Introduction
β3-Adrenoreceptors (β3ARs) are involved in adipocyte
metabolism, gut relaxation and vascular vasodilation (1). Mirabegron is a selective β3AR agonist
approved for overactive bladder treatment (2-4).
Although the role of β3AR in adipose, intestinal and vascular
tissues is well established, its existence and function in the
heart remain unclear. In human cardiomyocytes, β3ARs couple with
the inhibitory G protein to exhibit a negative inotropic effect,
which counterbalances the effects of β1- and β2-adrenergic
stimulation in heart failure by increasing nitric oxide production
(5,6). However, the differential expression
of β3ARs can lead to distinct effects in tissues and cells. β3ARs
can stimulate the L-type Ca2+ current (ICa-L)
in human atrial cells and enhance atrial tissue contractility
through the cyclic adenosine monophosphate (cAMP)-dependent protein
kinase pathway (7).
In clinical trials, the most commonly reported
cardiovascular adverse events in patients receiving mirabegron for
overactive bladder are hypertension, tachycardia and atrial
fibrillation (AF) (8-10).
The number of older patients with AF risk receiving mirabegron
treatment has increased recently. However, the mechanisms
underlying the potential arrhythmogenic effects of mirabegron
remain unclear. Mirabegron might change cardiac
electrophysiological characteristics due to its effects on
ICa-L activation through the cAMP-dependent pathway,
possibly resulting in Ca2+ dysregulation (11-15).
The left atrium (LA), the most critical ‘substrate’ of AF (16,17),
is vulnerable to oxidative stress and has higher arrhythmogenesis
compared with the right atrium (RA) (18). Moreover, our previous studies
indicate that the LA is more susceptible to hydrogen sulfide- and
chronic obstructive pulmonary disease-related atrial
arrhythmogenesis than is the RA (19,20).
The differential arrhythmogenic effects between the LA and RA
facilitate the maintenance of atrial arrhythmogenesis. Since
mirabegron alters cardiac electrophysiological characteristics,
resulting in arrhythmogenesis, the effects of mirabegron may differ
between the LA and RA. Mirabegron may induce atrial
arrhythmogenesis through differential arrhythmogenic effects
between the LA and RA. Therefore, the present study aimed to
investigate the differences in the effects of mirabegron on the
electrophysiological activities of the LA and RA and clarify the
underlying mechanisms.
Materials and methods
Rabbit atrial tissue preparations
All the experimental procedures were approved by the
Institutional Animal Care and Use Committee of Taipei Veterans
General Hospital, Taipei, Taiwan (approval no. IACUC-2021-011).
Furthermore, the experimental protocols conformed to the
institutional guideline for the care and use of laboratory animals
as well as the Guide for the Care and Use of Laboratory Animals,
published by the US National Institutes of Health (21). Male New Zealand white rabbits
(n=36; weight, 2.5-3.5 kg; age, 6-8 months) used in the present
study were purchased from Animal Health Research Institute (Council
of Agriculture, Executive Yuan). All of the rabbits were housed in
a temperature- and humidity-controlled environment (20-22˚C; 50-70%
humidity) with a 12 h light/dark cycle, raised in stainless steel
cages and had free access to food and water. Rabbits were
anesthetized with an intramuscular injection of a mixture of
zoletil (10 mg/kg; Virbac) and xylazine (5 mg/kg; Bayer AG) and
sacrificed with an overdose of inhaled isoflurane (5% in oxygen;
Panion & BF Biotech, Inc.) from a precision vaporizer (22,23).
The anesthesia dosage was confirmed to be adequate on the basis of
the absence of corneal reflexes and motor responses to pain
stimuli. The hearts were excised through midline thoracotomy. The
tissue preparations (1x1.5 cm2) of the LA and RA were
separated from the LA and RA appendages, respectively.
