Introduction
Rheumatoid arthritis (RA) is a chronic, autoimmune,
inflammatory disease. The pathological hallmarks of synovium in RA
are synovial hyperplasia, pannus formation, cartilage damage and
bone erosion (1). Abnormal
expansion of fibroblast-like synoviocytes (FLS) leads to synovial
hyperplasia, which is the leading pathophysiology of RA. Making up
two thirds of the hyperplastic synovial membrane, FLS secrete
several adhesion molecules and inflammatory cytokines, and this
also leads to bone erosion and cartilage damage. Migration of FLS
to unaffected cartilage drives the progression of polyarthritis in
RA (2). It has been reported that
there is a large amount of prostaglandin E2 (PGE2) in
the synovial fluid of RA patients that triggers anomalous
proliferation and migration of FLS (3).
Guanine nucleotide binding protein, also known as
the G protein, is a ubiquitous signal molecule that plays an
important role in transmembrane signal transduction. G protein
couples with the corresponding G protein-coupled receptor (GPCR).
The binding of ligand to GPCR induces the dissociation of G protein
into Gα and Gβγ dimers. Gβγ subunit plays multifunctional roles,
such as anchoring the G protein to the cell membrane and conducting
signal transduction. GPCRs and their effector molecules bind G
protein subunits before targeting to the cell surface. Interacting
with various effectors, including G protein-coupled receptor kinase
2 (GRK2), adenylyl cyclase II, and PI-3 kinase (4), Gβγ is important in recruiting GRKs to
activated receptors and regulating cellular functions.
GRK2 is associated with GPCR desensitization.
PGE2 binds to prostaglandin receptors (EPs). In the
physiologic state, EP4 primarily couples to the Gαs subunit to
mediate increases in the cAMP concentrations. In a pathological
environment, EP4 perceives continuous stimulation from abnormal
levels of inflammatory cytokines such as PGE2, causing
an overactive inflammatory response. Previous studies have shown
that in RA animal models, the PGE2-EP4-GRK2 signaling
pathway plays an important role in the occurrence and development
of arthritis. PGE2 binds to the EP4 receptor, inducing
excessive GRK2 translocation to the cell membrane and EP4
desensitization, thereby promoting downregulation of cAMP, FLS
dysfunction and synovium hyperplasia with the mechanism of
decreased protective Gαs signaling (5,6).
Paeoniflorin-6'O-benzene sulfonate (CP-25; patent
number in China: ZL201210030616.4) is a structurally modified
compound from paeoniflorin with anti-inflammatory and
immunomodulatory effects in RA animal models and other immune
diseases (7-10).
It was found that CP-25 ameliorated FLS abnormal proliferation and
migration by inhibiting GRK2 translocation (11) and re-sensitizing EP4(6), which was an important mechanism of
CP-25 in improving FLS dysfunction.
The GPCR-Gβγ-GRK complex is crucial for the
regulation of receptor function and GRK catalysis, and this has
been studied in adrenergic and opioid receptors, but has not been
reported in EP4 receptors. GRK2 binds both delta opioid receptor
and Gβγ to form a receptor-GRK2-Gβγ complex, which is required for
GRK-mediated receptor phosphorylation and desensitization (12). In heart failure animal model, GRK2
translocates to β-adrenergic receptor after binding with Gβγ to
phosphorylate the agonist-occupied receptor, thus cardiac
contractile function collapses as β-adrenergic receptor signaling
is attenuated (13). It remains
unclear whether Gβγ is involved in PGE2-EP4-GRK2
signaling. Hence, in the present study, the role of Gβγ and the
GRK2-Gβγ interaction in regulating FLS proliferation and migration
was mainly explored. In addition, the specific mechanism of CP-25
in restoring dysfunction of EP4 was investigated.
Materials and methods
Sample collection
RA synovial tissue samples were obtained from six
patients (4 females, 2 males; age range, 52-73 years) with RA
undergoing surgical synovectomy and knee replacement. Trauma
synovium samples were obtained from six patients (3 females, 3
males; age range, 28-50 years) with traumatic synovitis undergoing
arthroscopic synovectomy. The sample collection period was from
December 2017 to December 2018. Patients had been diagnosed with RA
at least three years prior to surgery and were treated using the
standard of care (non-steroidal anti-inflammatory drugs and
disease-modifying anti-rheumatic drugs DMARDs). Informed consent
was obtained from all patients. The study protocol was approved
(approval no. 20131321) by the Biomedical Ethics Committee of Anhui
Medical University (Hefei, China).
Animals
A total of 12 male Sprague Dawley rats (body weight,
180-200 g) were obtained from SPF Animal Laboratory of Anhui
Medical University (Hefei, China). All rats were housed in
pathogen-free animal houses at 20-26˚C under a 12-h light/dark
cycle and had free access to sterile water and food. All
experiments were conducted in accordance with the principles of the
laboratory animal care guidelines and were approved (approval no.
20131321) by Biomedical Ethics Committee of Anhui Medical
University (Hefei, China).
Drugs and reagents
PGE2 was purchased from Cayman Chemical
Company. Gallein was purchased from Santa Cruz Biotechnology, Inc.
mSIRK was purchased from Sigma-Aldrich; Merck KGaA. GRK2i was
purchased from Tocris Bioscience. The Cell Counting Kit-8 (CCK-8)
kit was purchased from Beyotime Institute of Biotechnology. The
Transwell inserts (8-µm) were purchased from Corning, Inc. CP-25
(purity >98%) was provided by the Institute of Clinical
Pharmacology, Anhui Medical University (Hefei, China). Gβ1 siRNA(h)
and the control siRNA(h) were purchased from Santa Cruz
Biotechnology, Inc. Lipofectamine® 2000 and fetal bovine
serum (FBS) were purchased from Invitrogen; Thermo Fisher
Scientific, Inc. Dulbecco's Modified Eagle's Medium (DMEM) was
purchased from Hyclone; Cytiva.
Induction of adjuvant-induced
arthritis (AA)
There were six rats in model group, and the other
six rats were in control group. The method of AA rat preparation
was consistent with that described in a previous study (5). A total of 0.1 ml of complete Freund's
adjuvant (CFA) emulsion was injected into the right hind metatarsal
footpad. The rats were assessed every three days for the signs of
arthritis by two independent observers. If tissue necrosis or
ulcerated masses were present (no signs of healing within 3 days)
and signs of deterioration such as inability to reach food or water
for ~12 h, weight loss >20% of starting weight or any sign of
suffering not relieved by pharmacologic analgesia, the rat would be
euthanized. The rats in model group were sacrificed for the AA-FLS
at day 30 after immunization under anesthesia using isoflurane
(induction 3-4%; maintenance 1-2%). Subsequently, all the rats were
euthanized via the inhalation of carbon dioxide. The displacement
of the euthanasia chamber was between 30-70% of the chamber volume
per min. Death would be confirmed by an appropriate method, such as
ascertaining cardiac and respiratory arrest or noting fixed and
dilated pupils of an animal.
Cell culture and identification
The RA-FLS and AA-FLS were obtained from the knee
synovium of the RA patients and AA rats. Human rheumatoid FLS
(MH7A) were purchased from Jennio Biotech Co., Ltd. All cells were
cultured in DMEM containing 20% FBS, 1% penicillin and 1%
streptomycin at 37˚C and 5%-CO2 containing incubator.
The RA-FLS and AA-FLS were cultured using Anti-Vimentin (cat. no.
sc-373717; Santa Cruz Biotechnology, Inc.) and Anti-CD55 (cat. no.
26580-1-AP; Proteintech) at a dilution ratio of 1:1,000 for 30 min
and then detected using immunofluorescence. Mycoplasma testing was
carried out for the cell lines used.
