Introduction
Lung cancer ranks first in incidence among
malignancies and remains the most common cause of cancer-associated
mortality in China. Lung adenocarcinoma (LA) is the most common
type of primary lung cancer, accounting for >40% of all lung
cancer cases (1). Although efforts
have been made to understand the molecular characteristics of LA
and develop novel screening and treatment strategies for the
disease, most patients are diagnosed at an advanced stage due to a
lack of effective diagnostic approaches (2,3).
Hence, searching for novel biomarkers to diagnose and treat LA is
an urgent concern.
MicroRNAs (miRNAs/miRs) are a group of small
non-coding RNAs that negatively regulate gene expression by
directly targeting the 3'-untranslated region (UTR) of mRNA. miRNAs
have important roles in maintaining normal physiological conditions
in the human body. Abnormal miRNA expression has been indicated to
be related to multiple human diseases, including lung cancer
(4-6).
A previous study by our group identified that miR-382-3p was
downregulated in the extracellular vesicles from the peripheral
blood of 153 patients with LA (7).
However, the mechanisms of how miR-382-3p downregulation is related
to LA have remained elusive.
Activation of PI3K/Akt signaling has been confirmed
as a major signaling event in carcinogenesis (8). Tsurutani et al (9) examined phosphorylated (p)AKT levels
in 300 non-small cell lung cancer (NSCLC) specimens and 100
adjacent lung tissue specimens and determined that AKT activation
was specific for NSCLC tumors vs. the adjacent tissue (73.4 vs. 0%;
P<0.05). They also observed that AKT activation was more
frequent in adenocarcinomas than in squamous cell carcinomas of the
lung (78.1 vs. 68.5%; P=0.04) and was associated with shorter
overall survival for all stages of the disease (log-rank P=0.04).
Yoshizawa et al (10)
reported that pAKT is overexpressed in 78% of NSCLCs. Once again,
pAKT was detected more frequently in adenocarcinomas (43%) than in
squamous cell carcinomas (36%), but this observation was not
statistically significant. The 5-year survival rate was
significantly lower in patients with pAKT-positive tumors. Lysine
modification has been reported to be critical for the
phosphorylation, subcellular localization, stability and activity
of Akt. Furthermore, Akt has been indicated to be modified via
SUMOylation at multiple sites, which is required for its kinase
activity and biological function (11).
SUMOylation is a modification process that involves
the addition of small ubiquitin-like modifier (SUMO) to the
acceptor lysine of target proteins. This modification process
consists of three enzymatic cascade steps: An activation step
[heterodimer E1 enzymes SUMO1 activating enzyme subunit 1 (SAE1)
and SAE2], a conjugation step (E2 enzyme Ubc9) and a substrate
modification step (cooperation of the E2 and E3 enzymes) (12). Dysregulated SUMOylation processes
are usually found in cancers as one of the most important
physiological mechanisms in cellular response to stress (13).
It has been reported that SAE1 overexpression is
related to poor prognosis of patients with multiple types of
cancer, suggesting that SUMOylation may be involved in the
progression of malignant tumors. In addition, the mRNA level of
SAE1 was positively correlated with lymph node metastasis in
patients with LA (14). However,
the mechanism by which SAE1 contributes to the progression of LA
remains elusive. The present study aimed to investigate the
relationship between miR-382-3p dysregulation and LA
carcinogenesis. The expression of miR-382-3p in tumor and non-tumor
control tissue samples from 78 patients with LA was examined and
the biological function and the underlying mechanisms of miR-382-3p
during the carcinogenesis of LA were identified.
Materials and methods
Patients
A cohort consisting of 78 patients with LA (age,
54-71 years; females/males, 33/45) and 59 healthy controls (age,
45-74 years; females/males, 26/33) was included in this study. All
participants were recruited at Fuxing Hospital, Capital Medical
University (Beijing, China) between February 2017 and July 2018.
Inclusion criteria: The LA cases were diagnosed as stage I/II
according to the eighth edition of the Union for International
Cancer Control Tumor-Node-Metastasis system and confirmed via
histopathological analysis (6).
Exclusion criteria: Patients with history of thoracic surgery; with
other tumors; complicated with severe liver dysfunction and kidney
dysfunction; with acute pulmonary infection, connective tissue
diseases, infectious diseases, metabolic diseases, hematopoietic
dysfunction and patients with mental illness or a family history of
mental illness, pregnant or lactating women.
The present study was approved by the Human Basic
and Clinical Research Ethics Committee of Fuxing Hospital (Beijing,
China; approval no. 2018FXHEC-KY-19). Written informed consent was
obtained from all the participants. The privacy of the subjects was
protected, according to the Declaration of Helsinki.
Xenograft mouse model
A total of 24 female NOD-SCID mice (age, 5 weeks;
body weight, 15-20 g) were purchased from the Charles River
Laboratories, Inc. All mice were housed at 23-25˚C with 50-60%
humidity, a 12-h light/dark cycle and food and water ad
libitum. To establish the xenograft mouse model, a total of
5x106 A549-miR-382-3p or H1299-miR-382-3p cells in 100
µl PBS together with an equal volume of Matrigel®
basement membrane matrix were subcutaneously injected into the
right flanks of the mice (3 mice in each group). Tumors were
measured every 7 days with a caliper and the tumor volume
(mm3) was calculated as 0.5 x length x
width2. Regarding the tumor burden, a marked increase in
tumor size (≥10% body weight) was applied as a humane endpoint.
Mice were euthanized using CO2 with the flow rate for
CO2 set at 30% chamber volume displaced per minute.
Death of the mice was verified by observation of respiratory
arrest, cessation of heartbeat (lack of activity for ≥5 min) and
pupillary response to light. All animal protocols were performed
strictly according to the relevant National Institutes of Health
Guidelines for the Care and Use of Laboratory Animals (15,16).
All experiments involving animals were pre-approved by the
Institutional Animal Care and Use Committee (IACUC) of Fuxing
Hospital (Beijing, China).
Cell culture
The normal human lung fibroblast cell lines MRC-9
(CCL-212) and WI-38 (CCL-75) cells were purchased from the American
Type Culture Collection and cultured in Eagle's minimum essential
medium (Hyclone; Cytiva) containing 10% fetal bovine serum
(Hyclone; Cytiva), 100 IU/ml penicillin and 100 IU/ml streptomycin.
Human embryonic kidney 293T cells (1101HUM-PUMC000091) and the
human non-small cell lung carcinoma cells lines A549
(1101HUM-PUMC000002), H1299 (1101HUM-PUMC000469), Calu-3
(1101HUM-PUMC000032), NCI-H1975 (1101HUM-PUMC000252), NCI-H2087
(1101HUM-PUMC000253) and SK-LU-1 (1101HUM-PUMC000237) were obtained
from China Infrastructure of Cell Line Resources and cultured in
Dulbecco's Modified Eagle's medium (Hyclone; Cytiva) containing 10%
fetal bovine serum (Hyclone; Cytiva), 100 IU/ml penicillin and 100
IU/ml streptomycin. All cells were maintained at 37˚C in a
humidified atmosphere containing 5% CO2. For functional
studies, A549 (p53 wild-type) and H1299 (p53 deleted) cells were
selected due to their miR-382-3p levels and p53 expression.