Electropharmacological measurements were obtained within 2 h after
the separation. Tissue preparations (1-1.5 cm) of the RA and LA
were superfused at 37˚C with normal Tyrode's solution composed of
NaCl (137 mM), KCl (4 mM), NaHCO3 (15 mM),
NaH2PO4 (0.5 mM), MgCl2 (0.5 mM),
CaCl2 (2.7 mM) and dextrose (11 mM) at a constant rate
of 3 ml/min. Tyrode's solution was saturated with a mixture of 97%
O2 and 3% CO2 and its pH was adjusted to 7.4
with NaOH.
Electropharmacological
experiments
The transmembrane action potentials of the RA and LA
were recorded using machine-pulled glass capillary microelectrodes
filled with KCl (3 M) (20). The
microelectrodes were connected to an FD223 electrometer (World
Precision Instruments) under 150-mg tension. The electrical events
were simultaneously displayed on a Gould 4072 oscilloscope (Gould)
and a Gould TA11 recorder (Gould). Electrical stimuli were applied
using a Grass S88 stimulator (Grass Instruments) through a Grass
SIU5B stimulus isolation unit. The action potential parameters were
measured by applying 2-Hz electrical stimuli. The action potential
amplitude (APA) was calculated as the difference between the
resting membrane potential (RMP) and the peak of action potential
depolarization. The action potential duration (APD) at
repolarization extents of 90, 50 and 20% of the APD were measured
and designated as APD90, APD50 and
APD20, respectively. Burst firing was defined as the
occurrence of accelerated spontaneous activities (faster than the
basal rate) with sudden onset and termination. The same RA and LA
tissue preparations were sequentially treated with different
concentrations (0.01, 0.1 and 1 µM) of mirabegron (Avara
Pharmaceutical Technologies) in Tyrode's solution for 30 min to
investigate the electrophysiological effects of mirabegron with and
without 1 µM KT5823 (an inhibitor of cAMP-dependent protein kinase;
MilliporeSigma).
Ionic current measurements
Single LA and RA myocytes from rabbits were
enzymatically dissociated, as described previously (24). Whole-cell patch clamp recordings of
the single LA and RA myocytes were obtained before and after the
administration of mirabegron (0.1 µM) with or without KT5823 (1 µM)
using an Axopatch 1D amplifier (Axon Instruments) at 35±1˚C. The
ionic currents were recorded at ~3-5 min after rupture or
perforation to obtain measurements before ion channel activity
decay over time. A small hyperpolarizing step from a holding
potential of -50 mV to a test potential of -55 mV for 80 msec was
delivered at the beginning of each experiment. The area under the
capacitive current curve was divided using the applied voltage step
to obtain the total cell capacitance. In general, 60-80% series
resistance was electronically compensated. Ionic currents were
recorded in the current- and voltage-clamp modes.
The Na+ current (INa) was
measured during depolarization from a holding potential of -120 mV
to testing potentials ranging from -80-0 mV in 10-mV increments for
40 msec at a 3-Hz frequency and at room temperature (25±1˚C). The
external solution contained NaCl (5 mM), CsCl (133 mM),
MgCl2 (2 mM), CaCl2 (1.8 mM), nifedipine
(0.002 mM), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
(HEPES; 5 mM) and glucose (5 mM) adjusted to a pH of 7.3 with KOH.
The micropipettes were filled with a solution containing CsCl (133
mM), NaCl (5 mM), ethylene glycol-bis (β-aminoethyl
ether)-N,N,N',N'-tetraacetic acid (EGTA; 10 mM), tetraethylammonium
chloride (TEACl; 20 mM), MgATP (5 mM) and HEPES (5 mM) titrated to
a pH of 7.3 with CsOH.
