Introduction
Cardiovascular disease (CVD) is a prime cause of
death worldwide, accounting for 17.3 million deaths per year
(1). Acute myocardial infarction
is the most serious type of CVD. There are >3 million patients
with acute ST-segment elevation myocardial infarction each year.
The most effective treatment for these patients is timely and
effective reperfusion therapy (2).
Reperfusion therapy improves the myocardial blood supply and is
accompanied by a series of pathophysiological reactions, including
peroxidation, inflammation, intracellular calcium overload, and
finally irreversible apoptosis and necrosis. Myocardial injury
resulting from reperfusion is termed reperfusion injury (3), and it presents a clinical problem
that urgently needs to be solved.
During myocardial ischemia-reperfusion (I/R),
myocardial nicotinamide adenine dinucleotide phosphate (NADPH)
oxidase enzyme 4 (NOX4) expression is upregulated, and myocardial
metabolic activity is enhanced, producing a large amount of
reactive oxygen species (ROS) which contributes to myocardial
injury (4). ROS can trigger a
variety of signal transduction pathways, including enzyme-coupled
receptor signaling pathways and G protein-coupled receptor
signaling pathways, among which the MAPK signaling pathways play a
key role in numerous cell activities (such as proliferation,
differentiation, survival and death) (5). The inhibition of overactivated p38
MAPK can significantly reduce experimental myocardial I/R injury
(6).
Plumbagin (PLB;
5-hydroxy-2-methyl-naphthalene-1,4-dione) is a major bioactive
compound extracted from the roots of Plumbago zeylanica that
acts as an inhibitor of NOX4(7).
PLB not only inhibits adenosine diphosphate (ADP)-induced platelet
aggregation in the cardiovascular system but also suppresses NOX4
expression, which can significantly improve the redox state
imbalance of myocardial I/R (8-10).
The aforementioned studies suggest that PLB can alleviate
myocardial I/R injury and has robust potential for application in
the treatment of CVD. As an organic hydroperoxide, tertiary butyl
hydrogen peroxide (TBHP) is more stable than hydrogenperoxide
(H2O2), and it selectively inhibits
mitochondrial function, induces membrane lipid peroxidation, and
promotes nucleotide degradation and adenosine formation (11). Based on the relatively well-defined
mechanisms of TBHP action, TBHP-treated H9c2 cells were selected as
a biological model likely to result in peroxidation injury in
response to oxidative stress. In the present study, the protective
effects of PLB in the prevention of TBHP-induced oxidative stress
and apoptosis were verified in H9c2 cardiomyocytes.
Materials and methods
Materials
PLB (cat. no. S4777) was obtained from Selleck
Chemicals. Anti-microtubule-associated protein 1 light chain 3
(LC3)-II/LC3-I (cat. no. PAB34124), anti-NOX4 (PAB30655),
anti-phosphorylated (p)-p38 MAPK (PAB43139-P) and anti-p38 MAPK
(PAB40560) were purchased from Bioswamp; Wuhan Bienle Biotechnology
Co., Ltd. Anti-cleaved caspase-3 (ab214430) and anti-GAPDH
(ab181602) were obtained from Abcam.
Cell culture and treatments
H9c2 cardiomyocytes were obtained from the American
Type Culture Collection (ATCC: CRL-1446). Cells were grown in DMEM
(product no. SH30022.01B; HyClone; Cytiva) supplemented with 10%
FBS (Gibco), 100 µg/ml penicillin (Sigma) and 100 µg/ml
streptomycin (Sigma) and maintained at 37˚C in a humidified 5%
CO2 incubator. Confluent cardiomyocytes were cultured in
DMEM supplemented with 2% FBS for an additional 12 h prior to
experimentation. For experiments, cells were preincubated with PLB
(5, 10 or 20 µM) for 24 h and TBHP (75 µM) for another 4 h. PLB was
dissolved in DMSO and then diluted with DMEM to a final
concentration of <0.1% DMSO. TBHP was dissolved in DMEM.
Cell viability assays
Cardiomyocytes were seeded in 96-well plates at a
density of 5x103 cells/well. Cells were pretreated with
PLB (5, 10, and 20 µM) for 24 h and then treated with TBHP for
another 4 h. The number of viable cells was determined using a Cell
Counting Kit-8 (CCK-8) assay. Briefly, the DMEM culture medium was
discarded, and 100 µl CCK-8 reagent (Beyotime Institute of
Biotechnology) was added to fresh DMEM. The 96-well plate was
placed in a CO2 incubator for 2 h. The optical density
(OD) values were determined at a wavelength of 450 nm. The cell
proliferation rate (%) was calculated as follows: (OD value of
experimental well - OD value of control well)/OD value of control
well x100%. The CCK-8 assay was repeated 3 times for
consistency.
Lactate dehydrogenase (LDH) and
creatine kinase (CK) leakage
Cytotoxicity was evaluated by detecting plasma
membrane damage using commercially available LDH-estimation (cat.
no. A020-1) and CK-estimation (cat. no. A032-1-1) kits (both from
Beyotime Institute of Biotechnology). For LDH and CK leakage
assays, H9c2 cells were grown in 24-well plates at a density of
3x105 cells/well, and cells were subjected to further
experiments after 24 h. The LDH and CK activities were measured
after 24 h of treatment per the manufacturer's protocols.
Estimation of intracellular ROS
production
ROS production was determined by detecting the
fluorescence intensity of dichlorofluorescin (DCF) via flow
cytometry. Cells were treated with 2'-7'dichlorofluorescin
diacetate (DCFH-DA) (10 µM) at 37˚C in the dark for 20 min. Cells
were then collected and suspended in PBS. The DCF fluorescence
intensity was analyzed using flow cytometry (ACEA NovoCyte, ACEA
Biosciences) at an excitation wavelength of 488 nm and an emission
wavelength of 519 nm. Each assay was performed 3 times. Data were
analyzed with NovoExpress 1.5 developed by ACEA Biosciences
Inc.