RNA extraction
Total RNA was extracted from tissues or cell samples
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
following the manufacturer's protocol. In brief, samples were
homogenized in 1 ml TRIzol mixed with 200 µl chloroform. After
centrifugation, the supernatants were transferred into a new tube
and the RNA was precipitated with isopropanol. The RNA pellets were
dissolved using RNase-free water after washing with 70% ethanol and
the RNA concentration and purity were determined using a
NanoDrop-2000 spectrophotometer (Thermo Fisher Scientific, Inc.).
RNA purity was determined through the absorbance ratio at 260
nm/280 nm and 260 nm/230 nm. Only high-purity RNA samples (260
nm/280 nm ~2.0 and 260 nm/230 nm between 1.9 and 2.2) were included
in this study.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cyclin D1 (CCND1), cyclin-dependent kinase (CDK2)
and p21 mRNA levels were determined via RT-qPCR. In brief, cDNA was
synthesized using a RevertAid first-strand cDNA synthesis kit
(Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol. The expression of candidate genes was quantified via qPCR
using SYBR-Green PCR master mix (Thermo Fisher Scientific, Inc.)
using 95˚C for 10 min as the initial denaturation, and with 40
cycles at 95˚C for 15 sec and 60˚C for 1 min. GAPDH was used as a
loading control.
For miRNA quantification, single-strand cDNA was
synthesized using a TaqMan MicroRNA Reverse Transcription Kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with
miRNA-specific primers. The levels of candidate miRNAs were
determined by using TaqMan Universal PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and miRNA-specific
TaqMan MGB probes (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The small nuclear RNA U6 was used as a loading control.
Each sample in each group was measured in triplicate
and the experiment was repeated at least three times. Relative gene
levels were compared using the 2-ΔΔCq method (17).
Primer sequences were as follows: CDK2 forward,
5'-GCCTAGCTTTCTGCCATTCT-3' and reverse,
5'-GTCCAAAGTCTGCTAGCTTGAT-3'; CCND1 forward,
5'-CCTCGGTGTCCTACTTCAAATG-3' and reverse,
5'-CACTTCTGTTCCTCGCAGAC-3'; p21 forward, 5'-GCGACTGTGATGCGCTAAT-3'
and reverse, 5'-GTGGTGTCTCGGTGACAAAG-3'; GAPDH forward,
5'-GGTGTGAACCATGAGAAGTATGA-3' and reverse,
5'-GAGTCCTTCCACGATACCAAAG-3', miR-382-3p forward,
5'-CTAATCATTCACGGACAA-3' and reverse, 5'-GTGCAGGGTCCGAGGT-3'; U6
forward, 5'-TGGAACGCTTCACGAATTTGCG-3' and reverse,
5'-GGA-ACGATACAGAGAAGATTAGC-3'.
Dual-luciferase assay
The full length of the 3'-UTR of SAE1 was amplified
by PCR using 293T cell genomic DNA as template. The primers
sequences were: SAE1-Nhe forward,
5'-CTAGCTAGCACTCAAGATTTGGCAGCCCCAGAGA-3' and SAE1-Xho reverse,
5'-CCGCTCGAGTCAGAATAGGAAAGAGACTGATTTATTGA-3'. The amplification
cycle using 95˚C 10 min as the initial denaturation with 25 cycles
at 95˚C for 10 min, 95˚C for 15 sec and 60˚C for 1 min. The product
was cloned downstream of the Firefly luciferase gene into a pmirGLO
plasmid (Promega Corporation), between Nhe I and Xho I sites, to
generate the reporter vector. The mutant vector generation was
processed using Fast MultiSite Mutagenesis System kit (TransGen
Biotech Co., Ltd.) and mutant primers (SAE1-Mu forward,
5'-GTTCTTTTTAAAAGAAATATAATAAAGTTACTTG-3', and 5'-SAE1-Mu reverse,
TTATTATATTTCTTTTAAAAAGAACCAAGGAAAA-3'. miR-382-3p mimics, inhibitor
and sequence-scrambled single- and double-strand control oligos
were purchased from Shanghai GenePharma Co., Ltd. 293T cells were
seeded into 48-well plates at 5x104 cells/well and
allowed to attach overnight for the luciferase reporter assays. The
reporter vector was co-transfected into the cells with miRNA mimics
or inhibitor using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol. In brief, the nucleotides were mixed with medium-diluted
Lipofectamine and then incubated at 25˚C for 20 min. The
nucleotide-Lipofectamine complexes were added to each well
containing cells and medium and gently mixed by rocking the plate
back and forth. The cells were incubated at 37˚C in a
CO2 incubator for 6 h and then the complexes were
removed. At 48 h after transfection, the cells were lysed and the
luciferase activity was analyzed using a
Dual-Luciferase® Reporter Assay System (Promega
Corporation). Experiments were performed in triplicate and the
results were expressed as the relative luciferase activity (Firefly
luciferase/Renilla luciferase).
The sequences were as follows: miR-382-3p mimics,
5'-AAUCAUUCACGGACAACACUU-3'; mimics control,
5'-AGUGUUGGACUAAGCGGAGGUA-3'; miR-382-3p inhibitor,
5'-AAGUGUUGUCCGUGAAUGAUU-3'; inhibitor control,
5'-UAACACGUCUAUACGCCCA-3'.
Immunoprecipitation (IP)
Cells were harvested and lysed with IP buffer
containing 20 mM Tris (pH 7.5), 150 nM NaCl, 1% Triton X-100, a
protease inhibitor cocktail and 20 mM N-ethylmaleimide. Next, 2 mg
of the cellular supernatant was incubated overnight with a complex
of anti-SUMO1 antibody (cat. no. ab32058; Abcam) and protein-A
beads to enrich SUMOylated proteins at 4˚C. After washing four
times with Tris-buffered saline (TBS), the protein complexes were
eluted with sample loading buffer for immunoblotting.
Protein extraction
Tumor and non-tumor control samples from 72 patients
with LA were used for protein extraction (for six patients, samples
were insufficient for protein extraction). To extract proteins from
tissue samples, the frozen tissue was mixed and homogenized in
ice-cold RIPA lysis and extraction buffer (Thermo Fisher
Scientific, Inc.) containing protease inhibitors (Thermo Fisher
Scientific, Inc.) and then centrifuged at 16,000 x g for 20 min at
4˚C.
To extract proteins from cells, the spent medium in
the culture dishes was discarded, and the cells were washed with
ice-cold PBS. The cells were then lysed by adding ice-cold RIPA
lysis and extraction buffer (Thermo Fisher Scientific, Inc.)
containing protease inhibitors (Thermo Fisher Scientific, Inc.).
The cells were collected using a cold plastic cell scraper and then
subjected to centrifugation at 16,000 x g for 20 min at 4˚C. The
total protein concentration was determined using a Pierce™ BCA
Protein Assay Kit (Thermo Fisher Scientific, Inc.) and the protein
was stored at -80˚C until use.