Histological analysis
Synovial tissue was fixed with 4% paraformaldehyde
solution at room temperature for 24 h and embedded in paraffin. The
paraffin sections (4 µm) were dewaxed in xylene, stained with
hematoxylin and eosin (H&E) at room temperature for 3 min and
30 sec, respectively, and were examined using standard light
microscopy.
Immunofluorescence assay
The synovial tissue samples were snap-frozen in OCT
medium. Cryostat sections (5 µm) were cut and mounted on adhesive
glass slides. Slides of the tissue samples and cells were fixed
with 4% paraformaldehyde at 4˚C for 10 min, permeabilized with 0.5%
Triton X-100 for 7 min at room temperature and blocked with 5%
bovine serum albumin (MilliporeSigma) for 1 h at 37˚C. After
incubating with primary antibodies (1:100 dilution) at 4˚C
overnight, the slides were incubated with anti-mouse-Alexa Fluor
594 (1:200 dilution; cat. no. ab150116; Abcam) and
anti-rabbit-Alexa Fluor 488 (1:200 dilution; cat. no. ab150073;
Abcam) for 1 h at 37˚C. DAPI (20 µl) was used to stain the nuclei
for 5 min at room temperature. The slides were then observed under
a fluorescence microscope (BX53; Olympus Corporation). The primary
antibodies were as follows: Gβ (cat. no. sc-166123), EP4 (1:1,000
dilution; cat. no. sc-55596; both from Santa Cruz Biotechnology,
Inc.) and GRK2 (cat. no. ab228705; Abcam).
Isolation of membranes from synovial
tissue
The synovial tissue was washed using PBS, cut into
pieces and added with a cold homogenization buffer (0.32 M sucrose,
5 mM Tris-HCl, pH 7.5, 120 mM KCl, 1 mM EDTA and 0.2 mM PMSF).
Samples were sonicated and centrifuged at 100,000 x g for 1 h at
4˚C. The supernatants were pooled and centrifuged at 100,000 x g
for 1 h. The pellets were resuspended in a cold extraction buffer
(20 mM HEPES, pH 7.5, 10% glycerol, 2% Triton X-100, 1 mM EDTA, 1
mM EGTA and 0.2 mM PMSF) and incubated at 4˚C overnight. The
supernatants containing the cell membrane were collected after
centrifuging at 7,800 x g for 30 min at 4˚C.
Isolation of proteins from the cell
cytoplasm and the membrane
RA-FLS, AA-FLS and MH7A cells were lysed with lysis
buffer (cat. no. P0013; Beyotime Institute of Biotechnology) and
centrifuged at 12,000 x g for 10 min at 4˚C. The supernatants were
centrifuged at 100,000 x g for 1 h at 4˚C. The precipitate was kept
as the membrane fraction, while the supernatant was collected as
the cytoplasm fraction.
Cell proliferation assay
Cell proliferation was examined using a CCK-8 assay.
RA-FLS, AA-FLA and MH7A cells were counted and seeded on to 96-well
plates at a concentration of 5x106/ml and incubated in
DMEM at 37˚C for 48 h. The cells were then treated with
PGE2 (2 µm) with or without different agonists or
antagonists and CP-25 (10-7, 10-6 and
10-5 mol/l) for 24 h. After treatment, the cells were
added to a 10-µl CCK-8 solution for each well. The plate was
incubated for 1 h at 37˚C. The optical density values of the plate
were measured at 490 nm on a Tecan Infinite M200 Microplate Reader
(Tecan Group, Ltd).
Cell migration assay
Cells were seeded into the upper wells of the
Transwell chambers at a concentration of 1x105/ml and
maintained in a medium containing 5% FBS. The lower chamber was
filled with DMEM containing 20% FBS. After 24 h, the upper chamber
was washed with PBS and fixed with 4% paraformaldehyde for 20 min
at room temperature and stained with 0.1% crystal violet for 15 min
at room temperature. All images were captured using a light
microscope. Cells were counted from at least four random
microscopic fields for each sample in three independent
experiments.
Small interfering RNA (siRNA)
The Gβ1 siRNA (cat. no. sc-41762) and negative
control siRNA (cat. no. sc-37007) were used to knockdown Gβ1 in
MH7A cells. The MH7A were transfected with 50 nM siRNA at 70%
confluency in 24-well plates using Lipofectamine 2000 at room
temperature according to the manufacturer's protocol. After 48 h,
silencing of Gβ1 was confirmed using western blot analysis.
Western blot analysis
Protein samples were collected and boiled in a
loading buffer for 10 min at 100˚C. The protein concentration was
detected using a BCA protein assay kit (Thermo Fisher Scientific,
Inc.). Equal amounts of protein (20 µg) were electrophoretically
separated using 10% SDS-PAGE and transferred to a PVDF membrane
(MilliporeSigma). The PVDF membrane was then incubated in blocking
solution (cat. no. P0205; Beyotime Institute of Biotechnology) at
room temperature for 1 h. Subsequently, the membrane was incubated
with primary antibodies at 4˚C overnight, followed by the
horseradish peroxidase-conjugated goat anti-mouse antibody (cat.
no. STAR207P;) or goat anti-rabbit antibody (cat. no. STAR208P;
both 1:10,000 dilution; Bio-Rad Laboratories, Inc.) at room
temperature for 2 h. The bands were visualized using
chemiluminescence detection reagents (MilliporeSigma) according to
the manufacturer's protocol. The following primary antibodies were
used: Gβ (1:1,000; cat. no. sc-166123), Gβ1 (1:1,000; cat. no.
sc-515764; both from Santa Cruz Biotechnology, Inc.), ATP1A1
(1:3,000; cat. no. 14418-1-AP; ProteinTech Group, Inc.), GRK2
(1:1,000; cat. no. sc-13143; Santa Cruz Biotechnology, Inc.), GRK2
(1:1,000; cat. no. ab228705; Abcam), EP4 (1:1,000; cat. no.
sc-55596), β-arrestin2 (1:1,000; cat. no. sc-13140; both from Santa
Cruz Biotechnology, Inc.), β-actin (1:5,000; cat. no. 60008-1-Ig)
and GAPDH (1:5,000; cat. no. 60004-1-Ig; both from ProteinTech
Group, Inc.). The protein bands were visualized using an enhanced
chemiluminescence detection system and quantified using ImageJ
software (version 1.46; National Institutes of Health).
Coimmunoprecipitation (Co-IP)
assay
To examine the association between GRK2 and Gβ in
the synovial tissue or in FLS, the tissue or cell lysate was
immunoprecipitated using an anti-GRK2, anti-EP4 or anti-Gβ antibody
and analyzed using western blot analysis. Briefly, the sample was
precleared for 2 h at 4˚C with 20 µl of protein A + G agarose beads
and subsequently incubated with 4 µl primary antibody for 2 h at
4˚C. Then the precleared lysates were associated with 20 µl of
protein A + G agarose beads and the primary antibody and incubated
overnight at 4˚C on the rotation shaker. Tubes were then
centrifuged at 14,000 x g for 5 sec at 4˚C. The pellets were washed
with lysis buffer for 10 min. The resin-bound immune complexes were
boiled and the following steps were according to the western blot
protocol.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from synovial tissue using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.), which
was then used to synthesize cDNA by RT using the PrimeScript™ RT
Master Mix Kit (TaKaRa Bio, Inc.) in a reaction volume of 20 µl
according to the manufacturer's protocol. Real-time qPCR assays
were performed with an Applied Biosystems 7500 fast real-time PCR
system with TB Green Premix Ex Taq II (TaKaRa Bio, Inc.). The
conditions of amplification were as follows: Initial denaturation
at 95˚C for 30 sec, followed by 40 cycles of annealing and
extension at 95˚C for 3 sec and 60˚C for 30 sec. The
oligonucleotide primer sequences were as follows: GNB1 forward,
5'-TCGTCTCTGGTGCTTGTGATGC-3' and reverse,
5'-GTCGTCTGAGCCAGTGGCAAAT-3'; and GNB2 forward,
5'-TGTTGCCGCTTCCTGGATGACA-3' and GNB2 reverse,
5'-CACTGTGTCCAGCAAAACCCAC-3'; GAPDH forward,
5'-GGAGCGAGATCCCTCCAAAAT-3' and reverse,
5'-GGCTGTTGTCATACTTCTCATGG-3'. GAPDH was used as an internal
reference and the 2-ΔΔCq method was utilized to
calculate the relative expression levels (14).