Measurement of apoptosis
Following treatment, cardiomyocytes
(1.5x105-1x106) were collected and
immobilized in 75% cold ethanol for 12 h at 4˚C. Immobilized cells
were double-stained with Annexin V-FITC (10 µl) and PI (10 µl; cat.
no. 556547, BD Biosciences) in the dark at room temperature for 30
min. The apoptotic rate of cardiomyocytes was analyzed using flow
cytometry (ACEA NovoCyte, ACEA Biosciences). Each test was repeated
3 times. Data were analyzed with NovoExpress 1.5 (ACEA Biosciences
Inc.).
Western blot analysis
Protein levels were analyzed in whole cardiomyocyte
lysates. H9c2 cell lysates were prepared with RIPA buffer
containing protease inhibitor cocktail (cat. no. PAB180006;
Bioswamp), and protein concentration were determined by BCA Protein
Assay Kit. A total of 30 µg of protein was separated by 10%
SDS-PAGE and transferred to a PVDF membrane (Millipore). Membranes
were blocked in 5% bovine serum albumin (Beyotime) for 2 h at room
temperature and then incubated with the following primary
antibodies overnight at 4˚C: NOX4, cleaved caspase-3, LC3-II/LC3-I,
p-p38 MAPK, p38 MAPK, and GAPDH (all 1:1,000). The following day,
the membranes were incubated with HRP-conjugated secondary antibody
(cat. no. SAB43714, Bioswamp, 1:10,000) at room temperature for 1.5
h. The secondary antibody was detected by ECL (Beyotime Institute
of Biotechnology). The bands were scanned and quantified by
densitometry analysis using Tanon GIS software (GIS 1D Ver.4.00,
Tanon, Shanghai, China).
Statistical analysis
Data are presented as the mean ± SD. The significant
differences between groups were assessed with SPSS version 13.0.
Comparisons of results were performed using one-way analysis of
variance (ANOVA) followed by Tukey's post hoc test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
PLB protects H9c2 cells from cell
death
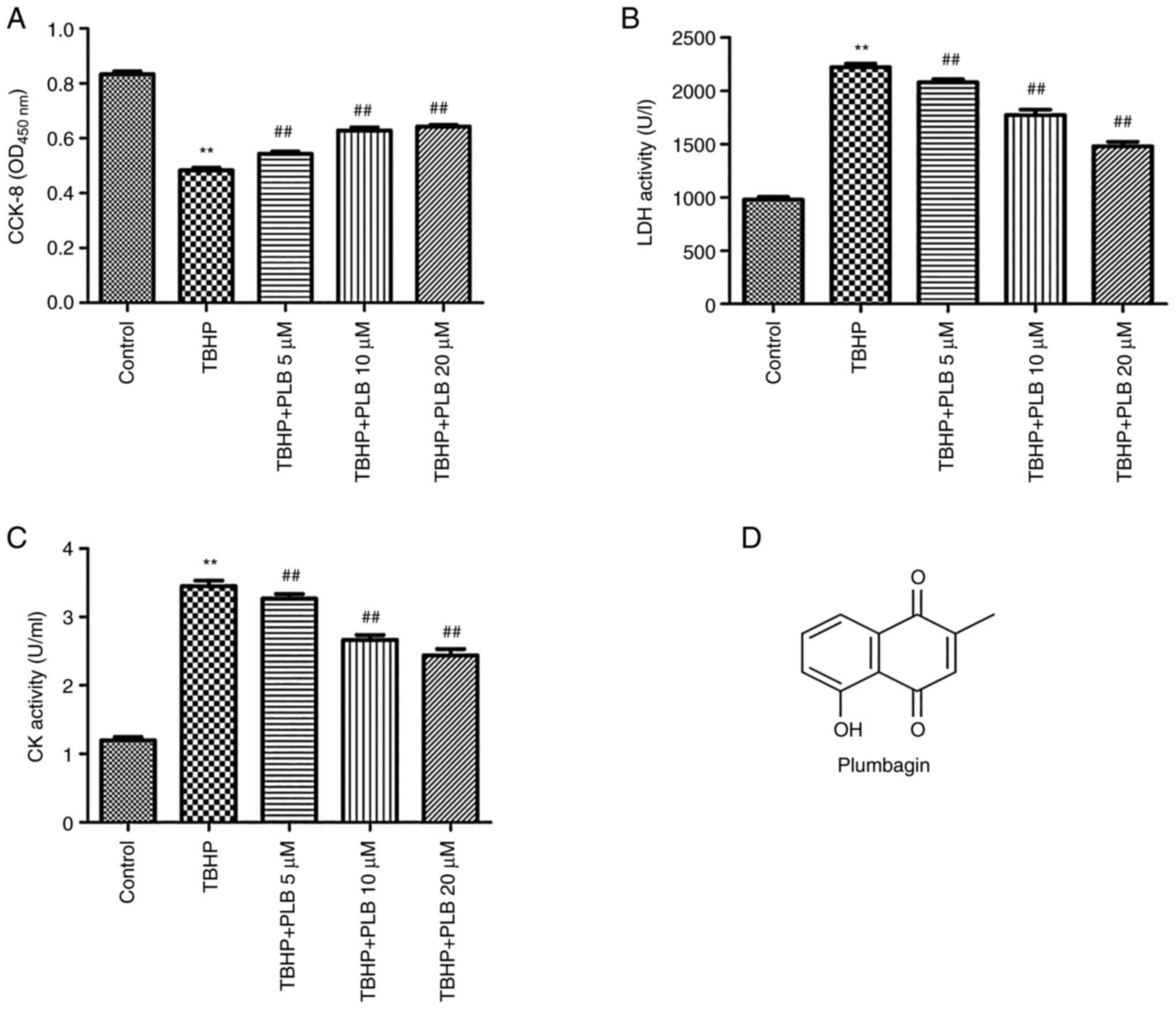
Cell viability was assessed using the CCK-8 assay,
as shown in Fig. 1A. TBHP
significantly reduced cell viability compared with the control
group (P<0.01). PLB (5, 10 or 20 µM) pretreatment attenuated the
TBHP-induced reduction in H9c2 cell viability (P<0.01).