ICa-L was measured as an inward current
during depolarization from a holding potential of -50 mV to test
potentials ranging from -40-+60 mV in 10-mV increments for 300 msec
at a 0.1-Hz frequency by using a perforated patch clamp with
amphotericin B. The external solution contained TEACl (20 mM), CsCl
(133 mM), HEPES (10 mM), MgCl2 (0.5 mM),
CaCl2 (1.8 mM) and glucose (10 mM) titrated to a pH of
7.4 with NaOH. Tetrodotoxin (10 µM) and 4-aminopyridine (2 mM) were
added to the external solution to block Na+ channels and
transient outward K+ current (Ito),
respectively. The micropipettes were filled with a solution
containing CsCl (130 mM), MgCl2 (1 mM),
Mg2ATP (5 mM), HEPES (10 mM), EGTA (10 mM), NaGTP (0.1
mM) and Na2 phosphocreatine (5 mM) titrated to a pH of
7.2 with CsOH. Steady-state inactivation of ICa-L was
evaluated using a standard protocol consisting of a 300-msec
prepulse and a 150-msec test pulse. The peak current elicited by
the test pulse was divided by the maximal current and plotted as a
function of prepulse voltage. Data points were fitted using a
Boltzmann function. Recovery from ICa-L inactivation was
assessed using a two-pulse protocol with a 200-msec prepulse and
test pulse (from -80-+10 mV) separated using varying time
intervals. Data points were fitted with a single-exponential
function (25).
Ito was estimated using a double-pulse
protocol. A 30-msec prepulse from -80--40 mV was used to inactivate
the Na+ channels and this was followed by a 300-msec
test pulse increasing to +60 mV in 10-mV increments at a 0.1-Hz
frequency. CdCl2 (200 µM) was added to the bath solution
for ICa-L inhibition. Ito was calculated as
the difference between the peak outward current and steady-state
current. The external solution contained NaCl (137 mM), KCl (5.4
mM), HEPES (10 mM), MgCl2 (0.5 mM), CaCl2
(1.8 mM) and glucose (10 mM) titrated to a pH of 7.4 with NaOH. The
micropipettes were filled with a solution containing KCl (20 mM),
K-aspartate (110 mM), MgCl2 (1 mM), MgATP (5 mM), HEPES
(10 mM), EGTA (0.5 mM), NaGTP (0.1 mM) and Na2
phosphocreatine (5 mM) titrated to a pH of 7.2 with KOH.
The ultrarapid component of the delayed rectifier
K+ current (IKur) was examined using a
double-pulse protocol, consisting of a 100-msec depolarizing
prepulse increasing to +40 mV from a holding potential of -50 mV,
which was followed by 150-msec voltage steps from -40-+60 mV in
10-mV increments at room temperature to provide an adequate
temporal resolution. IKur was measured as the
4-aminopyridine (1 mM)-sensitive current. The external solution
contained NaCl (137 mM), KCl (5.4 mM), HEPES (10 mM),
MgCl2 (0.5 mM), CaCl2 (1.8 mM) and glucose
(10 mM) titrated to a pH of 7.4 with NaOH. The micropipettes were
filled with a solution containing KCl (20 mM), K-aspartate (110
mM), MgCl2 (1 mM), MgATP (5 mM), HEPES (10 mM), EGTA
(0.5 mM), NaGTP (0.1 mM) and Na2 phosphocreatine (5 mM)
titrated to a pH of 7.2 with KOH.
The delayed rectifier K+ current
(IKr-tail) was measured as the outward peak tail current
density after 3 sec of prepulse increasing from a holding potential
of -40 mV to a voltage between -40 and +60 mV in 10-mV steps at a
0.1-Hz frequency in the presence of chromanol 293B (30 µM) and
CdCl2 (200 µM) in normal Tyrode's solution. The
micropipettes were filled with a solution containing KCl (120 mM),
MgCl2 (5 mM), CaCl2 (0.36 mM), EGTA (5 mM),
HEPES (5 mM), glucose (5 mM), K2ATP (5 mM),
Na2CrP (5 mM) and NaGTP (0.25 mM) adjusted to a pH of
7.2 with KOH.