Statistical analysis
Data were analyzed using the GraphPad Prism v7
statistical software (GraphPad Software, Inc.). Values are
expressed as the mean ± standard deviation. An Unpaired Student's
t-test was used to analyze the data between two independent groups.
A one-way analysis of variance followed by Bonferroni's post hoc
test was used to analyze the data from multiple groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
The GRK2-Gβγ interaction is
overactivated in the synovium tissue of patients with RA
It was previously revealed that PGE2
levels were increased in the RA synovium and AA rats. It was also
found that after PGE2 stimulation, the membrane
expression of GRK2 was increased in AA-FLS, which was related to
FLS dysfunction (5). In an attempt
to identify whether Gβγ was involved in the GRK2 translocation to
the membrane, the expression of Gβ and the interaction between GRK2
and Gβ in the synovial tissues were evaluated using western blot
analysis, Co-IP and double-stained immunofluorescence. The
pathological feature of the synovial tissue in patients with RA was
first assessed using H&E staining and expression of relevant
proteins was detected. Compared with the trauma patients, the RA
patients had synovial hyperplasia and inflammatory cell
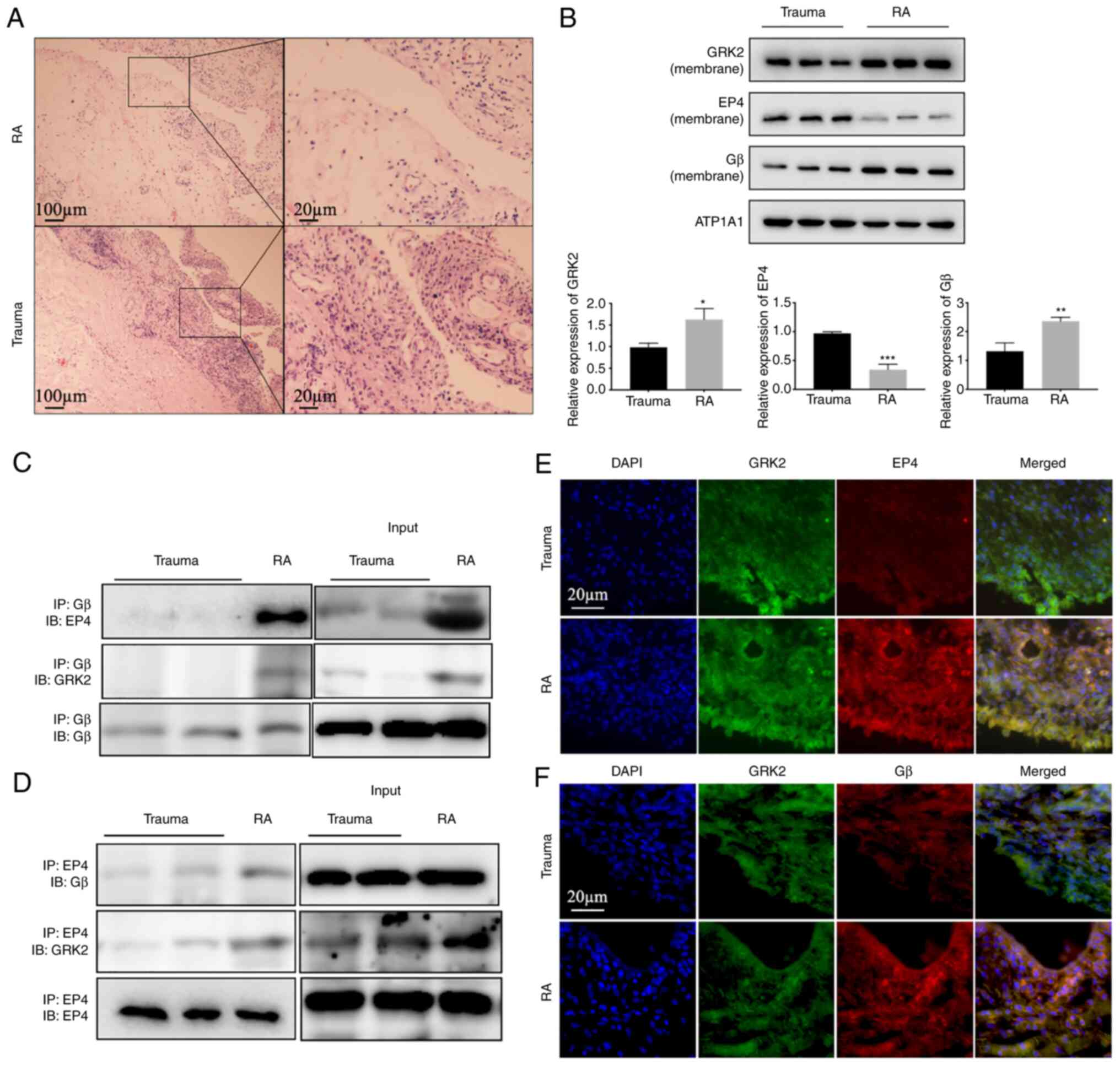
infiltration (Fig. 1A). The RA
group showed a significantly increased expression of GRK2 (Fig. S1). In the RA group, membrane
expression of GRK2 and Gβ was increased, whereas EP4 was decreased
(Fig. 1B). The aforementioned
results indicated GRK2 and Gβ translocation to the membrane and EP4
desensitization in the RA synovium. The results of the Co-IP and
immunofluorescence demonstrated that GRK2-Gβ, EP4-Gβ and GRK2-EP4
interactions were enhanced in the RA group compared with those in
the trauma group (Fig. 1C-F).
These results suggested that Gβγ was involved in GRK2 translocation
and EP4 desensitization in RA.
CP-25 inhibits proliferation and
migration of RA-FLS and AA-FLS by downregulating the GRK2-Gβγ
interaction
Since FLS is the major component of hyperplastic
pannus in the RA synovium, the primary synoviocytes from RA
patients and AA rats were isolated and identified as cells positive
for labeling with Vimentin and CD55 by immunofluorescence (Fig. S2). Before the experimental
endpoint, none of the rats succumbed. After the stimulation of
PGE2 (2 µM) for 24 h, the proliferation and migration in
both RA-FLS and AA-FLS was enhanced. CP-25 (10-6 and
10-5 mol/l) significantly inhibited proliferation and
migration induced by PGE2 in both primary cells.
Equivalent effects were observed in the Gβγ inhibitor and the
gallein (10 µM) group (Fig. 2A and
B), which indicated that
inhibition of both Gβγ and CP-25 may decrease the abnormal FLS
proliferation migration in the RA condition. To identify whether
Gβγ subunits were involved in the protection of CP-25, the membrane
expression of Gβ and its interaction with GRK2 were examined. It
was revealed that membrane expression of GRK2 and Gβ was increased,
whereas EP4 was decreased in the PGE2 group. CP-25
(10-6 mol/l) restored membrane expression of GRK2, Gβ
and EP4 (Fig. 2C). The Co-IP assay
showed enhanced EP4-Gβ, GRK2-Gβ, and GRK2-EP4 interactions in the
PGE2 group. CP-25 inhibited translocation of GRK2 and
Gβ, and reduced the binding of GRK2 and Gβ (Fig. 2D). These results suggested that
CP-25 inhibited proliferation and migration of RA-FLS and AA-FLS by
downregulating GRK2-Gβγ interaction.