Additionally, TBHP increased LDH and CK activities in H9c2
cardiomyocytes, and these increases were reduced by PLB
pretreatment (P<0.01; Fig. 1B
and C). The chemical structure of
PLB is presented in Fig. 1D.
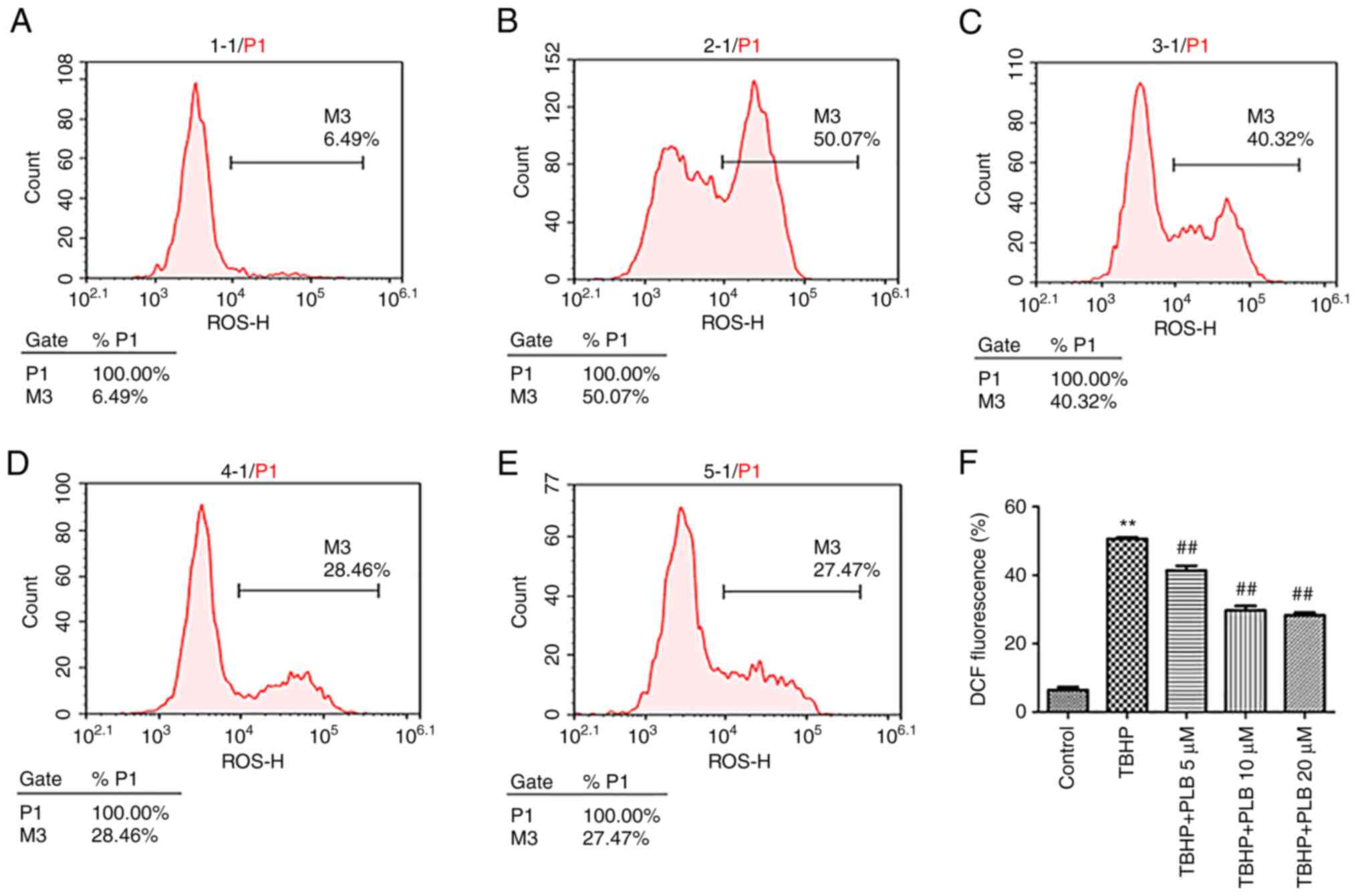
PLB alleviates TBHP-induced increase
of ROS
The ROS levels in H9c2 cells were quantified by
DCF-DA staining. TBHP treatment significantly increased
intracellular ROS levels in H9c2 cells, and pretreatment with PLB
(5, 10 and 20 µM) decreased ROS generation (P<0.01; Fig. 2).
