies (33,34,39).
Thus, it may be suggested that the dysregulation of MCU in
mitochondria induced by Aβ may be one of the pathological
mechanisms of AD, while targeting the expression of MCU may
contribute to the survival of neuronal cells in AD brains.
The neuroprotective effect of Ginkgo biloba
has been demonstrated by previous studies (40-42).
The mechanism of action of Ginkgo biloba may proceed through
multiple pathways, including antioxidative stress, regulation
function of mitochondria and anti-inflammation (40-42).
However, Ginkgo biloba contains numerous types of chemical
components, such as terpenoids, biflavones and flavonols (42). Furthermore, among the active
ingredients of Ginkgo biloba, terpenoids include the main
diterpene ginkgolides A, B, C, J, M, K and L (43). Different active components may
exert their functions through the regulation of different pathways.
Although the regulation of mitochondria by Ginkgo biloba
extracts has been revealed, there is a lack of knowledge regarding
which type of monomer contributes to the maintenance of
mitochondrial function. Thus, research on the monomers of Ginkgo
biloba extracts may contribute to demonstrating the
neuroprotective effect and its molecular mechanism. In the present
study, it was observed that GK treatment alleviated the increased
expression of MCU induced by Aβ in vitro, which then
decreased the Ca2+ levels in mitochondria and eventually
inhibited the apoptosis of cells. The cognitive ability of APP/PS1
mice was clearly improved, with decreased expression of MCU in the
neuronal cells of the mouse brain when GK was used to treat the AD
mice.
The present study had certain limitations. Only the
regulatory effect of GK on Ca2+ levels in mitochondria
by targeting MCU was investigated and further research is required
to investigate the effect of GK in AD pathology. For instance, an
MCU-knockout AD mouse model may require to be developed to
investigate the effect of GK treatment in AD mice with MCU deficit.
It is known that GK exerts multiple effects, including the
regulation of inflammation (11)
and inhibition of oxidative stress (12). Thus, further research is required
to explore the effect of GK on the inflammatory response and
oxidative stress in AD pathology. In addition, further cytology
experiments should be performed to detect the pathological changes
in an animal model, including the expression of ionophores, injury
and repair of neurons, as well as activities of intracellular
signal pathways. For instance, further research should investigate
the potential regulatory effect of GK through the glycogen synthase
kinase-3 (GSK-3)-related pathway, as Zhou et al (10) reported that GK attenuated neuronal
injury through the GSK-3β-dependent pathway.
In conclusion, the present study indicated that GK
inhibited MCU expression, decreasing Ca2+ levels in
mitochondria. It is suggested that GK treatment may be a potential
therapeutic strategy for AD.
Supplementary Material
Schematic figure of the molecular
mechanism of GK on calcium channel activity and a molecular
structure of GK. MCU, mitochondrial Ca2+ uniporter; GK,
ginkgolide K; ROS, reactive oxygen species.
Effect of GK on the expression of Aβ,
tau protein and phosphorylated tau protein in SH-SY5Y cells.
SH-SY5Y cells were cultured in vitro and then treated with
GK (25-100 μg/ml) for 72 h, while cells treated with DMSO were used
as controls. The expression was examined using western blot
analysis. The results indicated no significant difference in the
expression levels on Aβ, tau protein and phosphorylated tau protein
among different treatment groups. GK, ginkgolide K; Aβ, amyloid
β.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL obtained funding for the present study, performed
the majority of the experiments and wrote the manuscript. JL
obtained funding for the present study, designed the experiments,
performed parts of the experiments and wrote the manuscript. QL and
XZ performed certain parts of the experiments. YS was involved in
the conception of this study. HL and JL confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The procedures and experiments in this study were
approved by the Committee on Ethics of Animal Experiments, Beijing
Geriatric Hospital (Beijing, China; no. 2019134).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hasan TF, Hasan H and Kelley RE: Overview
of acute ischemic stroke evaluation and management. Biomedicines.
9(1486)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Huynh TV, Davis AA, Ulrich JD and Holtzman
DM: Apolipoprotein E and Alzheimer's disease: The influence of
apolipoprotein E on amyloid-β and other amyloidogenic proteins. J
Lipid Res. 58:824–836. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Michaelson DM: APOE ε4: The most prevalent
yet understudied risk factor for Alzheimer's disease. Alzheimers
Dement. 10:861–868. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Rub U, Stratmann K, Heinsen H, Turco DD,
Seidel K, Dunnen WD and Korf HW: The brainstem tau cytoskeletal
pathology of Alzheimer's Disease: A brief historical overview and
description of its anatomical distribution pattern, evolutional
features, pathogenetic and clinical relevance. Curr Alzheimer Res.