Measurements of Ca2+
transients (Ca2+i) and intracellular
Ca2+
Intracellular Ca2+ was measured in single
LA myocytes, as described previously (26). In brief, LA myocytes were loaded
with fluorescent Ca2+ (10 µM) fluo-3/AM for 30 min at
room temperature. After intracellular hydrolysis of fluo-3/AM for
30 min, the excess extracellular dye was removed by changing the
bath solution. Fluo-3 fluorescence was triggered using a 488-nm
argon-ion laser line, with the emission recorded at >515 nm. The
LA myocytes were repetitively scanned at 2-msec intervals for line
scan imaging (8-bit). Fluorescence imaging was performed using a
laser scanning confocal microscope (Zeiss LSM 510; Zeiss GmbH) and
an inverted microscope (Axiovert 100; Zeiss GmbH). To exclude
variations in the fluorescence intensity due to different volumes
of injected dye and to correct for variations in dye
concentrations, the fluorescent signals were calculated by
normalizing the fluorescence (F) against the baseline fluorescence
(F0); in this manner, reliable information was obtained
on transient intracellular Ca2+ changes from baseline
values [ΔF=(F-F0)/F0]. The intracellular
Ca2+ changes, including transient Ca2+i, peak
systolic Ca2+i and diastolic Ca2+i, were
obtained during a 2-Hz field stimulation with 10-msec
twice-threshold-strength square-wave pulses. Through the addition
of 20 mM caffeine after electric stimulation at 2 Hz for at least
30 sec, the estimated sarcoplasmic reticulum (SR) Ca2+
content was measured as the total SR Ca2+ content from
the peak amplitude of the caffeine induced Ca2+i.
Statistical analysis
All continuous variables are expressed as means ±
standard deviations. Paired t test, one-way or two-way
repeated-measures analysis of variance with the Bonferroni post hoc
test was used to compare the difference between LA and RA or the
effects of mirabegron on the LA and RA tissue preparations and
isolated single myocytes. The McNemar test was used to compare the
incidence of burst firing before and after drug administration in
the LA and RA tissue preparations. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of mirabegron on atrial
electric activity
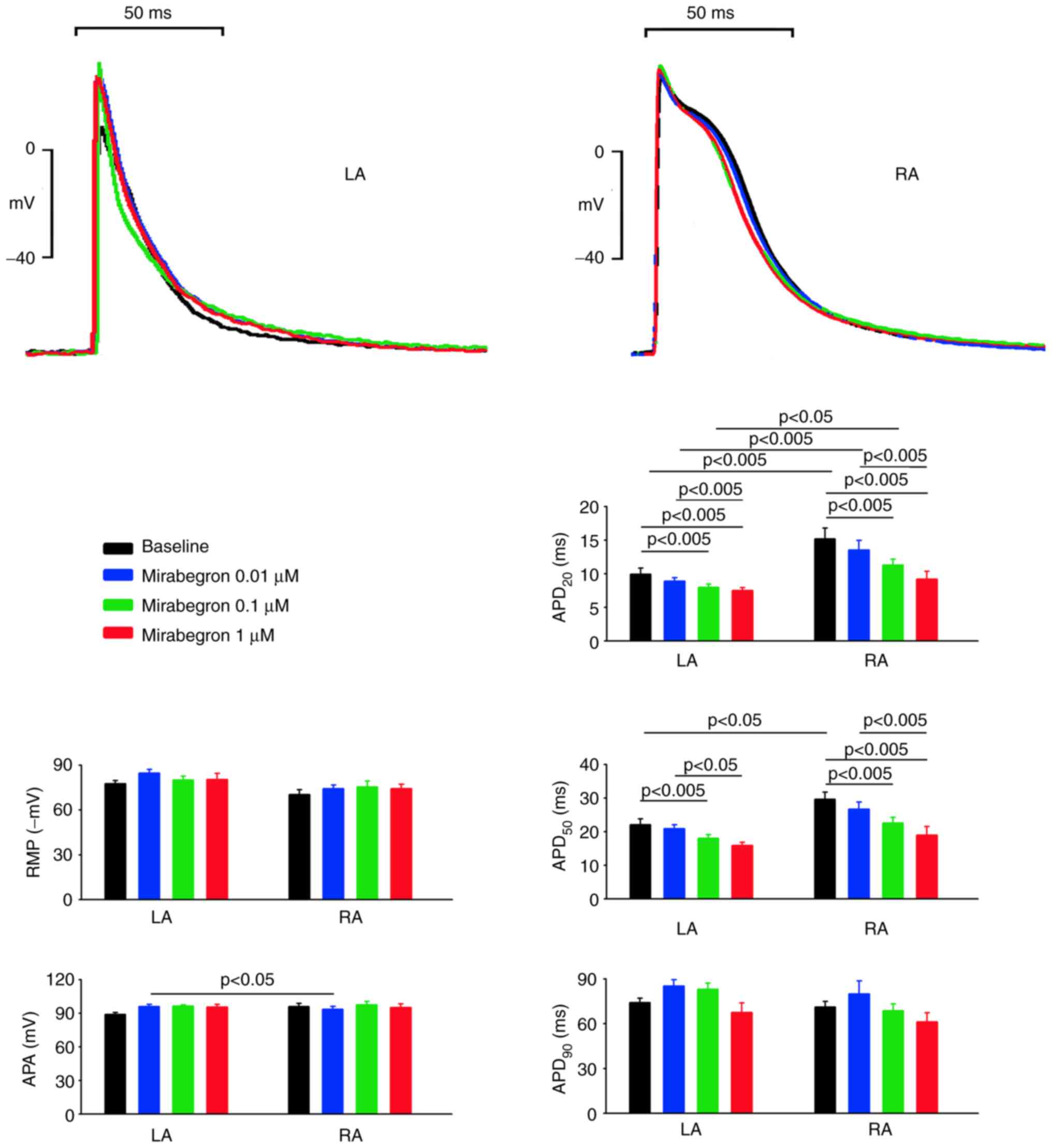
As shown in Fig. 1,
the baseline APA and RMP of the LA and RA were similar. However,