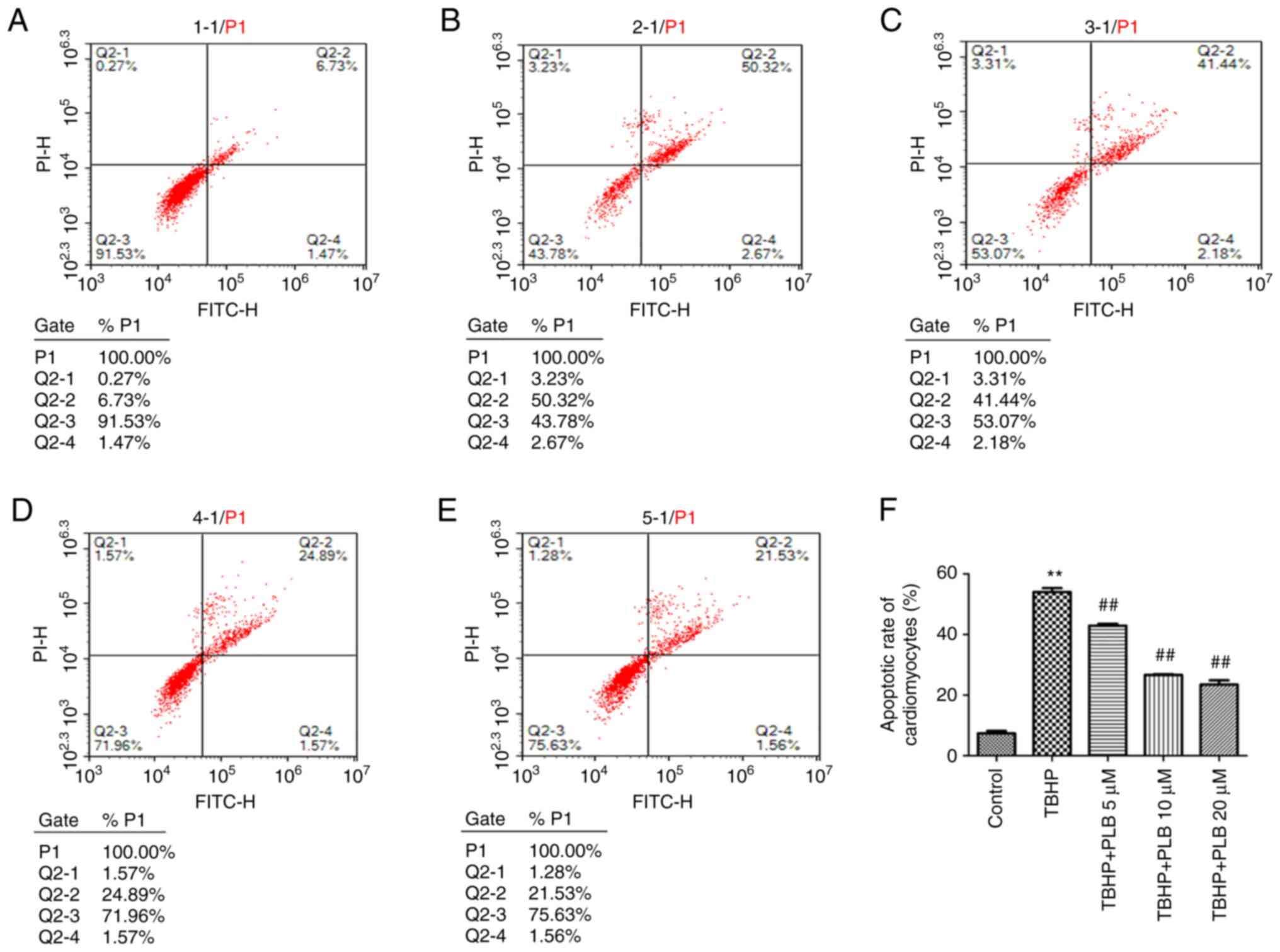
PLB prevents TBHP-induced
apoptosis
As revealed in Fig.
3, the apoptotic rate was significantly increased in the TBHP
group compared with the control group (P<0.01). Pretreatment
with PLB (5, 10 or 20 µM) significantly reduced the apoptotic rate
compared with TBHP treatment alone (P<0.01).
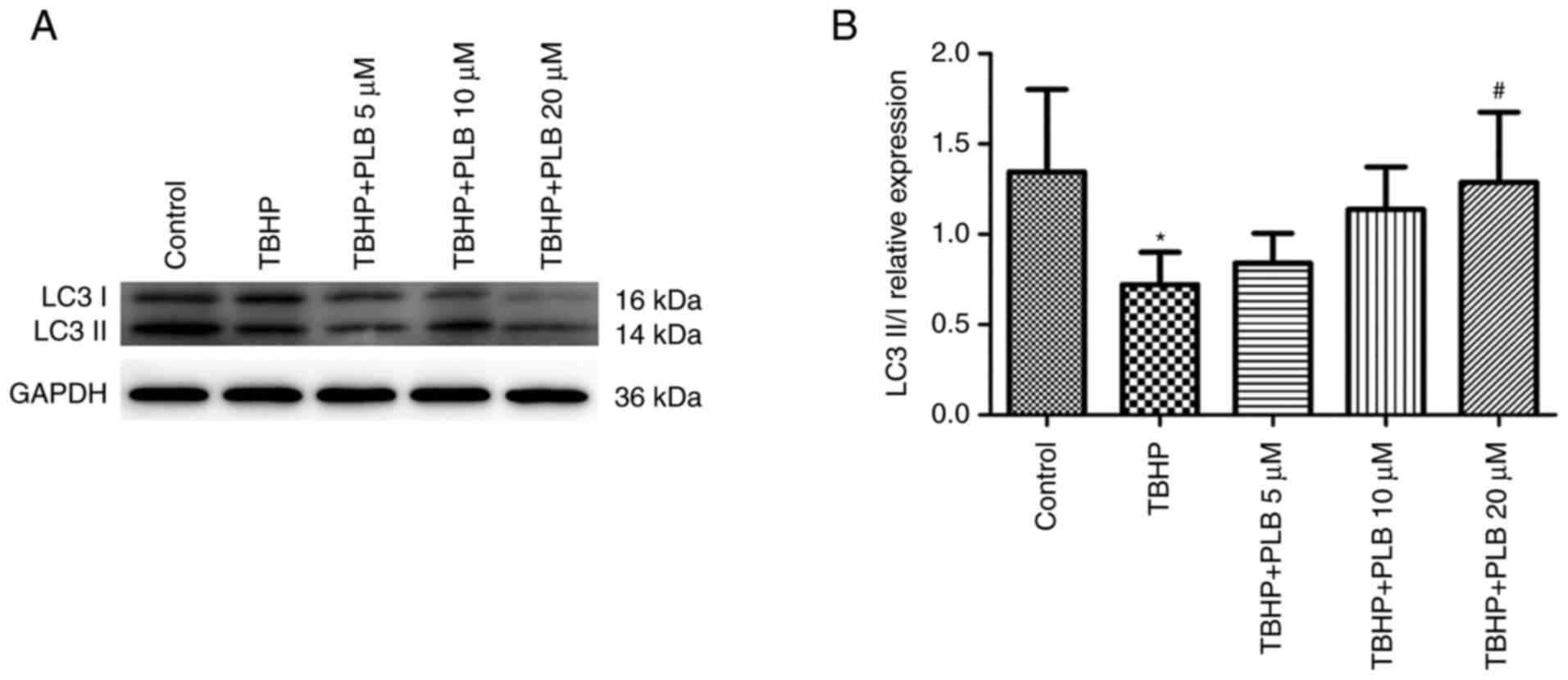
PLB promotes autophagy in H9c2
cells
LC3-II/LC3-I levels, a marker of active
autophagosomes, were analyzed by western blot analysis (Fig. 4). The ratio of LC3-II/LC3-I was
decreased in TBHP-treated H2c9 cells, and this decrease was
attenuated by PLB pretreatment compared with TBHP treatment alone
(P<0.05). These data indicated that PLB induces autophagy in
TBHP-treated H9c2 cells.