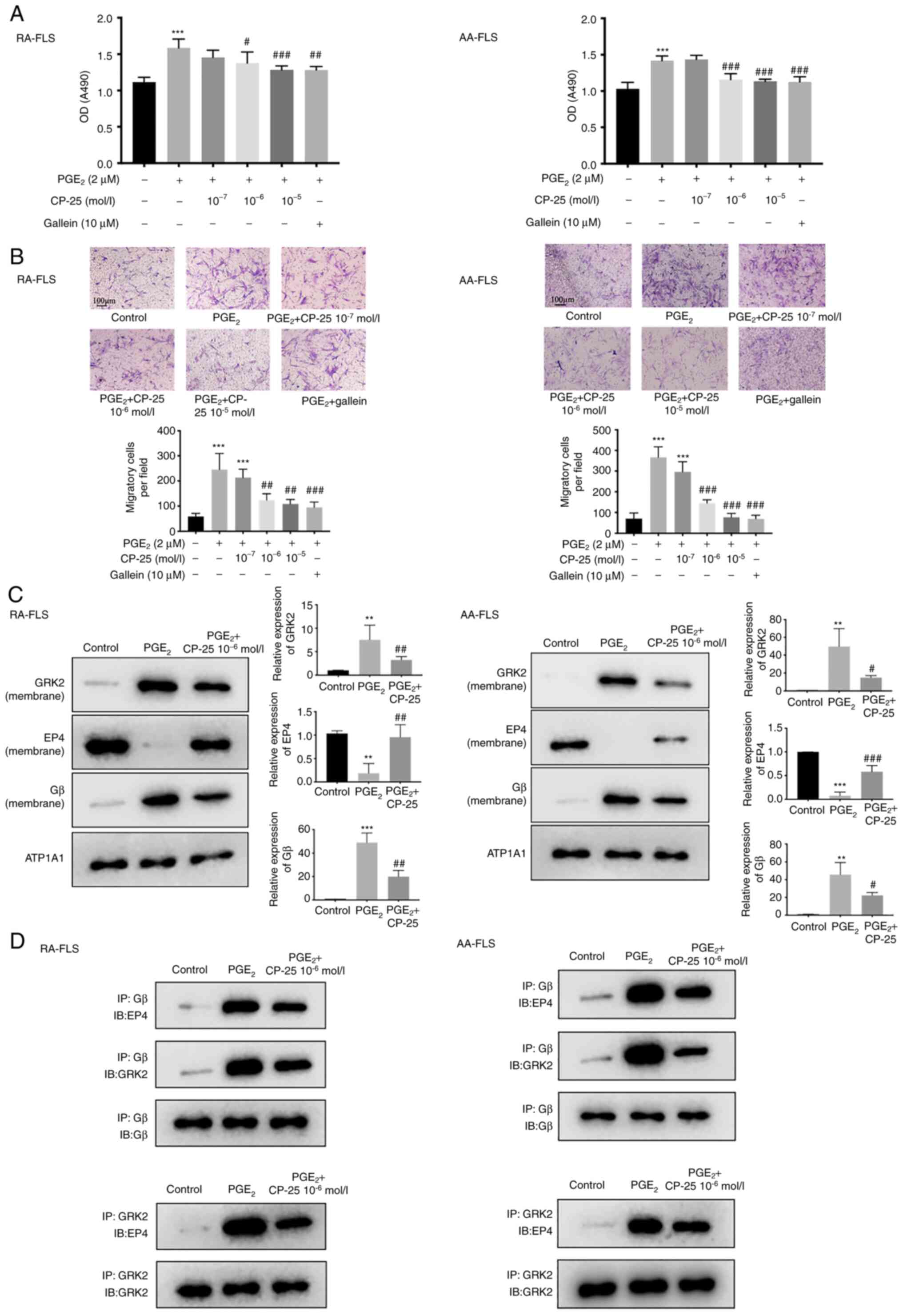 | Figure 2CP-25 inhibits proliferation and
migration of RA-FLS and AA-FLS by downregulating GRK2-Gβγ
interaction. (A and B) Inhibition of abnormal (A) proliferation and
(B) migration of RA-FLS and AA-FLS by CP-25 and gallein was
detected using Cell Counting Kit-8 and Transwell assays. (C)
Membrane expression of GRK2, EP4 and Gβ in RA-FLS and AA-FLS was
evaluated by western blotting. (D) Combination of EP4-Gβ, GRK2-Gβ,
and GRK2-EP4 in RA-FLS and AA-FLS using Co-immunoprecipitation.
Scale bars, 100 µm. **P<0.01 and
***P<0.001 vs. control group; #P<0.05,
##P<0.01 and ###P<0.001 vs.
PGE2 group (n=6). RA, rheumatoid arthritis; AA,
adjuvant-induced arthritis; FLS, fibroblast-like synoviocytes;
GRK2, G protein coupled receptor kinase 2; EP4, prostaglandin E4
receptor; PGE2, prostaglandin E2. |
CP-25 inhibits EP4-GRK2-Gβγ signaling
and promotes Gβγ-dependent EP4 re-sensitization in MH7A cells
In the next few experiments, drugs that interfered
with Gβγ in the immortal human synovial fibroblast cell line MH7A
were used to investigate whether Gβγ controlled the interaction
between GRK2 and EP4 and the role of CP-25.
First, excessive proliferation and migration induced
by PGE2 was observed in MH7A cells (Fig. 3A and B). GRK2i, gallein and CP-25 reversed the
abnormalities. The results were similar to that in the RA-FLS and
the AA-FLS. The proliferation and migration rate were further
enhanced in the mSIRK (Gβγ agonist) group compared with the
PGE2 group. PGE2 upregulated the membrane
expression of GRK2 and downregulated the membrane expression of
EP4. β-arrestin2, related to GPCR desensitization, increased in the
PGE2 group. Gβγ inhibitor, GRK2i, and gallein decreased
the membrane expression of GRK2 and β-arrestin2 and restored the
membrane expression of EP4. Membrane GRK2 was significantly
increased in the mSIRK group, whereas β-arrestin2 and EP4 remained
unchanged compared with the PGE2 group (Fig. 3C). Subsequently, it was identified
that the GRK2-EP4, EP4-Gβ, GRK2-Gβ and EP4-β-arrestin2 interactions
were enhanced in the PGE2 group, while they were
inhibited in the gallein group (Fig.
3E and G). Compared with the
PGE2 group, GRK2i decreased the combination of GRK2 and
EP4, while mSIRK enhanced the GRK2-EP4 interaction (Fig. 3D and F). CP-25 suppressed the GRK2-Gβ
interaction (Fig. 3E and G), decreased membrane expression of GRK2
and β-arrestin2 (Fig. 3C) and
blocked EP4 binding with GRK2 or β-arrestin2 (Fig. 3E), and thus restored membrane EP4
expression (Fig. 3C). These
results demonstrated that CP-25 inhibited PGE2-induced
EP4-GRK2-Gβγ signaling activation in MH7A. Exogenous stimulation of
PGE2 overactivated EP4-GRK2-Gβγ signaling and promoted
EP4 desensitization and dysfunction. CP-25 abrogated the binding of
GRK2 with Gβγ and caused Gβγ-dependent re-sensitization of EP4,
thus inhibiting abnormal proliferation and migration in the RA
condition. It is worthy to note that the total expression of Gβ
nearly remained unchanged between the control, PGE2,
CP-25 and gallein groups (Fig.
S3).