13:1178–1197. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Raffaello A, De Stefani D and Rizzuto R:
The mitochondrial Ca(2+) uniporter. Cell Calcium. 52:16–21.
2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Granatiero V, Pacifici M, Raffaello A, De
Stefani D and Rizzuto R: Overexpression of mitochondrial calcium
uniporter causes neuronal death. Oxid Med Cell Longev.
2019(1681254)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Qiu J, Tan YW, Hagenston AM, Martel MA,
Kneisel N, Skehel PA, Wyllie DJA, Bading H and Hardingham GE:
Mitochondrial calcium uniporter Mcu controls excitotoxicity and is
transcriptionally repressed by neuroprotective nuclear calcium
signals. Nat Commun. 4(2034)2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Moreira PI, Carvalho C, Zhu X, Smith MA
and Perry G: Mitochondrial dysfunction is a trigger of Alzheimer's
disease pathophysiology. Biochim Biophys Acta. 1802:2–10.
2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Fu YJ, Xiong S, Lovell MA and Lynn BC:
Quantitative proteomic analysis of mitochondria in aging PS-1
transgenic mice. Cell Mol Neurobiol. 29:649–664. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhou X, Wang HY, Wu B, Cheng CY, Xiao W,
Wang ZZ, Yang YY, Li P and Yang H: Ginkgolide K attenuates neuronal
injury after ischemic stroke by inhibiting mitochondrial fission
and GSK-3β-dependent increases in mitochondrial membrane
permeability. Oncotarget. 8:44682–44693. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang Y and Miao JM: Ginkgolide K promotes
astrocyte proliferation and migration after oxygen-glucose
deprivation via inducing protective autophagy through the
AMPK/mTOR/ULK1 signaling pathway. Eur J Pharmacol. 832:96–103.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Liu Q, Li X, Li L, Xu Z, Zhou J and Xiao
W: Ginkgolide K protects SHSY5Y cells against oxygenglucose
deprivationinduced injury by inhibiting the p38 and JNK signaling
pathways. Mol Med Rep. 18:3185–3192. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen M, Zou W, Chen M, Cao L, Ding J, Xiao
W and Hu G: Ginkgolide K promotes angiogenesis in a middle cerebral
artery occlusion mouse model via activating JAK2/STAT3 pathway. Eur
J Pharmacol. 833:221–229. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ma S, Liu X, Xun Q and Zhang X:
Neuroprotective effect of ginkgolide k against H2O2-induced PC12
cell cytotoxicity by ameliorating mitochondrial dysfunction and
oxidative stress. Biol Pharm Bull. 37:217–225. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kilkenny C, Browne W, Cuthill IC, Emerson
M and Altman DG: NC3Rs Reporting Guidelines Working Group. Animal
research: Reporting in vivo experiments: The ARRIVE guidelines. Br
J Pharmacol. 160:1577–1579. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Shao L, Dong C, Geng D, He Q and Shi Y:
Ginkgolide B protects against cognitive impairment in
senescence-accelerated P8 mice by mitigating oxidative stress,
inflammation and ferroptosis. Biochem Biophys Res Commun. 572:7–14.
2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Mulder GB and Pritchett K: The morris
water maze. Contemp Top Lab Anim Sci. 42:49–50. 2003.PubMed/NCBI
|
|
19
|
Su R, Su W and Jiao Q: NGF protects
neuroblastoma cells against beta-amyloid-induced apoptosis via the
Nrf2/HO-1 pathway. FEBS Open Bio. 9:2063–2071. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ding Y, Zhang H, Liu Z, Li Q, Guo Y, Chen
Y, Chang Y and Cui H: Carnitine palmitoyltransferase 1 (CPT1)
alleviates oxidative stress and apoptosis of hippocampal neuron in
response to beta-Amyloid peptide fragment Abeta25-35.
Bioengineered. 12:5440–5449. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Morris R: Developments of a water-maze
procedure for studying spatial learning in the rat. J Neurosci
Methods. 11:47–60. 1984.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jafari SMS and Hunger RE: IHC optical
density score: A new practical method for quantitative
immunohistochemistry image analysis. Appl Immunohistochem Mol
Morphol. 25:e12–e13. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Heggland I, Storkaas IS, Soligard HT,
Kobro-Flatmoen A and Witter MP: Stereological estimation of neuron
number and plaque load in the hippocampal region of a transgenic
rat model of Alzheimer's disease. Eur J Neurosci. 41:1245–1262.
2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Shen Y, Wang H, Sun Q, Yao H, Keegan AP,
Mullan M, Wilson J, Lista S, Leyhe T, Laske C, et al: Increased
Plasma beta-secretase 1 may predict conversion to Alzheimer's
disease dementia in individuals with mild cognitive impairment.