Immunoblotting
From each sample, 20 µg of protein were loaded and
separated using 10% SDS-PAGE and then blotted onto a polyvinylidene
fluoride membrane (EMD Millipore) via electrophoretic transfer. The
membranes were blocked with 5% nonfat milk (BD Biosciences) and
incubated with one of the primary antibodies overnight at 4˚C.
After incubation with the corresponding horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:5,000 dilution) for
another 2 h at room temperature, the signals were detected using an
enhanced chemiluminescence kit (Thermo Fisher Scientific, Inc.).
The GAPDH signal was used for normalization. The following
antibodies were used: Rabbit anti-SAE1 polyclonal antibody (cat no.
13585S; 1:1,000 dilution; Cell Signaling Technology, Inc.), rabbit
anti-SUMO1 polyclonal antibody (cat no. 4930S; 1:2,000 dilution;
Cell Signaling Technology, Inc.), rabbit anti-pAkt (Ser473)
polyclonal antibody (cat no. 4060S; 1:2,000 dilution; Cell
Signaling Technology, Inc.), mouse anti-GAPDH monoclonal antibody
(cat no. sc-47724; 1:2,000 dilution; Santa Cruz Biotechnology,
Inc.). HRP conjugated goat anti-rabbit antibody (1:5,000; cat no.
7074; Cell Signaling Technology, Inc.), HRP conjugated goat
anti-mouse antibody (1:5,000; cat no. 7076; Cell Signaling
Technology, Inc.).
Small interfering (si)RNA
transfection
siRNA targeting SAE1 or control RNA (RiboBio Co.,
Ltd.) was transfected into cells by using Lipofectamine 2000
(Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol. In brief, the nucleotides were mixed with medium-diluted
Lipofectamine® and then incubated at 25˚C for 20 min.
The nucleotide-Lipofectamine® complexes were added to
each well containing cells and medium, with a final concentration
of 20 nM, and gently mixed by rocking the plate back and forth. The
cells were incubated at 37˚C in a CO2 incubator for 6 h
and then the complexes were removed. At 48 h after transfection,
the cells were subjected to a cell proliferation assay. The
sequence were as follows: siSAE1, 5'-AGACAACGATGGTCAAAAA-3'; and
siRNA control, 5'-GAGACTAAGAACGAACTAA-3'.
Cell proliferation assay
An MTT assay was employed to determine cell
viability. In brief, 2x103 A549 or H1299 cells were
seeded into each well of a 96-well plate and allowed to attach
overnight. siRNA targeting SAE1 or the control RNA was transfected
into the cells separately for 48 h and subsequently, 20 µl of MTT
(5 mg/ml; MilliporeSigma) was added to each well. After incubation
for 4 h, the medium was discarded followed by the addition of DMSO
to each well. The absorbance at 570 nm was recorded using
Multiskan™ FC Microplate Photometer (Thermo Fisher Scientific,
Inc.).
Cell apoptosis assay
Apoptotic cells were analyzed by flow cytometry
after staining with FITC-Annexin V and propidium iodide (cat no.
640914; BioLegend, Inc.). Briefly, cells were washed twice with
cold Cell Staining buffer, and then resuspended in Annexin V
Binding buffer at a concentration of 5x106 cells/ml. Add
5 µl of FITC-Annexin V and 10 µl of propidium iodide solution into
100 µl of cell suspension, and then the cells were gently vortexed
and incubated for 15 min at 25˚C in the dark. Add 400 µl of Annexin
V Binding Buffer to each tube and then the cells were subjected to
flow cytometry analysis using FACSCanto Plus instrument (BD
Biosciences). The results were analyzed using FlowJo software
(v.10.4.1; Tree Star, Inc.).
Immunohistochemistry
Paraffin-embedded sections were first deparaffinized
and then heated in a microwave oven with 10% citrate buffer for 10
min twice and cooled to room temperature. After blocking with 5%
BSA was for 30 min at 25˚C, the slides were incubated with rabbit
anti-Ki-67 polyclonal antibody (cat. no. ab16667; 1:500 dilution;
Abcam) or rabbit anti-SAE1 polyclonal antibody (cat. no. ab38434;
1:500 dilution; Abcam) at 4˚C overnight. After three washes with
TBS containing Tween-20 (TBST), the sections were incubated with
HRP-conjugated goat anti-rabbit secondary antibody (cat. no.
ab6721; 1:2,500 dilution; Abcam). The sections were washed with
TBST three times and the signals were detected using a DAB
Substrate kit following the manufacturer's protocol. Images were
obtained using a microscope (DM2000; Leica Microsystems GmbH). The
intensity of staining (0, negative; 1, weak; 2, moderate; 3,
strong) and percentage of tumor cells showing staining (0-100%)
were evaluated independently. An H-score of 0-300 was generated by
multiplying the intensity of positivity by the percentage of tumor
cells with staining.
Statistical analysis
Data were analyzed using the SPSS statistical
package (version 19.0; IBM Corporation). A paired t-test was used
to compare the relative miR-382-3p levels between tumors and
non-tumor tissues, miR-382-3p levels in established miR-382-3p
overexpressing cell lines and control cells, as well as miR-382-3p
and mRNA levels in miR-382-3p inhibitor or inhibitor
control-transfected cells. One-way ANOVA was used as a
multiple-comparisons test to analyze datasets containing more than
two groups with a subsequent Tukey's post-hoc test for comparison
between two groups. Pearson's correlation analysis was used to
determine the correlation coefficient between miR-382 levels and
SAE1 protein levels. The survival of different groups of patients
was analyzed using the Kaplan-Meier method with the log-rank test.
P<0.05 was considered to indicate statistical significance.