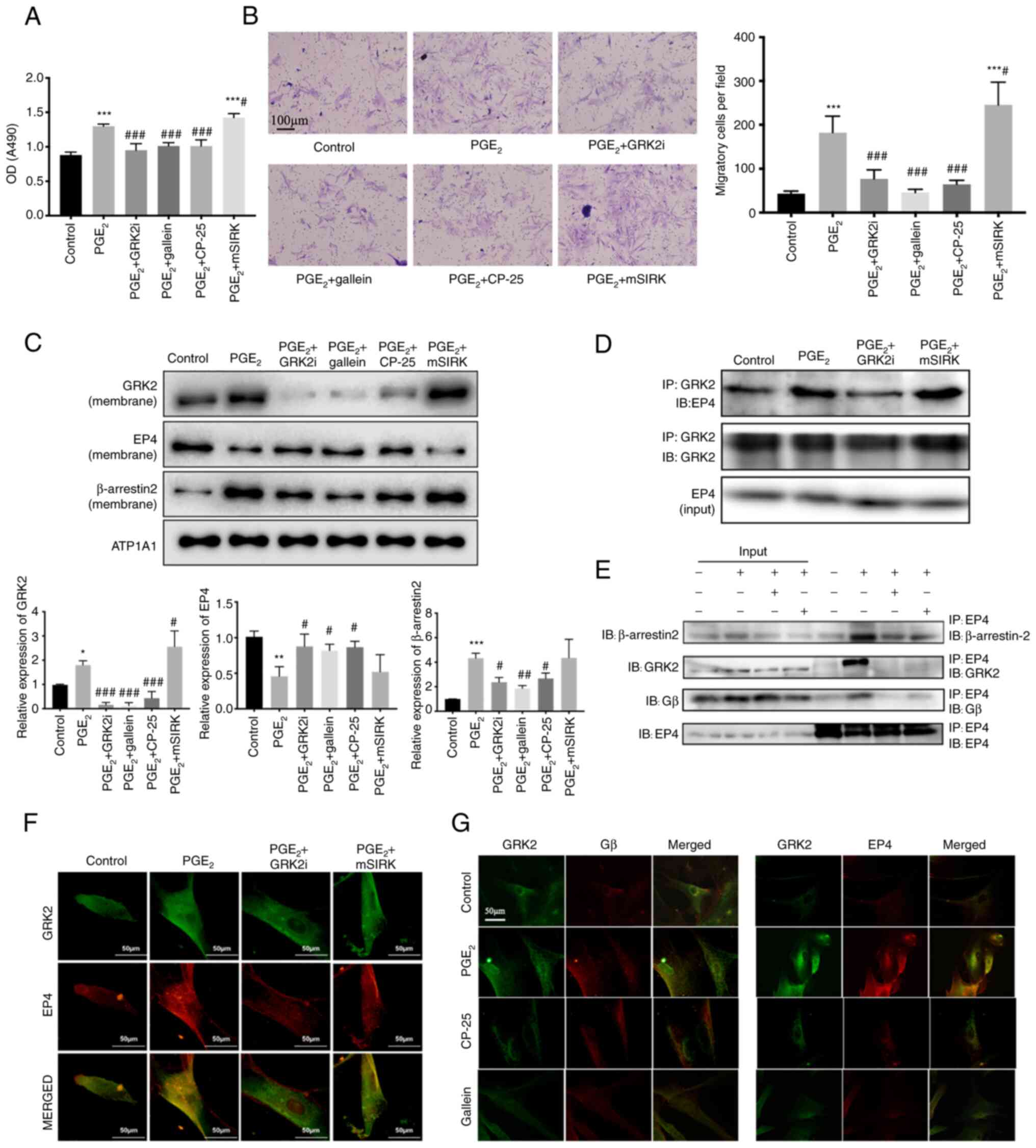 | Figure 3Gβγ on proliferation and migration of
MH7A cells and the involvement of EP4-GRK2-Gβγ signaling. MH7A
cells were treated with Gβγ antagonist (5 µM GRK2i and 10 µM
gallein), Gβγ agonist (1 µM mSIRK), CP-25 (10-6 mol/l)
and stimulated by PGE2 (2 µM). (A) Cell proliferation
was detected using Cell Counting Kit-8. (B) Cell migration was
detected using Transwell assay. Scale bars, 100 µm. (C) Membrane
expression of GKR2, EP4 and Gβ in PGE2-stimulated MH7A
cells was detected using western blotting. (D) Co-expression of
GRK2 and EP4 with GRK2i and mSIRK treatment detected by Co-IP. (E)
Co-expression of EP4-β-arrestin2, EP4-GRK2 and EP4-Gβ with CP-25
and gallein treatment was detected by Co-IP. (F) Co-localization of
GRK2 and EP4 with GRK2i and mSIRK treatment was evaluated by
immunofluorescence. Scale bars, 50 µm. (G) Co-localization of GRK2
and Gβ, EP4 and GRK2 in PGE2-stimulated MH7A cells was
evaluated by immunofluorescence. Scale bars=50 µm.
*P<0.05, **P<0.01 and
***P<0.001 vs. control group; #P<0.05,
##P<0.01 and ###P<0.001 vs.
PGE2 group (n=5). EP4, prostaglandin E4 receptor; GRK2,
G protein coupled receptor kinase 2; PGE2, prostaglandin
E2; Co-IP, Co-immunoprecipitation. |
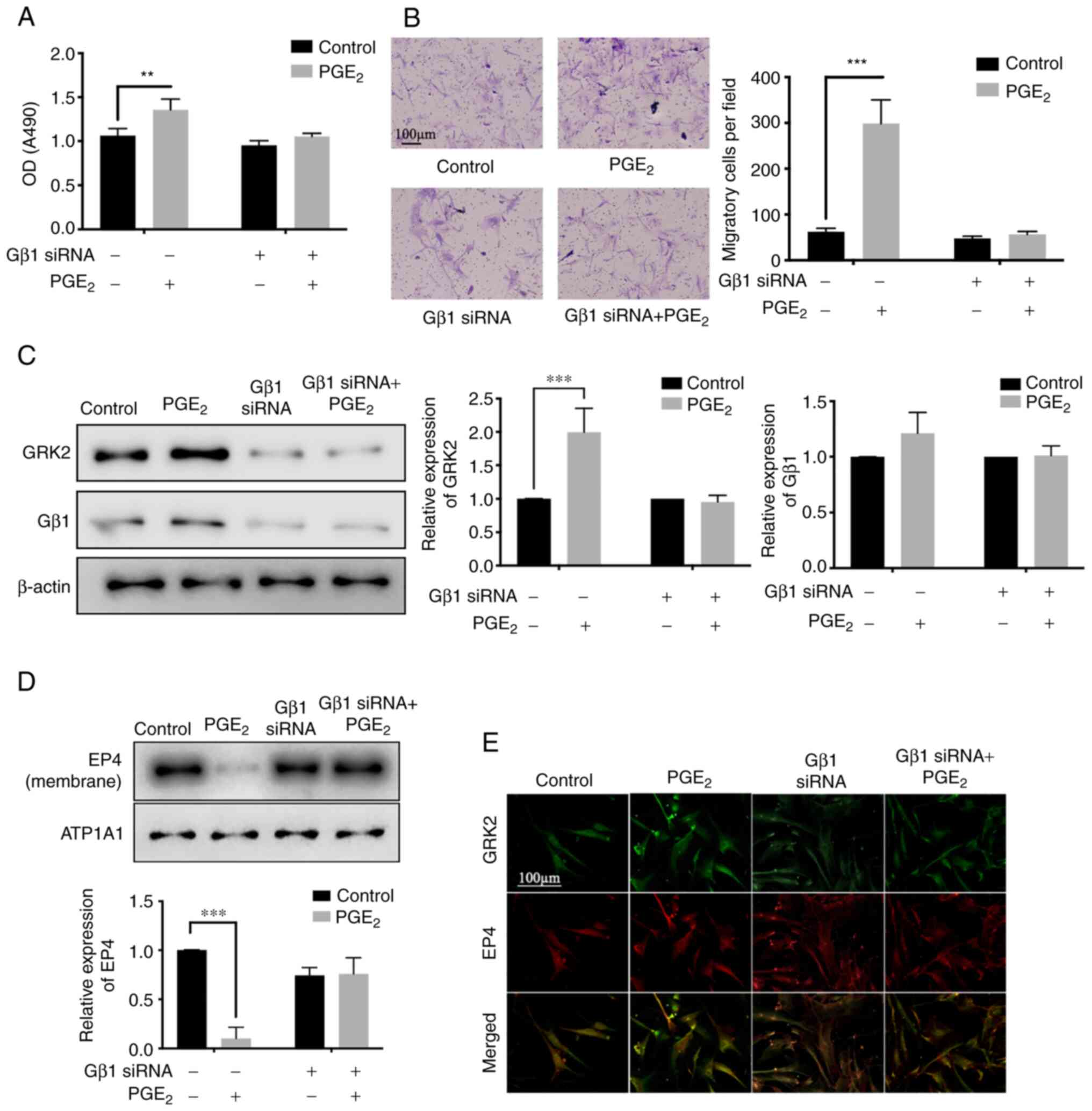
Gβγ knockdown inhibits the
PGE2-induced proliferation and migration by possibly
interfering with the GRK2-EP4 interaction
The mRNA expression of Gβ1 and Gβ2 in synovial
tissue of patients with trauma and RA was compared. It was observed
that the expression of Gβ1 mRNA was higher in the synovial tissue
of patients with RA (Fig. S4). To
further confirm the functions of Gβγ in MH7A cells, the Gβ1 siRNA
was transfected to knock down the endogenous Gβ1. It was found that
the knockdown of Gβ1 significantly decreased
PGE2-induced proliferation (Fig. 4A) and migration (Fig. 4B). In untransfected cells, there
was increased total expression of GRK2 (Fig. 4C), decreased membrane expression of
EP4 (Fig. 4D) and enhanced
GRK2-EP4 interaction after the PGE2 induction (Fig. 4E). These results suggested that Gβγ
knockdown inhibits PGE2-induced proliferation and
migration of MH7A cells, possibly by interfering with the GRK2-EP4
interaction. Collectively, Gβγ was involved in the anomalous
proliferation and migration by hindering the interaction between
GRK2 and EP4.