the baseline APD20 and APD50 but not
APD90 were shorter in the LA than in the RA. Mirabegron,
at 0.1 and 1 µM (but not 0.01 µM), reduced the APD20 and
APD50 of the LA, but it did not alter the APA, RMP, or
APD90 of the LA at 0.01, 0.1, or 1 µM. Similarly,
mirabegron at 0.1 and 1 µM (but not 0.01 µM), reduced the
APD20 and APD50 of the RA, but it did not
alter the APA, RMP, or APD90 of the RA at 0.01, 01, or 1
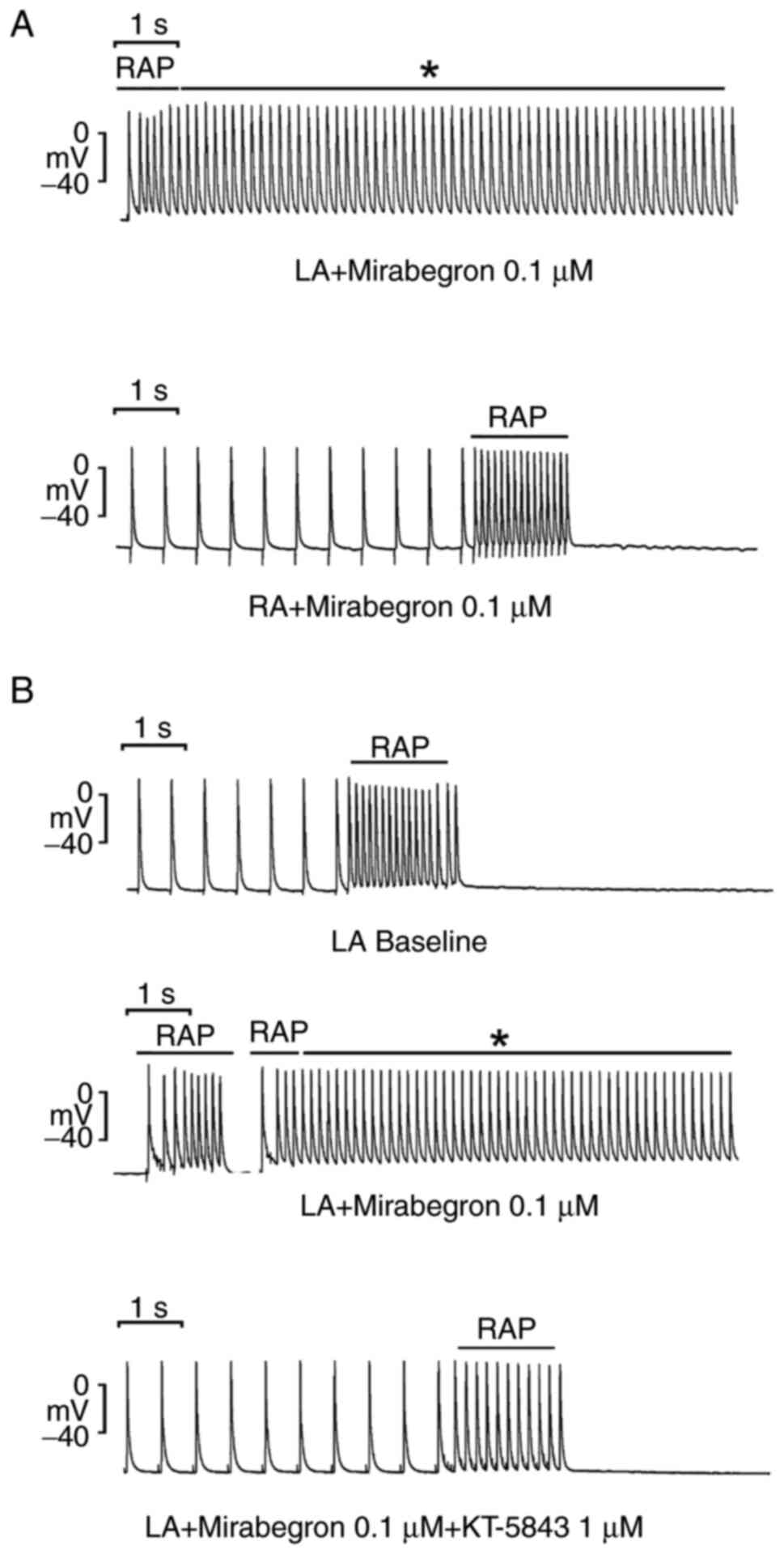
µM. Moreover, as shown in Fig. 2A,
under mirabegron treatment at 0.1 and 1 µM (but not 0.01 µM), 20-Hz
tachypacing induced burst firing in 7 of the 10 LAs (70% vs. 0% at
baseline, P<0.05). By contrast, 20-Hz tachypacing did not induce
burst firing in any of the 10 RAs under mirabegron treatment at
0.01, 01 and 1 µM. However, with the addition of KT5823 (1 µM),
burst firing was suppressed in 5 of the 6 LAs (83.3% vs. 0% at
baseline, P<0.05, Fig. 2B).
Effects of mirabegron on ionic
currents and Ca2+ homeostasis
The present study compared baseline ICa-L
and Ito in the LA and RA myocytes and found that the LA
myocytes had a smaller ICa-L but a larger Ito
than the RA myocytes (Fig.
S1).