Results
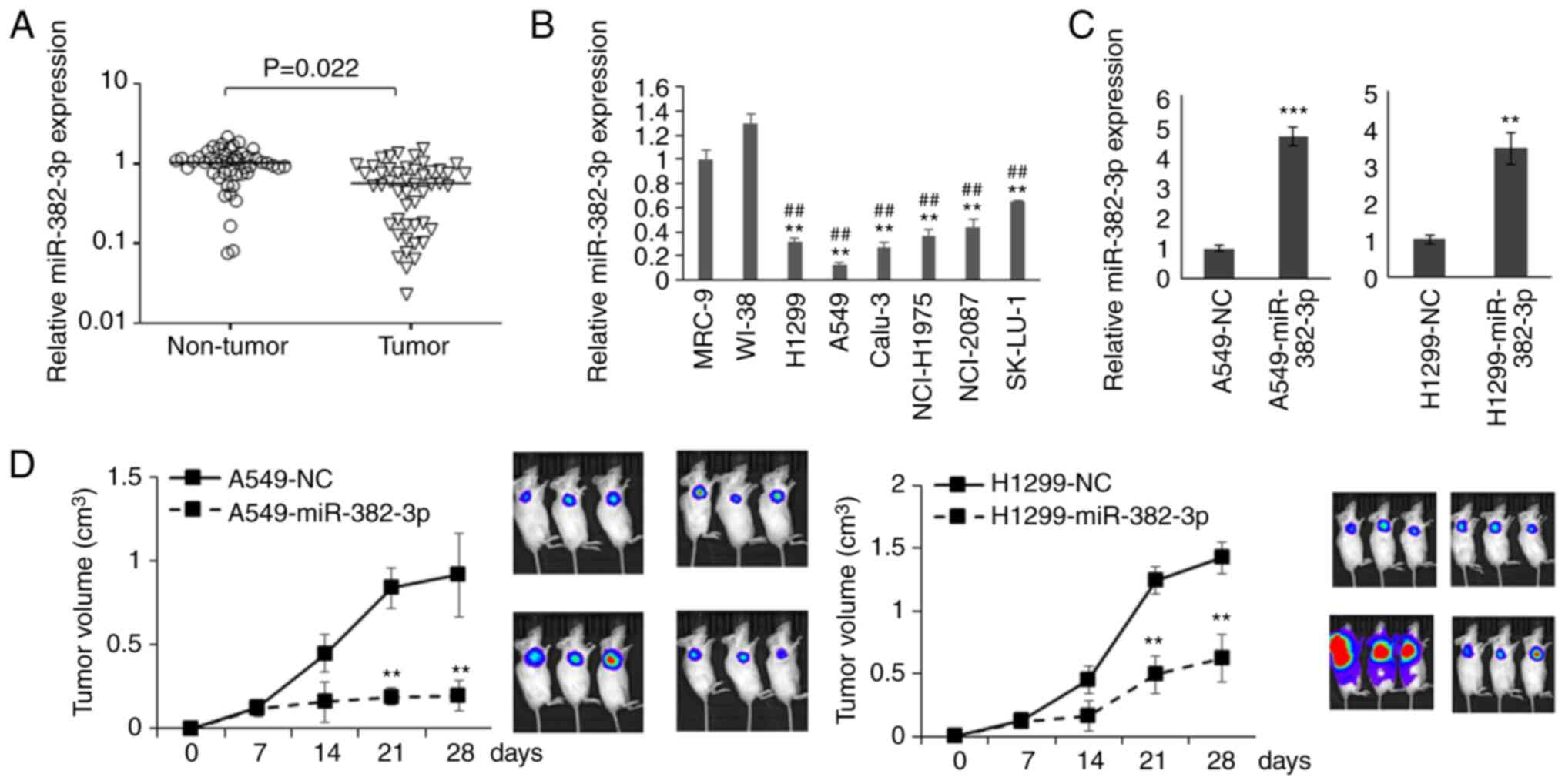
miR-382-3p is downregulated in
patients with LA and functions as a tumor suppressor in LA
To investigate whether miR-382-3p is related to the
carcinogenesis of LA, miR-382-3p levels in tumor samples and
adjacent non-tumor tissues from 78 patients with LA (age,
63.00±8.52 years old; male:female, 45/33; Table I) were analyzed. miR-382-3p levels
in normal lung fibroblasts and six LA cell lines were also
examined. As presented in Fig. 1A
and B, the levels of miR-382-3p
were significantly decreased in LA tumors and cell lines,
suggesting that miR-382-3p downregulation may contribute to the
carcinogenesis of LA.
 | Table ICharacteristics of the patients with
lung adenocarcinoma included in this study (n=78). |
Table I
Characteristics of the patients with
lung adenocarcinoma included in this study (n=78).
| Variable | Value |
|---|
| Sex | |
|
Male | 45 (57.7) |
|
Female | 33 (42.3) |
| Age, years | 63.00±8.52 |
| Smoking | |
|
No | 22 (28.2) |
|
Yes | 56 (71.8) |
| Clinical stage | |
|
IA | 16 (20.5) |
|
IB | 22 (28.2) |
|
IIA | 18 (23.1) |
|
IIB | 22 (28.2) |
To understand the function of miR-382-3p in LA
cells, A549 cells that had the lowest expression of miR-382-3p and
H1299 cells lacking the expression of p53 protein were used to
construct miR-382-3p-overexpressing cell lines to generate a
xenograft mouse model (Fig. 1C).
As presented in Fig. 1D and, tumor
growth was significantly repressed by miR-382-3p.
miR-382-3p inhibits SAE1 expression by
targeting its 3'-UTR
To further investigate how miR-382-3p represses LA
growth, potent miR-382-3p targets were predicted using TargetScan
(http://www.targetscan.org/vert_72/)
and the predicted targets were subjected to gene ontology analysis
using DAVID (https://david.ncifcrf.gov/). It was observed that 18
genes were involved in protein ubiquitination modification and 6
genes were functionally enriched in protein SUMOylation
modification. The level of SAE1 protein was examined and it was
indicated that 57% (41 out of 72) of patients had increased SAE1
expression in their tumor samples (Fig. 2A and B), and that SAE1 expression levels were
negatively correlated with the miR-382-3p levels in tumors
(Fig. 2C). Subsequently, the
survival data of patients with LA were analyzed online (http://kmplot.com/analysis/index.php?p=background) and
it was revealed that patients with low SAE1 expression had a
significantly longer survival time (Fig. 2E), suggesting that SAE1 may
function as an oncogene that promotes the progression of LA.