Discussion
FLS is a prominent component of the hyperplastic
synovium. Inflammatory environments induce FLS to migrate, and this
results in bone damage and cartilage erosion. Compared with normal
synovial tissue, there are high expression levels of GRK2 in
patients with RA and in RA animal models (6,7).
Accordingly, GRK2 upregulation and translocation to the cell
membrane play a role in the mediation of the pathological
progression of RA. In the present study, the protein expression of
EP4-GRK2-Gβγ signaling was detected using western blot analysis and
protein-protein interaction was evaluated using Co-IP and
immunofluorescence. Increased GRK2 membrane expression and enhanced
interactions of GRK2-Gβ, EP4-Gβ, and GRK2-EP4 were revealed in the
collected synovial tissue of patients with RA.
To mimic the chronic inflammatory environment of RA,
RA-FLS, AA-FLS and MH7A cells were stimulated with PGE2
for 24 h. Similar results as in the synovial tissue of patients
with RA were observed, suggesting that Gβγ was involved in synovial
hyperplasia. The enhanced GRK2-Gβγ and the downregulated expression
of EP4 in the FLS were significantly associated with synovium
hypertrophy. The aforementioned mechanisms promoted the occurrence
and development of RA. To further identify the role of Gβγ in RA,
Gβγ interference with pharmacological and genetic methods was
conducted. Gβγ agonist, mSIRK, promoted abnormalities by
PGE2. However, the membrane expression of EP4 and
β-arrestin2 were unchanged compared with the PGE2 alone
group. It was hypothesized that the desensitized EP4 reached a
saturated state. There may be other mechanisms independent of EP4
signaling that account for the extra elevated proliferation and
migration after the mSIRK treatment. For example, the PI3K/AKT
pathway was activated by Gβγ (15). Intervention abrogating GRK2-Gβγ or
a Gβγ blocker and Gβγ knockdown mitigated the GRK2 translocation
and resulted in a decreased membrane expression of EP4. This
finally alleviated the abnormal proliferation and migration of FLS
in the RA model. As revealed by the present data, the activation of
Gβγ was involved in FLS proliferation and migration.
In the present study, FLS were treated with CP-25 at
a concentration of 10-7,10-6 and
10-5 mol/l. The results showed that the 10-6
and 10-5 mol/l CP-25 significantly inhibited the
proliferation and migration after the stimulation of
PGE2, suggesting that CP-25 may provide a potential
novel strategy for RA treatment in terms of synovial hyperplasia.
In consistency with previous studies, CP-25 suppressed
proliferation (5) and migration
(15) in the AA-FLS and MH7A
cells. In addition, CP-25 inhibits the progress of AA rats by
reducing inflammation, immunity and joint injury (16). All of the previous studies by our
group proved that CP-25 may be an effective drug for RA (5-11,15,16).
In the present study, the effect of CP-25 on the interaction
between GRK2 and Gβγ was evaluated. A decreased GRK2-Gβγ
interaction and less EP4 desensitization was observed under the
CP-25 treatment. The results supported the hypotheses that the
interaction of Gβγ and GRK2 contributes to the effect of CP-25 on
relieving FLS dysfunction.
GRK2 is a family of protein kinases that regulates
the activity of GPCR by phosphorylating the intracellular domain.
The pleckstrin homology (PH) domain in the C-terminal of GRK2 is
considered to interact with free Gβγ subunits (17). Phosphorylation at Ser685
of GRK2 by PKA facilitates the GRK2-Gβγ interaction and enhances
the kinase activity of GRK2(18).
It was recently found that CP-25 downregulates phosphorylation at
Ser685 of GRK2 and directly binds to the kinase domain
of GRK2 in vitro and inhibits GRK2 activity by controlling
the key amino acid residue of Ala321 of GRK2 (7,19).
Other studies have revealed that when Ser670 in the
C-terminal domain of GRK2 was phosphorylated by MAPK, the binding
of Gβγ with GRK2 was impaired. Thus, GRK2 translocation to the
membrane and subsequent GPCR regulation are inhibited (20,21).
Manipulation of GRK2 represents a promising molecular basis for
treating RA. Previous studies by our group emphasized the
importance of inhibiting GRK2 in maintaining receptor function
(6,7). Approaches such as the genetic
deletion of Grk2 and systemic inhibition of GRK2 showed some
drawbacks or potential side effects differentiated from a
pharmacological approach such as a selective GRK2 inhibitor. For
instance, Grk2-knockout mice succumbed on day 15 of
embryonic development from cardiac hypoplasia and cardiac
dysfunction (21). GRK2 knockdown
also caused glomerular injury and renal damage when treating heart
disease (22). It is worth
exploring whether there exists a safer and efficacious influence on
the balanced receptor function and kinase activity with minimal
side effects, but does not just completely abolish the expression
of the receptor or kinase.
At present, small molecule targeting GPCR-Gβγ-GRK2
signaling is an area of increased research attention in pathologic
conditions such as heart failure and kidney dysfunction (23,24).
In this pathway, one approach is the pharmacological inhibition of
GRK2, such as paroxetine. The other strategy is to interfere with
GRK2-Gβγ or inhibit Gβγ. Preclinical evaluations have revealed a
cardioprotective effect for GRK2 genetic deletion or
pharmacological inhibition in cardiovascular diseases by binding to
and stabilizing GRK2, thus inhibiting downstream β-adrenergic
receptor (β-AR) signaling (25,26).