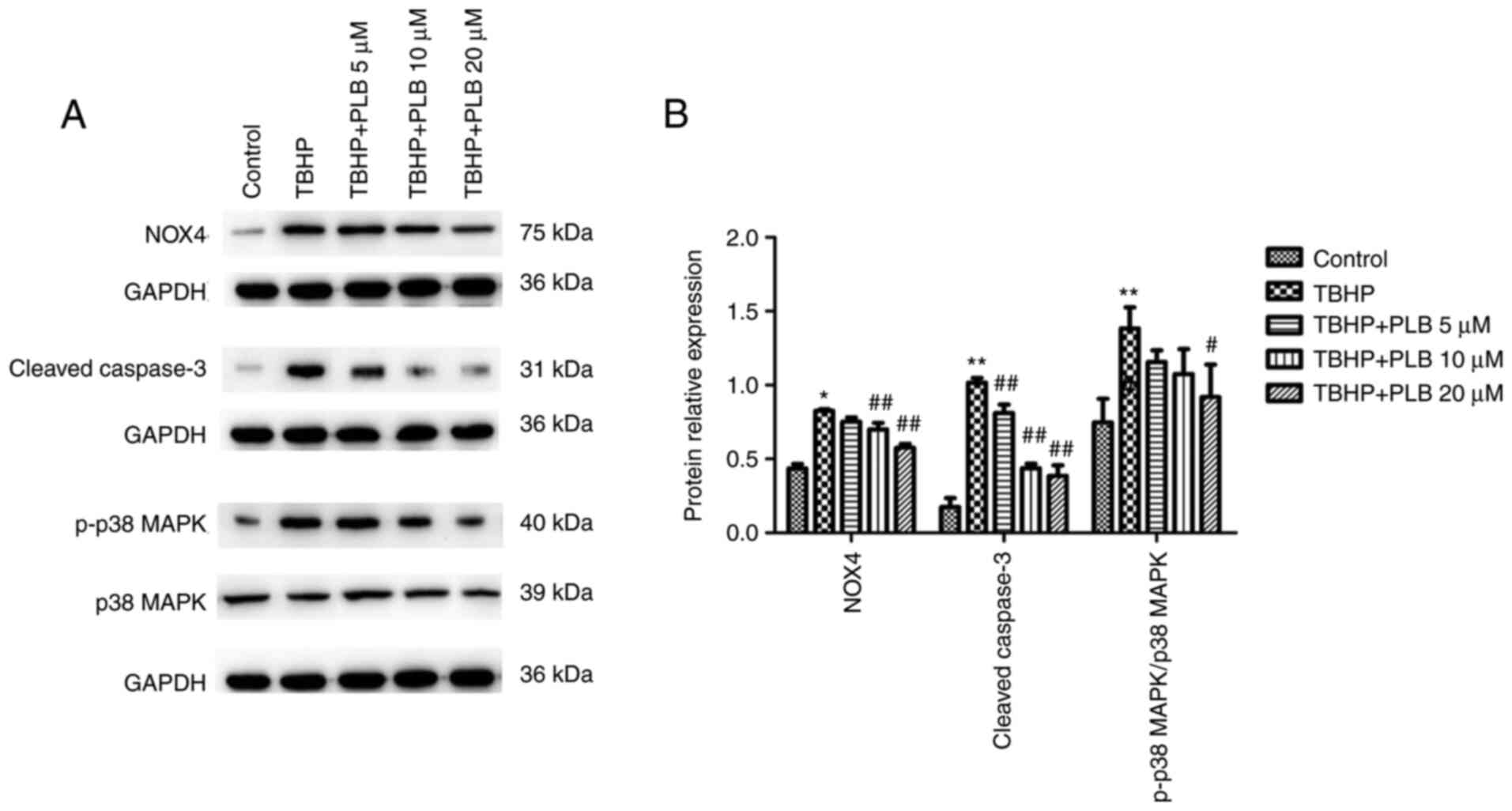
PLB suppresses the NOX4/p38 MAPK
pathway
NOX4, cleaved caspase-3, and p-p38/p38 MAPK protein
expression was significantly increased in the TBHP group compared
with the control group (P<0.05 and P<0.01). Pretreatment with
PLB (5, 10 or 20 µM) suppressed TBHP-induced NOX4, cleaved
caspase-3, and p-p38/p38 MAPK protein expression when compared with
the TBHP only group (P<0.01 and P<0.05; Fig. 5).
Discussion
Myocardial I/R injury is closely related to
oxidative stress. Under physiological conditions, small amounts of
oxygen free radicals can be quickly eliminated in the body.
However, when cells are ischemic and hypoxic, intracellular
metabolism becomes disordered, and oxygen free radical scavenging
capacity is insufficient. When blood supply is suddenly restored in
previously ischemic tissue, oxygen free radicals are produced at a
level that is unable to be quickly eliminated, causing damage to
myocardial tissues and surrounding cells (12-14).
PLB is a natural naphthoquinone compound that protects against
myocardial damage by modulating cardiac biomarkers, antioxidants,
and apoptotic signaling in doxorubicin-induced cardiotoxicity in
rats (15). PLB can also inhibit
NOX4 and regulate redox signals (16). Thus, in the present study, the
effect of PLB on oxidative stress-induced H9c2 cardiomyocyte injury
was investigated. Experimental doses of PLB were based on a
previous in vitro study (17).
TBHP is a pro-oxidant that increases membrane
permeability, lipid peroxidation, ATP consumption, protein thiol
group modification, and cytoplasmic calcium ion concentration
imbalance by generating tert-butoxy groups (18). TBHP also causes cytotoxicity,
mitochondrial-mediated apoptosis, and necrosis through the loss of
membrane integrity, characterized by the release of cytochrome
c, increased p53 expression, and mitochondrial membrane
transformation (19). In the
present study, the classic oxidant TBHP was selected to induce
oxidative stress damage in the H9c2 cell line and to explore the
cytoprotective effects of PLB on TBHP-induced cardiomyocyte injury.