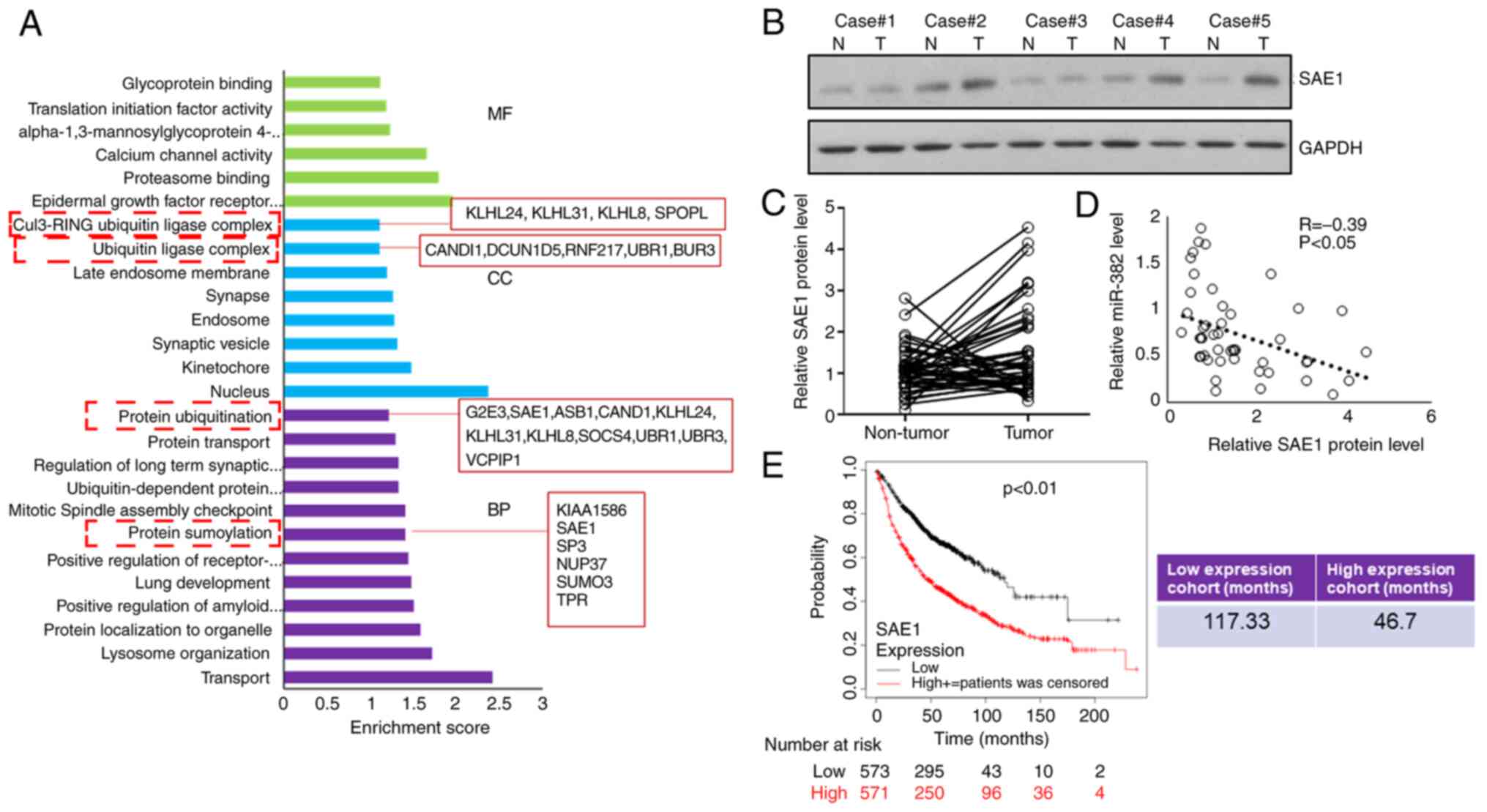 | Figure 2Expression of SAE1, a target of
miR-382-3p, is negatively correlated with miR-382-3p levels in
tumors. (A) Gene Ontology analysis of predicted miR-382-3p targets.
(B and C) SAE1 protein levels in tumor and adjacent non-tumor
control samples were examined by immunoblotting. (B) Representative
western blots and (C) comparison of SAE1 protein levels between
tumor and adjacent non-tumor control samples for each case. (D)
Correlation analysis of the SAE1 protein levels and miR-382-3p
levels in lung adenocarcinoma tumor samples. (E) Kaplan-Meier
curves depicting the overall survival of 1,044 patients with lung
cancer. Data were from Kaplan-Meier Plotter. SAE1, small
ubiquitin-like modifier 1 activating enzyme subunit 1; miR,
microRNA; MF, molecular function; CC, cellular component; BP,
biological process; T, tumor tissue; N, normal tissue. |
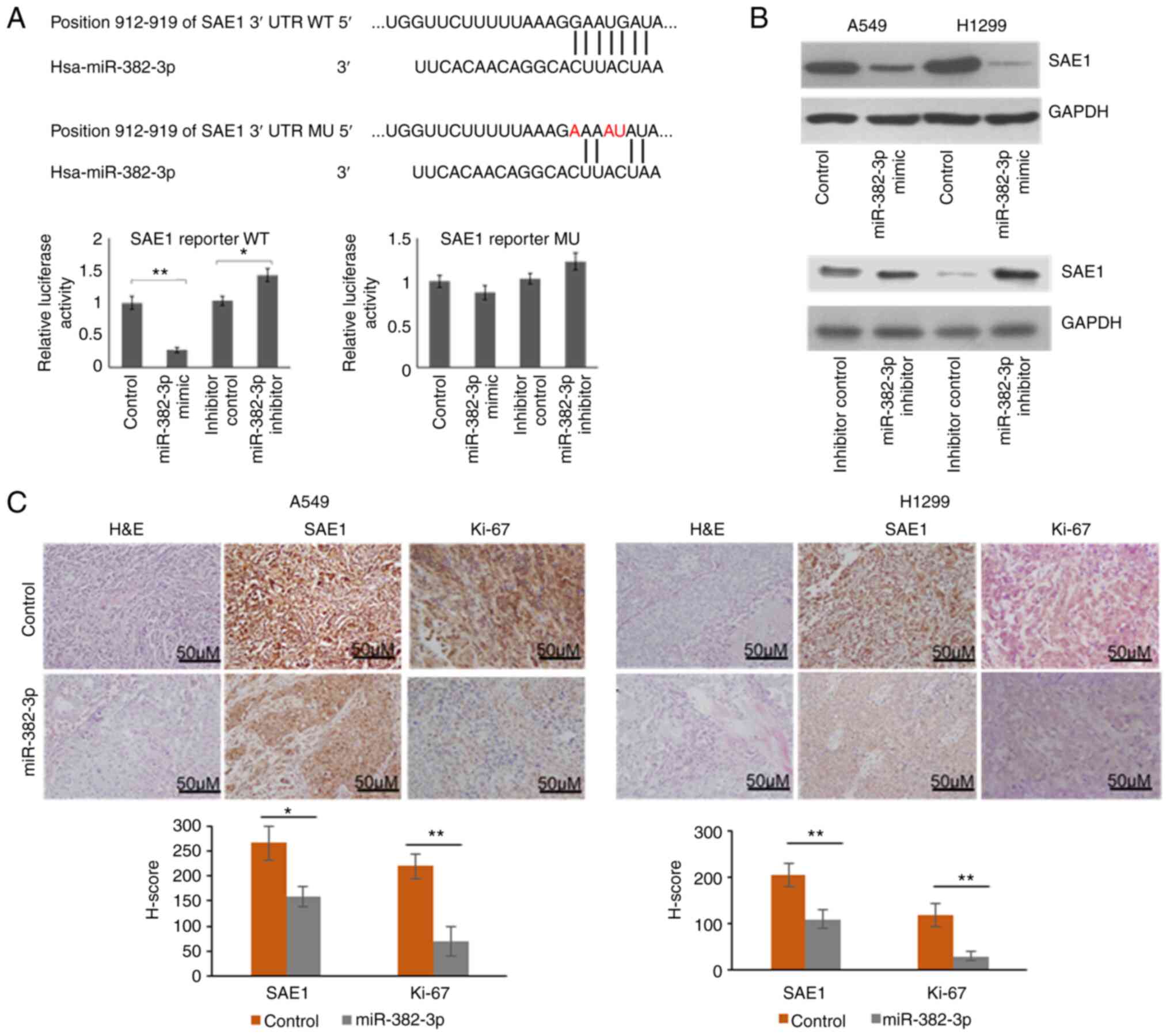
To explore whether SAE1 upregulation is directly
induced by miR-382-3p downregulation, an SAE1 reporter vector was
first constructed. A mutant vector containing 3 replaced
nucleotides was also generated to identify the target site of
miR-382-3p. As presented in Fig.
3A, relative luciferase activity was significantly repressed by
the miR-382-3p mimics and upregulated by the miR-382-3p inhibitor.