Furthermore, M119 and gallein hindered Gβγ binding to GRK2, leading
to decreased translocation to the membrane of GRK2 and thus
preserving the normal function of the β-adrenaline receptor so as
to mitigate heart failure progression and cardiac hypertrophy
(27). In the present study,
gallein and CP-25 suppressed the abnormal proliferation and
migration in PGE2-stimulated FLS by interfering with the
binding of Gβγ with GRK2. The present results are in a manner
analogous to those observed of the β-AR.
There are five different Gβ and 12 Gγ in humans. An
investigation of the binding preferences revealed that GRK2 binds
preferentially to Gβ1 and Gβ2(28). It was revealed that the expression
of Gβ1 mRNA was higher than Gβ2 in the synovial tissue of patients
with RA; hence, Gβ1 was silenced in MH7A cells. It has become clear
that Gβγ is a multifunctional protein complex that interacts not
only with the Gα subunit and GPCRs but also with intracellular
proteins such as PI3K, AC and GIRK (29). Targeting Gβγ signaling appears to
be a growing potential pharmacological intervention for treating
cancer cell migration (30). It
has been reported that the EP4 antagonist suppresses cancer cell
invasion and migration in prostate cancer (31). However, a balance should be
maintained in suppressing cancer cell growth and motility without
destroying the normal functions of cells. FLS displays cancerous
properties of proliferation and migration in RA synovial tissue
that resemble cancer-associated fibroblasts in tumors (32). FLS has its own physiological
function to maintain the homeostasis of joints (33). Excessive inhibition of the normal
function of FLS in rheumatoid or non-rheumatoid joints would cause
detrimental outcomes. This notion may be related to the soft
regulation of the inflammatory immune response (SRIIR) proposed by
our group. This emphasized that the normal function as well as gene
and protein expression or activity of the cell should not be
completely disabled by drugs. The most appropriate drug is
characterized by regulating and restoring abnormal activity to
achieve physiological levels with minimal adverse reactions. In the
present study, CP-25 likely is a SRIIR drug due to its ability to
re-sensitize EP4 and reduce the excessive proliferation and
migration in PGE2-stimulated FLS.
The long-term activation of GPCRs after ligand
exposure has been revealed to desensitize receptors and
downregulate downstream signals. GPCR desensitization is generally
explained by paradigms proposed in studies on the β2-AR (34,35).
Once the activated receptor is phosphorylated by GRK2/3, its
affinity for β-arrestin is enhanced. β-arrestin2 binds GPCRs that
are targeted for endocytosis and receptor internalization (35). Agonist-induced GRK2-Gβγ interaction
is a prerequisite for GRK2-mediated receptor desensitization and
downregulation. The PH domain of GRK2 competitively binds to Gβγ
and sequesters it from the Gα subunit. Evidence has demonstrated
that the Gβγ-mediated recruitment of PI3K in complex with GRK2 is
directly implicated in β-AR desensitization (36). In the present study, in response to
PGE2, GRK2 was passively pulled to the cell membrane by
the binding of Gβγ subunits, and then both simultaneously
translocated to the cell membrane. GRK2 was phosphorylated on the
membrane. β-arrestin2 was recruited to the membrane simultaneously,
inducing excessive desensitization and internalization of EP4.
Blocking Gβγ signaling, either with the small molecule Gβγ
selective inhibitor, gallein and GRK2i, or with Gβ1 siRNA or CP-25,
led to the decreased translocation of GRK2 and EP4
re-sensitization. In this regard, EP4 desensitization via the
GRK2-Gβγ interaction could be a novel therapeutic target to treat
RA. Although the present study was focused primarily on the
interaction between GRK2 and Gβγ, it was found that GRK2, Gβγ and
EP4 interacted with each other. It is a possibility that these
three proteins bind directly to form a ternary complex. Further
research is required to demonstrate the complex formed by EP4, GRK2
and Gβγ in a sequential order, as well as the association and
disassociation of it. Several issues, including the details of Gβγ
releases from Gα-GDP, the binding site of Gβγ with GRK2, and the
Gβγ downstream signal pathways, remain to be further confirmed.
The limitation of the present study is that Gβ
antibody was selected instead of Gβγ. Although distinct Gβ and Gγ
antibodies may reveal in an improved way the activity of Gβγ, in
the present study, main focus was addressed on the interaction
between Gβγ and GRK2; it is considered that one kind of Gβ
antibodies may not be optimal, but should be sufficient to draw the
conclusion of the present study. It is considered that future work
is needed to explore changes of different subunits of Gβ and
Gγ.
Collectively, it was revealed that GRK2-Gβγ
interaction was enhanced in RA synovial tissue and FLS.
PGE2-induced abnormal proliferation and migration in FLS
were associated with strengthened GRK2-Gβγ interaction and EP4
desensitization. Regulating EP4-GRK2-Gβγ signaling may be one of
the mechanisms that underlie the salutary effect of CP-25.
Additional in vivo studies are required to confirm the
therapeutic potential of the interfering GRK2-Gβγ interaction in
RA.
Supplementary Material
Total expression of GRK2, EP4 and Gβ
in synovial tissues of patients with RA detected by western blot
analysis. * * *P<0.001 vs. trauma group (n=6). GRK2,
G protein coupled receptor kinase 2; EP4, prostaglandin E4
receptor; RA, rheumatoid arthritis.
Validation of RA-FLS and AA-FLS by
immunofluorescence. Scale bars, 100 μm. RA, rheumatoid arthritis;
AA, adjuvant-induced arthritis; FLS, fibroblast-like
synoviocytes.
Expression of Gβ in MH7A cells
determined by western blot analysis (n=5). PGE2, prostaglandin
E2.
Relative expression of GNB1 mRNA and
GNB2 mRNA in synovial tissue of patients with RA was evaluated
using quantitative PCR. ***P<0.001 vs. trauma group
(n=6). RA, rheumatoid arthritis. GNB, guanine nucleotide-binding
protein subunit beta.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant no. 81330081).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ participated in the design of the study,
performed most of the experiments and wrote the manuscript. XY, CH
and DW performed experiments. YM and WW conceived the study and
revised the manuscript. YZ and DW confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study protocol was approved (approval no.
20131321) by the Biomedical Ethics Committee of Anhui Medical
University (Hefei, China). Informed consent was obtained from all
patients for the use of their samples in scientific research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Benson RA, McInnes IB, Brewer JM and
Garside P: Cellular imaging in rheumatic disease. Nat Rev
Rheumatol. 11:357–367. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ
and Xu J: Rheumatoid arthritis: Pathological mechanisms and modern
pharmacologic therapies. Bone Res. 6(15)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ma Y, Hong FF and Yang SL: Role of
prostaglandins in rheumatoid arthritis. Clin Exp Rheumatol.
39:162–172. 2021.PubMed/NCBI
|
|
4
|
Dupré DJ, Robitaille M, Rebois RV and
Hébert TE: The role of Gbetagamma subunits in the organization
assembly and function of GPCR signaling complexes. Annu Rev
Pharmacol Toxicol. 49:31–56. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Jia X, Chang Y, Wei F, Dai X, Wu YJ, Sun
XJ, Xu S, Wu HX, Wang C, Yang XZ and Wei W: CP-25 reverses
prostaglandin E4 receptor desensitization-induced fibroblast-like
synoviocyte dysfunction via the G protein-coupled receptor kinase 2
in autoimmune arthritis. Acta Pharmacol Sin. 40:1029–1039.