The electron carrier 1-methoxy PMS mediates CCK-8 reduction by
dehydrogenase in cell mitochondria to form a highly water-soluble
yellow formazan. The amount of formazan produced is directly
proportional to the number of living cells. Therefore, CCK-8
measurement can indirectly reflect the number of living cells. The
results of the present study demonstrated that TBHP reduced H9c2
proliferation rate, suggesting that TBHP-induced oxidative stress
decreased cell viability. Pretreatment with PLB reduced the
negative impact of TBHP on cellular proliferation and improved cell
viability in a dose-dependent manner. LDH and CK are important
diagnostic indicators of acute myocardial infarction, angina
pectoris, myocarditis, and other myocardial injuries. Following
ischemia and reperfusion, the myocardial cell membrane is damaged
and its permeability increases, causing leakage of intracellular
LDH and other enzymes. As a result, plasma LDH levels increase. CK
is an important enzyme in energy metabolism. When various tissues
and organs of the body are injured, intracellular CK leaks and cell
vitality is reduced, which can particularly be observed with
myocardial tissue damage that occurs during ischemia (20). PLB pretreatment reduces LDH and CK
activity induced by TBHP, suggesting that PLB may improve
TBHP-induced cardiomyocyte damage.
Excessive ROS production during the I/R period leads
to oxidative stress, an important pathogenic factor of myocardial
I/R injury (21). Under
physiological conditions, there is a certain amount of ROS in the
myocardium. When myocardial ischemia and hypoxia occur, the
function of the ROS scavenging system decreases and the function of
the generating system increases. Once the blood oxygen supply is
restored, ROS are produced in large quantities and accumulate
quickly, causing acute or chronic cardiomyocyte injury (22). During myocardial I/R, ROS is
produced in large quantities by myocardial cell mitochondria,
vascular endothelial cell purine oxidase and other oxidases,
neutrophil respiratory bursts, catecholamines, and other pathways
that allow the myocardial cell membrane and subcellular organelle
membrane to communicate. Increased permeability and loss of
membrane integrity result in membrane dysfunction, allowing a large
amount of Ca2+ to flood into cells and the direct attack
of cell structural proteins and nucleic acids by ROS. Consistent
with this, TBHP significantly increased intracellular ROS levels in
H9c2 cells in the present study, whereas TBHP-induced ROS
production was reduced by PLB pretreatment.
Apoptosis is a main pathogenic mechanism of I/R
injury (23,24). Oxidative stress leads to changes in
the metabolic and functional properties of mitochondria in ischemic
myocardium, thus activating the mitochondrial apoptotic pathway.
ROS cause oxidative damage to membrane proteins and lipids, leading
to mitochondrial dysfunction and cytochrome c release and
ultimately activating caspases, particularly caspase-3, that induce
apoptosis (25). In the present
study, it was determined that PLB reduced the TBHP-induced
apoptosis of H9c2 cells by reducing lysed caspase-3, indicating
that PLB prevents apoptosis by inhibiting the intrinsic apoptotic
pathway mediated by mitochondria.
Autophagy is a ubiquitous protein degradation
process that removes abnormal proteins and organelles to promote
energy recycling (26). The
specific process is divided into induction of macroautophagy,
formation of autophagosomes, autophagosome docking and fusion, and
autophagic body breakdown (27).
Autophagy occurs at a basic level to allow sustained metabolic
recycling of intracellular components in most tissues. Under
pathological conditions, autophagy can act as a cytoprotective
mechanism to degrade and recycle defective cytoplasmic proteins
(28). Autophagy inhibition in
cardiomyocytes has adverse effects (29). Additionally, the dual effect of
autophagy in CVD has been investigated. An increasing number of
investigations have confirmed that autophagy stimulates the
inflammatory response and is responsible for ceroid formation in
atherosclerosis (30-33).
LC3 plays a key role in autophagosome formation during autophagy.
Activated LC3-I conjugates with the target lipid
phosphatidylethanolamine (PE) on the outer membrane, forming
LC3-II. LC3-II is cleaved from LC3-I and released back to the
cytosol or degraded upon autophagosome maturation (34). Thus, expression of LC3-II/LC3-I has
been regarded as a classic autophagy marker. In the present study,
PLB induced autophagy in H9c2 cells, as evidenced by the increased
LC3-II/LC3-I ratio determined by western blot analysis. Crosstalk
between apoptosis (type 1 cell death) and autophagy (type 2 cell
death) occurs in myocardial injury. When autophagy is promoted,
cell death via apoptosis is inhibited (35). Likewise, results of the present
study demonstrated that TBHP enhanced apoptosis in H9c2 cells, and
PLB abolished this apoptosis when autophagy was induced.
MAPK activation constitutes a pattern of
intracellular signaling and participates in myocardial I/R injury
(36). The p38 MAPK pathway
regulates the cellular response to growth, apoptosis, and stress
signals in different cell models. Phosphorylation of threonine and
tyrosine residues in p38 leads to conformational changes, thereby
increasing accessibility of the p38 active site and enhancing
catalysis (37). Some researchers
have suggested that autophagy may be induced by activating p38 MAPK
and that upregulating autophagy via the p38 MAPK pathway may
protect H9c2 cells from oxidative stress (38,39).
In addition, the downstream effects of p38 MAPK pathway activation
depends on the assembly of Nox subunits into the NAPDH oxidase
complex responsible for ROS production (40). Considering that ROS leads to p38
MAPK phosphorylation and cytotoxicity, the effect of PLB treatment
on p38 MAPK phosphorylation was evaluated in the present study.
Compared with the use of TBHP alone, PLB treatment significantly
reduced TBHP-induced phosphorylation of p38 MAPK in H9c2 cells.