When 3 out of 7 nucleotides in the miR-382-3p targeting region were
mutated, luciferase activity was not repressed by miR-382-3p. These
results indicated that miR-382-3p repressed Firefly luciferase
expression by targeting the 3'-UTR region of SAE1. To further
identify whether endogenous SAE1 is regulated by miR-382-3p,
endogenous SAE1 expression was examined in A549 and H1299 cells
after transfection with miR-382-3p mimics or inhibitor. It was
observed that SAE1 expression was repressed by miR-382-3p and
upregulated by an miR-382-3p inhibitor, further indicating that
SAE1 is a direct target of miR-382-3p (Fig. 3B). SAE1 protein levels were then
examined in xenograft tumors generated from
miR-382-3p-overexpressing A549 and H1299. To understand the tumor
cell proliferation in vivo, the Ki-67 signal was examined by
immunohistochemistry. As presented in Fig. 3C, miR-382-3p-overexpressing tumors
had reduced SAE1 and Ki-67 levels.
Downregulation of miR-382-3p promotes
AKT SUMOylation and phosphorylation
To understand the role of SAE1 upregulation in LA, a
full-scale SUMOylation analysis was performed in both tumors and
adjacent non-tumor control samples. As presented in Fig. 4A, patients with increased SAE1
expression in the tumor (cases no. 4 and 5 of the 57% of patients
with increased SAE1 expression.) exhibited upregulated SUMOylation
modification and increased pAKT levels compared to normal
tissues.
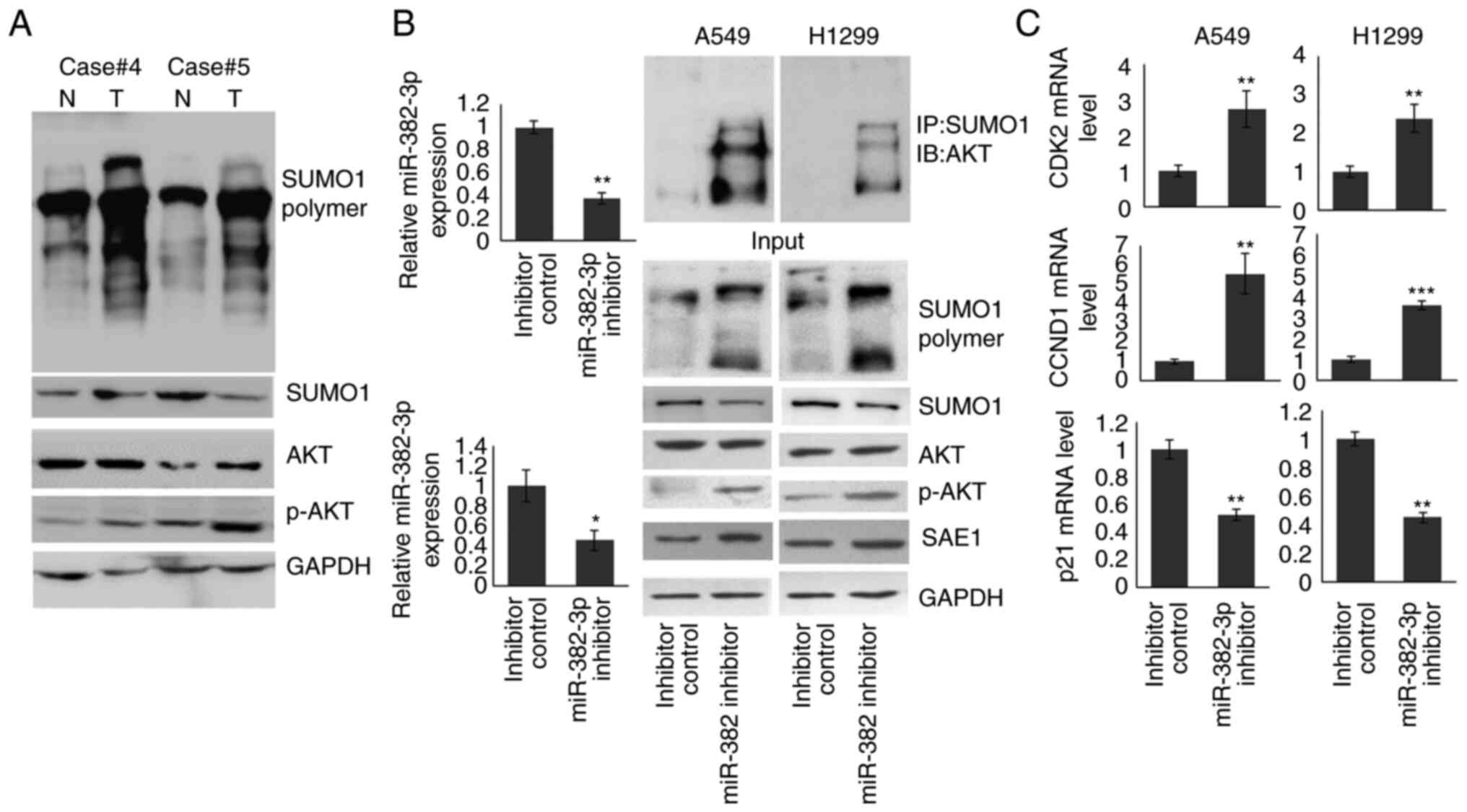 | Figure 4miR-382-3p inhibition upregulates AKT
SUMOylation and activates the AKT signaling pathway. (A)
Immunoblotting was used to detect the levels of full-scale
SUMOylation, SUMO1, AKT and p-AKT in tumor and non-tumor control
samples. (B) A549 and H1299 cells were transfected with miR-382-3p
inhibitor for 48 h. The cells were then subjected to RT-qPCR and
immunoblotting. (C) The expression of AKT signaling downstream
genes was examined by RT-qPCR. *P<0.05,
**P<0.01, ***P<0.001 vs. control. miR,
microRNA; RT-qPCR, reverse transcription-quantitative PCR; p-AKT,
phosphorylated AKT; T, tumor tissue; N, normal tissue; IP,
immunoprecipitation; IB, immunoblot; CCND1, cyclin D1; CDK2,
cyclin-dependent kinase 2; SUMO1, small ubiquitin-like modifier
1. |
To identify whether an increase in pAKT resulted
from downregulation of miR-382-3p, miR-382-3p was knocked down in
A549 and H1299 cells. Proteins modified by SUMO1 were enriched by
immunoprecipitation using anti-SUMO1 antibody, and the AKT protein
level was examined by immunoblotting. As indicated in Fig. 4B, endogenous miR-382-3p was reduced
to 37.1 and 40.7% in A549 and H1299 cells, respectively. SUMOylated
AKT levels were increased and pAKT was upregulated. To confirm the
activation of the AKT signaling pathway by the miR-382-3p
inhibitor, the mRNA levels of three downstream genes were examined.
As presented in Fig. 4C, CCND1 and
CDK2 expression were significantly increased in miR-382-3p
inhibitor-transfected A549 and H1299 cells, while p21 levels were
reduced. These results indicated that miR-382-3p inhibition
activated the AKT signaling pathway by promoting AKT SUMOylation
and phosphorylation.