2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang X, Zhao Y, Jia X, Wang C, Wu Y, Zhang
L, Chang Y and Wei W: CP-25 combined with MTX/ LEF ameliorates the
progression of adjuvant-induced arthritis by the inhibition on GRK2
translocation. Biomed Pharmacother. 110:834–843. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Han C, Li Y, Zhang Y, Wang Y, Cui D, Luo
T, Zhang Y, Liu Q, Li H, Wang C, et al: Targeted inhibition of GRK2
kinase domain by CP-25 to reverse fibroblast-like synoviocytes
dysfunction and improve collagen-induced arthritis in rats. Acta
Pharm Sin B. 11:1835–1852. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen J, Wang Y, Wu H, Yan S, Chang Y and
Wei W: A modified compound from paeoniflorin, CP-25, suppressed
immune responses and synovium inflammation in collagen-induced
arthritis mice. Front Pharmacol. 9(563)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tu J, Guo Y, Hong W, Fang Y, Han D, Zhang
P, Wang X, Körner H and Wei W: The regulatory effects of
paeoniflorin and its derivative paeoniflorin-6'-O-benzene sulfonate
CP-25 on inflammation and immune diseases. Front Pharmacol.
10(57)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shu J, Zhang XZ, Han L, Zhang F, Wu Y,
Tang XY, Wang C, Tai Y, Wang QT, Chen JY, et al:
Paeoniflorin-6'-O-benzene sulfonate alleviates collagen-induced
arthritis in mice by downregulating BAFF-TRAF2-NF-κB signaling:
Comparison with biological agents. Acta Pharmacol Sin. 40:801–813.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang BJ, Wang YY, Jia CY, Li SS, Wang XW,
Xu Y, Chen AY, Xu HP, Wang C, Yang ZY, et al:
Paeoniflorin-6'-o-benzene sulfonate ameliorates the progression of
adjuvant-induced arthritis by inhibiting the interaction between
Ahr and GRK2 of fibroblast-like synoviocytes. Int Immunopharmacol.
108(108678)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li J, Xiang B, Su W, Zhang X, Huang Y and
Ma L: Agonist-induced formation of opioid receptor-G
protein-coupled receptor kinase (GRK)-G beta gamma complex on
membrane is required for GRK2 function in vivo. J Biol Chem.
278:30219–30226. 2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rudomanova V and Blaxall BC: Targeting
GPCR-Gβγ-GRK2 signaling as a novel strategy for treating
cardiorenal pathologies. Biochim Biophys Acta Mol Basis Dis.
1863:1883–1892. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang DD, Jiang MY, Wang W, Zhou WJ, Zhang
YW, Yang M, Chen JY and Wei W: Paeoniflorin-6'-O-benzene sulfonate
down-regulates CXCR4-Gβγ-PI3K/AKT mediated migration in
fibroblast-like synoviocytes of rheumatoid arthritis by inhibiting
GRK2 translocation. Biochem Biophys Res Commun. 526:805–812.
2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chang Y, Jia X, Wei F, Wang C, Sun X, Xu
S, Yang X, Zhao Y, Chen J, Wu H, et al: CP-25, a novel compound,
protects against autoimmune arthritis by modulating immune
mediators of inflammation and bone damage. Sci Rep.
6(26239)2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Homan KT and Tesmer JJG: Molecular basis
for small molecule inhibition of G protein-coupled receptor
kinases. ACS Chem Biol. 10:246–256. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Murthy KS, Mahavadi S, Huang J, Zhou H and
Sriwai W: Phosphorylation of GRK2 by PKA augments GRK2-mediated
phosphorylation, internalization, and desensitization of VPAC2
receptors in smooth muscle. Am J Physiol Cell Physiol.
294:C477–C487. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Han CC, Liu Q, Zhang Y, Li YF, Cui DQ, Luo
TT, Zhang YW, Wang XM, Wang C, Ma Y and Wei W: CP-25 inhibits
PGE2-induced angiogenesis by down-regulating
EP4/AC/cAMP/PKA-mediated GRK2 translocation. Clin Sci (Lond).
134:331–347. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Penela P, Ribas C and Mayor F Jr:
Mechanisms of regulation of the expression and function of G
protein-coupled receptor kinases. Cell Signal. 15:973–981.
2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jaber M, Koch WJ, Rockman H, Smith B, Bond
RA, Sulik KK, Ross J Jr, Lefkowitz RJ, Caron MG and Giros B:
Essential role of beta-adrenergic receptor kinase 1 in cardiac
development and function. Proc Natl Acad Sci USA. 93:12974–12979.
1996.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tutunea-Fatan E, Abd-Elrahman KS,
Thibodeau JF, Holterman CE, Holleran BJ, Leduc R, Kennedy CRJ, Gros
R and Ferguson SSG: GRK2 knockdown in mice exacerbates kidney
injury and alters renal mechanisms of blood pressure regulation.
Sci Rep. 8(11415)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hullmann J, Traynham CJ, Coleman RC and
Koch WJ: The expanding GRK interactome: Implications in
cardiovascular disease and potential for therapeutic development.
Pharmacol Res. 110:52–64. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kamal FA, Travers JG, Schafer AE, Ma Q,
Devarajan P and Blaxall BC: G protein-coupled receptor-G-protein
βγ-subunit signaling mediates renal dysfunction and fibrosis in
heart failure. J Am Soc Nephrol. 28:197–208. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Schumacher SM, Gao E, Zhu W, Chen X,
Chuprun JK, Feldman AM, Tesmer JJ and Koch WJ: Paroxetine-mediated
GRK2 inhibition reverses cardiac dysfunction and remodeling after
myocardial infarction. Sci Transl Med. 7(277ra31)2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Murga C, Arcones AC, Cruces-Sande M,
Briones AM, Salaices M and Mayor Jr F: G protein-coupled receptor
kinase 2 (GRK2) as a potential therapeutic target in cardiovascular
and metabolic diseases. Front Pharmacol. 10(112)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Casey LM, Pistner AR, Belmonte SL,
Migdalovich D, Stolpnik O, Nwakanma FE, Vorobiof G, Dunaevsky O,
Matavel A, Lopes CMB, et al: Small molecule disruption of G
signaling inhibits the progression of heart failure. Circ Res.
107:532–539. 2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Daaka Y, Luttrell LM and Lefkowitz RJ:
Switching of the coupling of the beta2-adrenergic receptor to
different G proteins by protein kinase A. Nature. 390:88–91.
1997.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Senarath K, Kankanamge D, Samaradivakara
S, Ratnayake K, Tennakoon M and Karunarathne A: Regulation of G
protein βγ signaling. Int Rev Cell Mol Biol. 339:133–191.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tang X, Sun Z, Runne C, Madsen J, Domann
F, Henry M, Lin F and Chen S: A critical role of Gbetagamma in
tumorigenesis and metastasis of breast cancer. J Biol Chem.
286:13244–13254. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Xu S, Zhang Z, Ogawa O, Yoshikawa T,
Sakamoto H, Shibasaki N, Goto T, Wang L and Terada N: An EP4
antagonist ONO-AE3-208 suppresses cell invasion, migration, and
metastasis of prostate cancer. Cell Biochem Biophys. 70:521–527.
2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Liu Y, Pan YF, Xue YQ, Fang LK, Guo XH,
Guo X, Liu M, Mo BY, Yang MR, Liu F, et al: uPAR promotes
tumor-like biologic behaviors of fibroblast-like synoviocytes
through PI3K/Akt signaling pathway in patients with rheumatoid
arthritis. Cell Mol Immunol. 15:171–181. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Nygaard G and Firestein GS: Restoring
synovial homeostasis in rheumatoid arthritis by targeting
fibroblast-like synoviocytes. Nat Rev Rheumatol. 16:316–333.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rajagopal S and Shenoy SK: GPCR
desensitization: Acute and prolonged phases. Cell Signal. 41:9–16.
2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Smith JS and Rajagopal S: The β-Arrestins:
Multifunctional regulators of G protein-coupled receptors. J Biol
Chem. 291:8969–8977. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kamal FA, Smrcka AV and Blaxall BC: Taking
the heart failure battle inside the cell: Small molecule targeting
of Gβγ subunits. J Mol Cell Cardiol. 51:462–467. 2011.PubMed/NCBI View Article : Google Scholar
|