Scavenging ROS fails to effectively prevent CVD
progression (41). Large-scale
multicenter clinical studies (HOPE, SECURE, GISSI and HPS)
(42-44)
have not confirmed the therapeutic value of vitamin E (a free
radical scavenger) in slowing atherosclerosis or in reducing major
cardiovascular events (45). This
may be due to the fact that simply administering antioxidant
vitamins fails to eliminate the root cause of large amounts of
ROS-oxidase, especially NOX (46).
The NOX family is a tissue-specific homolog of NADPH oxidase, which
exists on the plasma membrane of various nonphagocytic cells. Among
the NOX family members, NOX1 and NOX5 are mainly expressed in
smooth muscle and endothelial cells, respectively. NOX2 and NOX4
are found in endothelial cells, adventitia fibroblasts, and
vascular smooth muscle cells, and NOX4 is the main source of
intracellular ROS (47). The
expression and activity of NOX4 are increased in myocardial I/R,
leading to increased ROS and representing an important mechanism of
myocardial injury (48). The
results of the present study demonstrated that PLB suppressed NOX4
expression induced by TBHP in H9c2 cells, indicating that PLB may
help reduce ROS production. Even though H9c2 cardiomyocytes have
been widely used to investigate the molecular mechanisms underlying
the cellular response to oxidative stress, it may not be fully
consistent with other models. Thus, further studies based on in
vivo models are still ongoing.
In summary, the present study suggested that PLB
alleviated TBHP-induced cytotoxicity by reducing ROS-induced
apoptosis and modulating autophagy in cardiomyocytes.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Science and
Technology Project of the Changjiang River Administration of
Navigational Affairs (grant no. 201810010) and Science Foundations
of Hubei Provincial Health Commission (grant no. WJ2021M040).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QZ and TW designed the study and wrote the
manuscript. HF, WG and FC performed some of the experiments and
collected the main data. FH conducted the statistical analysis. TW
conceived the study. QZ and TW confirm the authenticity of all the
raw data. All authors have read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Syama HP, Arya AD, Dhanya R, Nisha P,
Sundaresan A, Jacob E and Jayamurthy P: Quantification of phenolics
in Syzygium cumini seed and their modulatory role on tertiary
butyl-hydrogen peroxide-induced oxidative stress in H9c2 cell lines
and key enzymes in cardioprotection. J Food Sci Technol.
54:2115–2125. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Arslan F, Bongartz L, Ten Berg JM, Jukema
JW, Appelman Y, Liem AH, de Winter RJ, van 't Hof AWJ and Damman P:
2017 ESC guidelines for the management of acute myocardial
infarction in patients presenting with ST-segment elevation:
Comments from the Dutch ACS working group. Neth Heart J.
26:417–421. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/Reperfusion injury following acute
myocardial infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017(7018393)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Siu KL, Lotz C, Ping P and Cai H: Netrin-1
abrogates ischemia/reperfusion-induced cardiac mitochondrial
dysfunction via nitric oxide-dependent attenuation of NOX4
activation and recoupling of NOS. J Mol Cell Cardiol. 78:174–185.
2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhou QL, Teng F, Zhang YS, Sun Q, Cao YX
and Meng GW: FPR1 gene silencing suppresses cardiomyocyte apoptosis
and ventricular remodeling in rats with ischemia/reperfusion injury
through the inhibition of MAPK signaling pathway. Exp Cell Res.
370:506–518. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yong R, Chen XM, Shen S, Vijayaraj S, Ma
Q, Pollock CA and Saad S: Plumbagin ameliorates diabetic
nephropathy via interruption of pathways that include NOX4
signalling. PLoS One. 8(e73428)2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang Q, Liao X and Wu F: The
naphthoquinone plumbagin suppresses ADP-induced rat platelet
aggregation through P2Y1-PLC signaling pathway. Pak J Pharm Sci. 30
(2(Suppl.)):S573–S578. 2017.PubMed/NCBI
|
|
9
|
Guida M, Maraldi T, Resca E, Beretti F,
Zavatti M, Bertoni L, La Sala GB and De Pol A: Inhibition of
nuclear Nox4 activity by plumbagin: Effect on proliferative
capacity in human amniotic stem cells. Oxid Med Cell Longev.
2013(680816)2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang SX, Wang J, Shao JB, Tang WN and
Zhong JQ: Plumbagin mediates cardioprotection against myocardial
ischemia/reperfusion injury through Nrf-2 signaling. Med Sci Monit.
22:1250–1257. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Luo C, Li Y, Wang H, Feng Z, Li Y, Long J
and Liu J: Mitochondrial accumulation under oxidative stress is due
to defects in autophagy. J Cell Biochem. 114:212–219.
2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zorov DB, Juhaszova M, Yaniv Y, Nuss HB,
Wang S and Sollott SJ: Regulation and pharmacology of the
mitochondrial permeability transition pore. Cardiovasc Res.
83:213–225. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kurian GA, Rajagopal R, Vedantham S and
Rajesh M: The role of oxidative stress in myocardial ischemia and
reperfusion injury and remodeling: Revisited. Oxid Med Cell Longev.
2016(1656450)2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li Z, Chinnathambi A, Ali Alharbi S and
Yin F: Plumbagin protects the myocardial damage by modulating the
cardiac biomarkers, antioxidants, and apoptosis signaling in the
doxorubicin-induced cardiotoxicity in rats. Environ Toxicol.
35:1374–1385. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ding Y, Chen ZJ, Liu S, Che D, Vetter M
and Chang CH: Inhibition of Nox-4 activity by plumbagin, a
plant-derived bioactive naphthoquinone. J Pharm Pharmacol.
57:111–116. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang Q, Zhao S, Zheng W, Fu H, Wu T and
Hu F: Plumbagin attenuated oxygen-glucose
deprivation/reoxygenation-induced injury in human SH-SY5Y cells by
inhibiting NOX4-derived ROS-activated NLRP3 inflammasome. Biosci
Biotechnol Biochem. 84:134–142. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chang G, Zhang D, Yu H, Zhang P, Wang Y,
Zheng A and Qin S: Cardioprotective effects of exenatide against
oxidative stress-induced injury. Int J Mol Med. 32:1011–1020.