Inhibition of miR-382-3p promotes
proliferation and inhibits apoptosis in LA cells by upregulating
SAE1
To further understand the biological function of
miR-382-3p and SAE1 in lung cancer carcinogenesis, A549 and H1299
cells were transfected with a miR-382-3p inhibitor with or without
siRNAs specifically targeting SAE1. SAE1 knockdown was confirmed
using immunoblotting (Fig. S1).
As presented in Fig. 5A, cell
viability was increased in the miR-382-3p knockdown A549 and H1299
cells. Although the cell viability was still slightly higher in
miR-382-3p and SAE1 double knockdown cells, the difference was not
significant. A parallel effect was observed on cell apoptosis. As
presented in Fig. 5B, the number
of apoptotic cells was reduced in miR-382-3p knockdown cells, while
SAE1 and miR-382-3p double-knockdown cells had a similar proportion
of apoptotic cells as the control cells. Furthermore, miR-382-3p
knockdown A549 and H1299 cells had an increased S-phase population
(Fig. 5C and D). SAE1 knockdown partially restored
miR-382-3p inhibitor function (Fig.
5C and D).
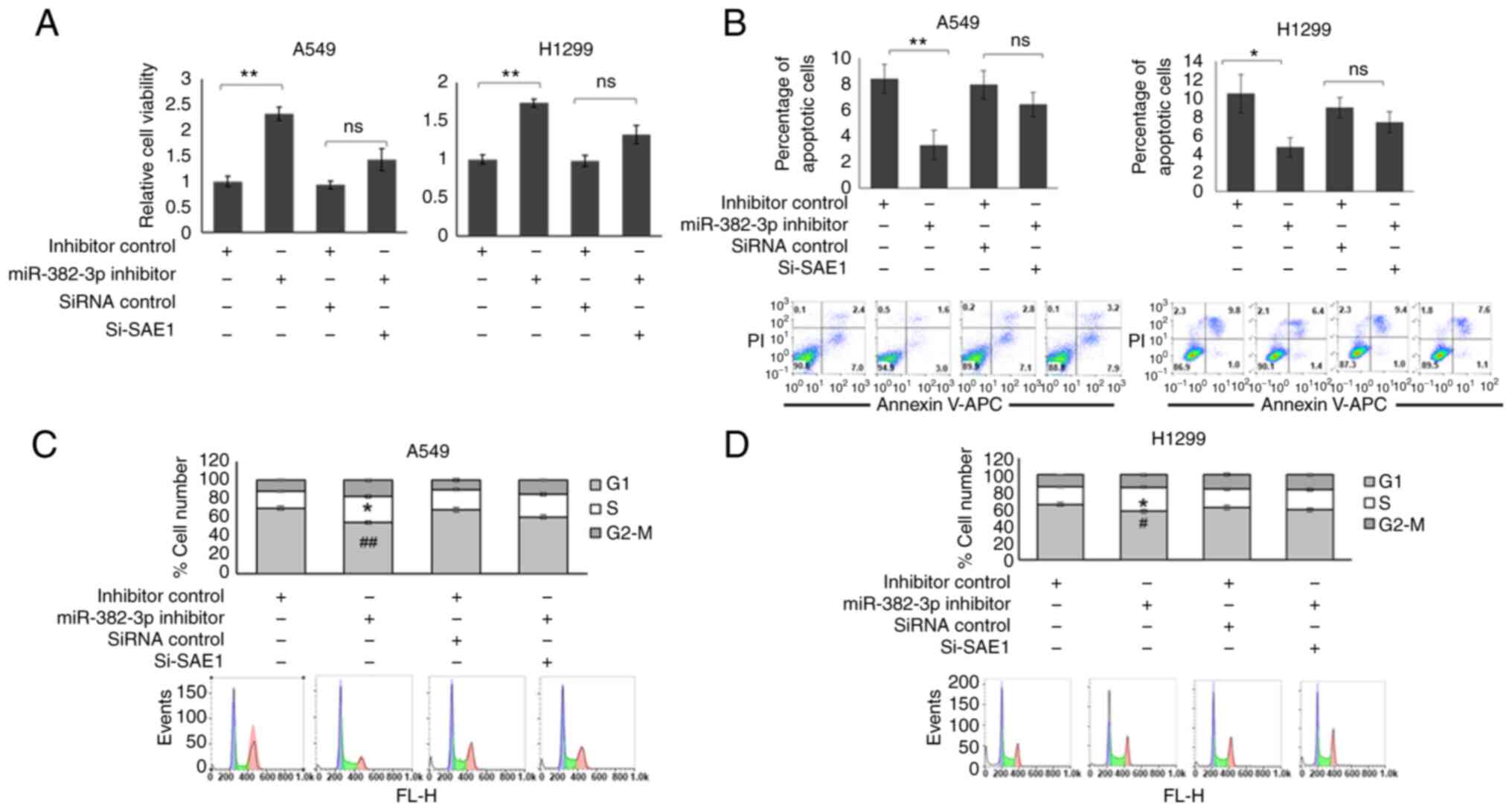 | Figure 5miR-382-3p inhibition promotes
proliferation and inhibits apoptosis in lung adenocarcinoma cell
lines. A549 and H1299 cells were transfected with miR-382-3p
inhibitor, with or without SAE1-specific siRNA for 48 h. (A) The
cells were subjected to MTT assays to examine proliferation and
viability. (B) Flow cytometric analysis was used to detect cell
viability and apoptosis. *P<0.05,
**P<0.01. (C and D) Cell cycle analysis of (C) A549
and (D) H1299 cells. *P<0.05 S phase vs. inhibitor
control; #P<0.05, ##P<0.01 G2-M phase
vs. inhibitor control. ns, no significance; miR, microRNA; PI,
propidium iodide; APC, allophycocyanin; siRNA, small interfering
RNA; si-SAE1, siRNA targeting SAE1; SAE1, small ubiquitin-like
modifier 1 activating enzyme subunit 1; FL-H,
fluorescence-height. |
Discussion
miR-382-3p expression has been known to be
downregulated in the peripheral blood of patients with LA, but its
direct targets and biological functions have remained elusive
(7). In the present study, it was
confirmed that miR-382-3p was also downregulated in tumor samples
from patients with LA. The direct targets of miR-382-3p were
predicted using bioinformatics tools and their interaction was
confirmed using a dual-luciferase assay and immunoblotting. It was
identified that SAE1, a key component of the SUMO activation
complex, is a direct target of miR-382-3p. Knockdown of miR-382-3p
in LA cells upregulated SAE1 protein levels and promoted the
SUMOylation of AKT, which further promoted cell proliferation and
inhibited apoptosis by upregulating AKT downstream genes.
miR-382-5p is the predominant product of the gene
encoding miR-382, which acts as a tumor suppressor and is involved
in the pathogenesis of various human cancers, including
hepatocellular carcinoma, ovarian cancer and osteosarcoma (18-20).