Biol Psychiatry. 83:447–455. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Butterfield DA, Castegna A, Lauderback CM
and Drake J: Evidence that amyloid beta-peptide-induced lipid
peroxidation and its sequelae in Alzheimer's disease brain
contribute to neuronal death. Neurobiol Aging. 23:655–664.
2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Carrillo-Mora P, Luna R and Colin-Barenque
L: Amyloid beta: Multiple mechanisms of toxicity and only some
protective effects? Oxid Med Cell Longev.
2014(795375)2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Arispe N, Pollard HB and Rojas E: Giant
multilevel cation channels formed by Alzheimer disease amyloid
beta-protein [A beta P-(1-40)] in bilayer membranes. Proc Natl Acad
Sci USA. 90:10573–10577. 1993.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Abramov AY, Canevari L and Duchen MR:
Changes in intracellular calcium and glutathione in astrocytes as
the primary mechanism of amyloid neurotoxicity. J Neurosci.
23:5088–5095. 2003.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tatebayashi Y, Takeda M, Kashiwagi Y,
Okochi M, Kurumadani T, Sekiyama A, Kanayama G, Hariguchi S and
Nishimura T: Cell-cycle-dependent abnormal calcium response in
fibroblasts from patients with familial Alzheimer's disease.
Dementia. 6:9–16. 1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Peterson C, Ratan RR, Shelanski ML and
Goldman JE: Altered response of fibroblasts from aged and Alzheimer
donors to drugs that elevate cytosolic free calcium. Neurobiol
Aging. 9:261–266. 1988.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cavallucci V, Ferraina C and D'Amelio M:
Key role of mitochondria in Alzheimer's disease synaptic
dysfunction. Curr Pharm Des. 19:6440–6450. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Perez MJ, Ponce DP, Aranguiz A, Behrens MI
and Quintanilla RA: Mitochondrial permeability transition pore
contributes to mitochondrial dysfunction in fibroblasts of patients
with sporadic Alzheimer's disease. Redox Biol. 19:290–300.
2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Calvo-Rodriguez M, Hou SS, Snyder AC,
Kharitonova EK, Russ AN, Das S, Fan Z, Muzikansky A, Garcia-Alloza
M, Serrano-Pozo A, et al: Increased mitochondrial calcium levels
associated with neuronal death in a mouse model of Alzheimer's
disease. Nat Commun. 11(2146)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jadiya P, Kolmetzky DW, Tomar D, Meco AD,
Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Praticò D and
Elrod JW: Impaired mitochondrial calcium efflux contributes to
disease progression in models of Alzheimer's disease. Nat Commun.
10(3885)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Calvo-Rodriguez M and Bacskai BJ: High
mitochondrial calcium levels precede neuronal death in vivo in
Alzheimer's disease. Cell Stress. 4:187–190. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ferreiro E, Oliveira CR and Pereira CMF:
The release of calcium from the endoplasmic reticulum induced by
amyloid-beta and prion peptides activates the mitochondrial
apoptotic pathway. Neurobiol Dis. 30:331–342. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Nikseresht Z, Ahangar N, Badrikoohi M and
Babaei P: Synergistic enhancing-memory effect of D-serine and
RU360, a mitochondrial calcium uniporter blocker in rat model of
Alzheimer's disease. Behav Brain Res. 409(113307)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Calvo-Rodriguez M, Hernando-Perez E, Nuñez
L and Villalobos C: Amyloid β oligomers increase ER-mitochondria Ca
2+ cross talk in young hippocampal neurons and exacerbate
aging-induced intracellular Ca 2+ remodeling. Front Cell Neurosci.
13(22)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Singh SK, Srivastav S, Castellani RJ,
Plascencia-Villa G and Perry G: Neuroprotective and antioxidant
effect of ginkgo biloba extract against ad and other neurological
disorders. Neurotherapeutics. 16:666–674. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Shi C, Liu J, Wu F and Yew DT: Ginkgo
biloba extract in Alzheimer's disease: From action mechanisms to
medical practice. Int J Mol Sci. 11:107–123. 2010.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Nowak A, Kojder K, Zielonka-Brzezicka J,
Wróbel J, Bosiacki M, Fabiańska M, Wróbel M, Sołek-Pastuszka J and
Klimowicz A: The use of ginkgo biloba L. as a neuroprotective agent
in the Alzheimer's disease. Front Pharmacol.
12(775034)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Feng Z, Sun Q, Chen W, Bai Y, Hu D and Xie
X: The neuroprotective mechanisms of ginkgolides and bilobalide in
cerebral ischemic injury: A literature review. Mol Med.
25(57)2019.PubMed/NCBI View Article : Google Scholar
|