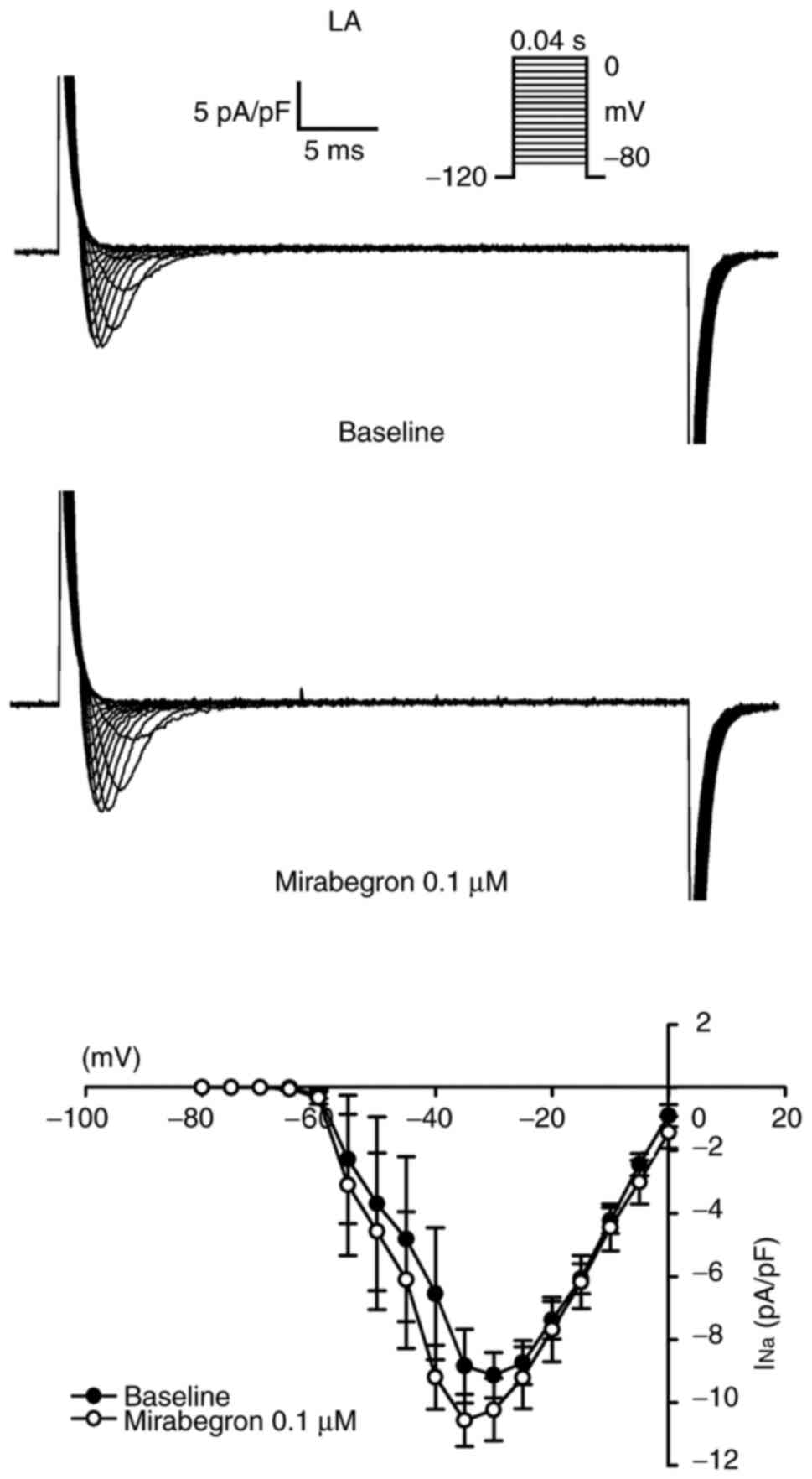
The effects of mirabegron on INa,
ICa-L, Ito, IKur and
IKr-tail in the LA myocytes were investigated. As shown
in Fig. 3, INa in the
LA myocytes remained unchanged after mirabegron (0.1 µM) treatment.