However, the role of miR-382-3p in the lung has remained to be
fully elucidated. In the present study, it was identified for the
first time, to the best of our knowledge, that the downregulation
of miR-382-3p in patients with LA contributes to the carcinogenesis
of LA via activation of the AKT signaling pathway. miR-382-3p
overexpression repressed tumor growth in xenograft mouse models,
providing a candidate for LA treatment.
miRNAs are a group of non-coding RNAs that are
initially transcribed by RNA polymerase II to form primary
transcripts (pri-miRNAs) (21).
Pri-miRNAs are then processed through a two-step cleavage mechanism
involving two RNase III-class enzymes, Drosha and Dicer, and become
mature 21-23 nt-long miRNAs (22).
miRNA duplexes are loaded onto Argonaute family proteins and are
subsequently unwound to form mature effector complexes that
regulate the expression of target mRNAs. One miRNA typically
regulates the expression of multiple targets simultaneously. In the
present study, only SAE1 was identified as one of the targets of
miR-382-3p. Although the importance of SAE1 in the regulatory
effect of miR-382-3p on cell proliferation, apoptosis and cell
cycle has been confirmed through knockdown experiments, the full
roles of miR-382-3p downregulation in the carcinogenesis and
processes associated with LA still require to be fully
elucidated.
Cells control miRNA levels in multiple ways,
including transcription, maturation and degradation. miR-382-3p was
identified to be related to the carcinogenesis of LA in the present
study, but how miR-382-3p exerts its regulatory roles in lung
cancer cells remains to be further investigated.
In conclusion, the present study determined that
downregulation of miR-382-3p contributes to the carcinogenesis of
LA by upregulating SAE1 and promoting AKT SUMOylation.
Supplementary Material
siRNA targeting SAE1 to knockdown
endogenous SAE1 protein levels successfully. Si-SAE1 or siRNA
control was transfected into A549 or H1299 for 48 h and the SAE1
protein level was detected by immunoblotting. siRNA, small
interfering RNA; SAE1, small ubiquitin-like modifier 1 activating
enzyme subunit 1.
Acknowledgements
Not applicable.
Funding
Funding: This research was supported by Beijing Gold-Bridge
Project (grant no. ZZ19058) and Beijing Xicheng District Excellent
Talent Training Funding Project (grant no. 202038).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HF was responsible for the conception and design of
the study. HF, ZW and WW were involved in data acquisition. HF, ZW
and WW were involved in the development of the study methodology,
analysis and interpretation of the data. HF was involved in the
writing, reviewing and revision of the article and analyzed the
relevant literature. All the authors have read and approved the
final manuscript. HF, ZW and WW confirmed the authenticity of the
raw data.
Ethics approval and consent to
participate
The present study was approved by the Human Basic
and Clinical Research Ethics Committee of Fuxing Hospital (Beijing,
China; approval no. 2018FXHEC-KY-19). All participants provided
written informed consent according to the principles of the
Declaration of Helsinki prior to sampling. All experiments
involving animals were pre-approved by the IACUC of Fuxing Hospital
(Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Li Q, Liu M, Ma F, Luo Y, Cai R, Wang L,
Xu N and Xu B: Circulating miR-19a and miR-205 in serum may predict
the sensitivity of luminal A subtype of breast cancer patients to
neoadjuvant chemotherapy with epirubicin plus paclitaxel. PLoS One.
9(e104870)2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Maes OC, Chertkow HM, Wang E and Schipper
HM: MicroRNA: Implications for Alzheimer disease and other human
CNS disorders. Curr Genomics. 10:154–168. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Xu J, Li Y, Wang F, Wang X, Cheng B, Ye F,
Xie X, Zhou C and Lu W: Suppressed miR-424 expression via
upregulation of target gene Chk1 contributes to the progression of
cervical cancer. Oncogene. 32:976–987. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Farazi TA, Hoell JI, Morozov P and Tuschl
T: MicroRNAs in human cancer. Adv Exp Med Biol. 774:1–20.
2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fang H, Liu Y, He Y, Jiang Y, Wei Y, Liu
H, Gong Y and An G: Extracellular vesicle-delivered miR-505-5p, as
a diagnostic biomarker of early lung adenocarcinoma, inhibits cell
apoptosis by targeting TP53AIP1. Int J Oncol. 54:1821–1832.
2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Tsurutani J, Fukuoka J, Tsurutani H, Shih
JH, Hewitt SM, Travis WD, Jen J and Dennis PA: Evaluation of two
phosphorylation sites improves the prognostic significance of Akt
activation in non-small-cell lung cancer tumors. J Clin Oncol.
24:306–314. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yoshizawa A, Fukuoka J, Shimizu S, Shilo
K, Franks TJ, Hewitt SM, Fujii T, Cordon-Cardo C, Jen J and Travis
WD: Overexpression of phospho-eIF4E is associated with survival
through AKT pathway in non-small cell lung cancer. Clin Cancer Res.
16:240–248. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li R, Wei J, Jiang C, Liu D, Deng L, Zhang
K and Wang P: Akt SUMOylation regulates cell proliferation and
tumorigenesis. Cancer Res. 73:5742–5753. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kessler BM, Bursomanno S, McGouran JF,
Hickson ID and Liu Y: Biochemical and mass spectrometry-based
approaches to profile SUMOylation in human cells. Methods Mol Biol.
1491:131–144. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Seeler JS and Dejean A: SUMO and the
robustness of cancer. Nat Rev Cancer. 17:184–197. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Inamura K, Shimoji T, Ninomiya H,
Hiramatsu M, Okui M, Satoh Y, Okumura S, Nakagawa K, Noda T,
Fukayama M and Ishikawa Y: A metastatic signature in entire lung
adenocarcinomas irrespective of morphological heterogeneity. Hum
Pathol. 38:702–709. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Orlans FB: Regulation of animal
experimentation: United States of America. Acta Physiol Scand
Suppl. 554:138–152. 1986.PubMed/NCBI
|
|
16
|
Guide for the Care and Use of Laboratory
Animals. Washington, DC, 1996.
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang S, Ge W, Zou G, Yu L, Zhu Y, Li Q,
Zhang Y, Wang Z and Xu T: MiR-382 targets GOLM1 to inhibit
metastasis of hepatocellular carcinoma and its down-regulation
predicts a poor survival. Am J Cancer Res. 8:120–131.
2018.PubMed/NCBI
|
|
19
|
Tan H, He Q, Gong G, Wang Y, Li J, Wang J,
Zhu D and Wu X: miR-382 inhibits migration and invasion by
targeting ROR1 through regulating EMT in ovarian cancer. Int J
Oncol. 48:181–190. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Xu M, Jin H, Xu CX, Sun B, Song ZG, Bi WZ
and Wang Y: miR-382 inhibits osteosarcoma metastasis and relapse by
targeting Y box-binding protein 1. Mol Ther. 23:89–98.
2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kim VN, Han J and Siomi MC: Biogenesis of
small RNAs in animals. Nat Rev Mol Cell Biol. 10:126–139.
2009.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Okamura K: Diversity of animal small RNA
pathways and their biological utility. Wiley Interdiscip Rev RNA.
3:351–368. 2012.PubMed/NCBI View
Article : Google Scholar
|