2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
T MM, Anand T and Khanum F: Attenuation of
cytotoxicity induced by tBHP in H9C2 cells by Bacopa monniera and
Bacoside A. Pathophysiology. 25:143–149. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Callegari GA, Novaes JS, Neto GR, Dias I,
Garrido ND and Dani C: Creatine kinase and lactate dehydrogenase
responses after different resistance and aerobic exercise
protocols. J Hum Kinet. 58:65–72. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Inafuku H, Kuniyoshi Y, Yamashiro S,
Arakaki K, Nagano T, Morishima Y and Kise Y: Determination of
oxidative stress and cardiac dysfunction after ischemia/reperfusion
injury in isolated rat hearts. Ann Thorac Cardiovasc Surg.
19:186–194. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ruiz-Ginés JA, López-Ongil S,
González-Rubio M, González-Santiago L, Rodríguez-Puyol M and
Rodríguez-Puyol D: Reactive oxygen species induce proliferation of
bovine aortic endothelial cells. J Cardiovasc Pharmacol.
35:109–113. 2000.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jeremias I, Kupatt C, Martin-Villalba A,
Habazettl H, Schenkel J, Boekstegers P and Debatin KM: Involvement
of CD95/Apo1/Fas in cell death after myocardial ischemia.
Circulation. 102:915–920. 2000.PubMed/NCBI View Article : Google Scholar
|
|
24
|
McClintock DS, Santore MT, Lee VY,
Brunelle J, Budinger GR, Zong WX, Thompson CB, Hay N and Chandel
NS: Bcl-2 family members and functional electron transport chain
regulate oxygen deprivation-induced cell death. Mol Cell Biol.
22:94–104. 2002.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bi YM, Wu YT, Chen L, Tan ZB, Fan HJ, Xie
LP, Zhang WT, Chen HM, Li J, Liu B and Zhou YC:
3,5-Dicaffeoylquinic acid protects H9C2 cells against oxidative
stress-induced apoptosis via activation of the PI3K/Akt signaling
pathway. Food Nutr Res. 62(1423)2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and Autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim I, Rodriguez-Enriquez S and Lemasters
JJ: Selective degradation of mitochondria by mitophagy. Arch
Biochem Biophys. 462:245–253. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Gottlieb RA and Mentzer RM: Autophagy
during cardiac stress: Joys and frustrations of autophagy. Annu Rev
Physiol. 72:45–59. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Peng S, Xu LW, Che XY, Xiao QQ, Pu J, Shao
Q and He B: Atorvastatin inhibits inflammatory response, attenuates
lipid deposition, and improves the stability of vulnerable
atherosclerotic plaques by modulating autophagy. Front Pharmacol.
9(438)2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Guo FX, Wu Q, Li P, Zheng L, Ye S, Dai XY,
Kang CM, Lu JB, Xu BM, Xu YJ, et al: The role of the
LncRNA-FA2H-2-MLKL pathway in atherosclerosis by regulation of
autophagy flux and inflammation through mTOR-dependent signaling.
Cell Death Differ. 26:1670–1687. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hassanpour M, Rahbarghazi R, Nouri M,
Aghamohammadzadeh N, Safaei N and Ahmadi M: Role of autophagy in
atherosclerosis: Foe or friend? J Inflamm (Lond).
16(8)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang H, Ge S, Ni B, He K, Zhu P, Wu X and
Shao Y: Augmenting ATG14 alleviates atherosclerosis and inhibits
inflammation via promotion of autophagosome-lysosome fusion in
macrophages. Autophagy. 17:4218–4230. 2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nishida K, Yamaguchi O and Otsu K:
Crosstalk between autophagy and apoptosis in heart disease. Circ
Res. 103:343–351. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tao H, Nuo M and Min S: Sufentanil
protects the rat myocardium against ischemia-reperfusion injury via
activation of the ERK1/2 pathway. Cytotechnology. 70:169–176.
2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Treusch S, Albert FW, Bloom JS, Kotenko IE
and Kruglyak L: Genetic mapping of MAPK-mediated complex traits
Across S. cerevisiae. PLoS Genet. 11(e1004913)2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zheng YH, Tian C, Meng Y, Qin YW, Du YH,
Du J and Li HH: Osteopontin stimulates autophagy via integrin/CD44
and p38 MAPK signaling pathways in vascular smooth muscle cells. J
Cell Physiol. 227:127–135. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lv XC and Zhou HY: Resveratrol protects
H9c2 embryonic rat heart derived cells from oxidative stress by
inducing autophagy: Role of p38 mitogen-activated protein kinase.
Can J Physiol Pharmacol. 90:655–662. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Tormos AM, Taléns-Visconti R, Nebreda AR
and Sastre J: p38 MAPK: A dual role in hepatocyte proliferation
through reactive oxygen species. Free Radic Res. 47:905–916.
2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Harrison DG, Gongora MC, Guzik TJ and
Widder J: Oxidative stress and hypertension. J Am Soc Hypertens.
1:30–44. 2007.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sleight P: The HOPE Study (Heart Outcomes
Prevention Evaluation). J Renin Angiotensin Aldosterone Syst.
1:18–20. 2000.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Stone NJ: The Gruppo Italiano per lo
Studio della Sopravvivenza nell'Infarto Miocardio
(GISSI)-Prevenzione Trial on fish oil and vitamin E supplementation
in myocardial infarction survivors. Curr Cardiol Rep. 2:445–451.
2000.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Collins R, Peto R and Armitage J: The
MRC/BHF heart protection study: Preliminary results. Int J Clin
Pract. 56:53–56. 2002.PubMed/NCBI
|
|
45
|
Violi F, Nocella C, Loffredo L, Carnevale
R and Pignatelli P: Interventional study with vitamin E in
cardiovascular disease and meta-analysis. Free Radic Biol Med.
178:26–41. 2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Schramm A, Matusik P, Osmenda G and Guzik
TJ: Targeting NADPH oxidases in vascular pharmacology. Vascul
Pharmacol. 56:216–231. 2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Takac I, Schröder K and Brandes RP: The
Nox family of NADPH oxidases: Friend or foe of the vascular system?
Curr Hypertens Rep. 14:70–78. 2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018.PubMed/NCBI View Article : Google Scholar
|