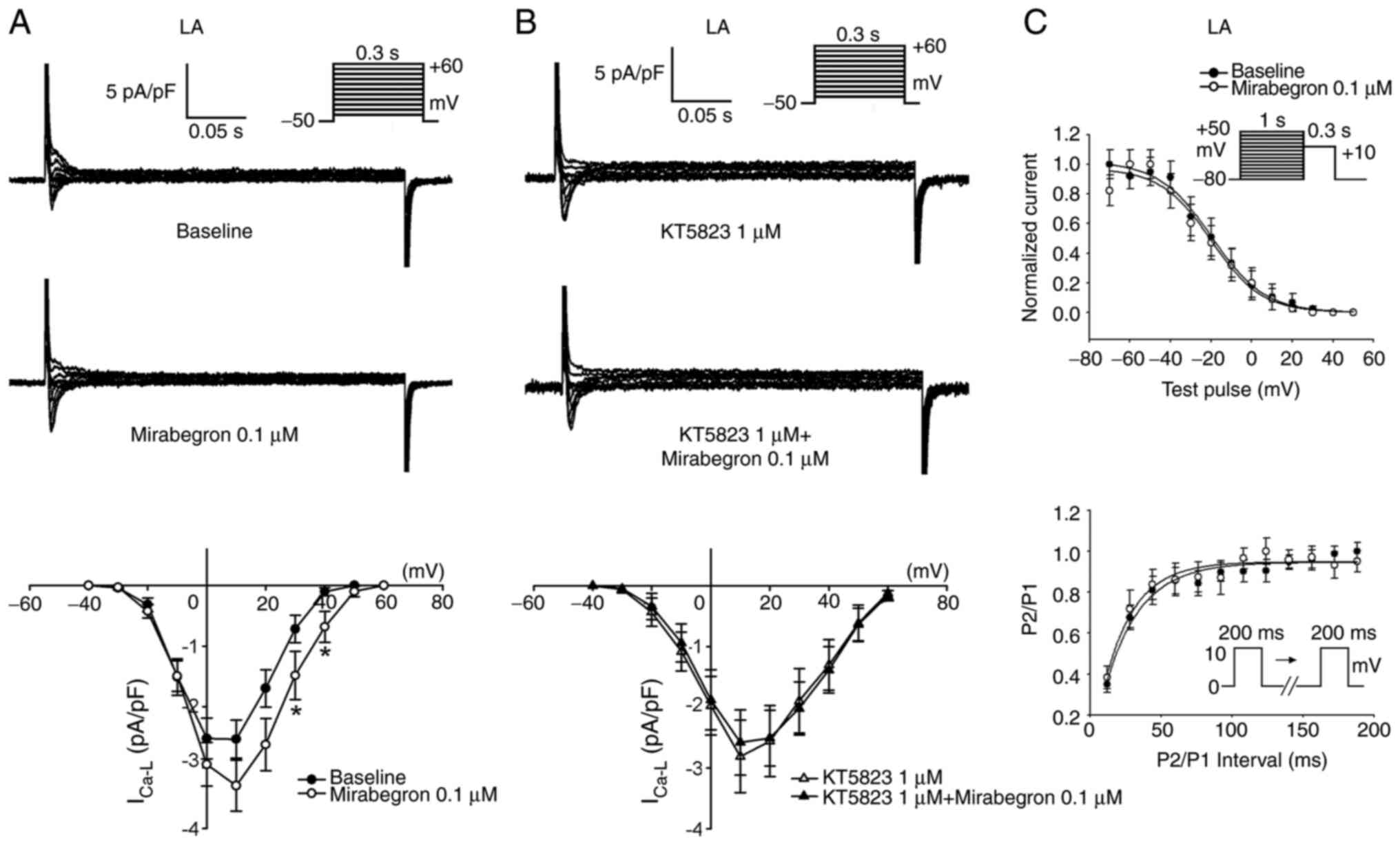
Moreover, mirabegron (0.1 µM) significantly increased
ICa-L in the LA myocytes (Fig. 4A). KT5823 (1 µM) pretreatment
attenuated the effects of mirabegron on ICa-L (Fig. 4B). Additionally, as shown in
Fig. 4C, mirabegron (0.1 µM) did
not alter the voltage-dependent inactivation of ICa-L
(-32.4±5.3 mV vs. -29.5±4.2 mV, P>0.05) and time constants of
recovery from inactivation of ICa-L (40.5±21.4 msec vs.
53.1±13.4 msec, P>0.05).
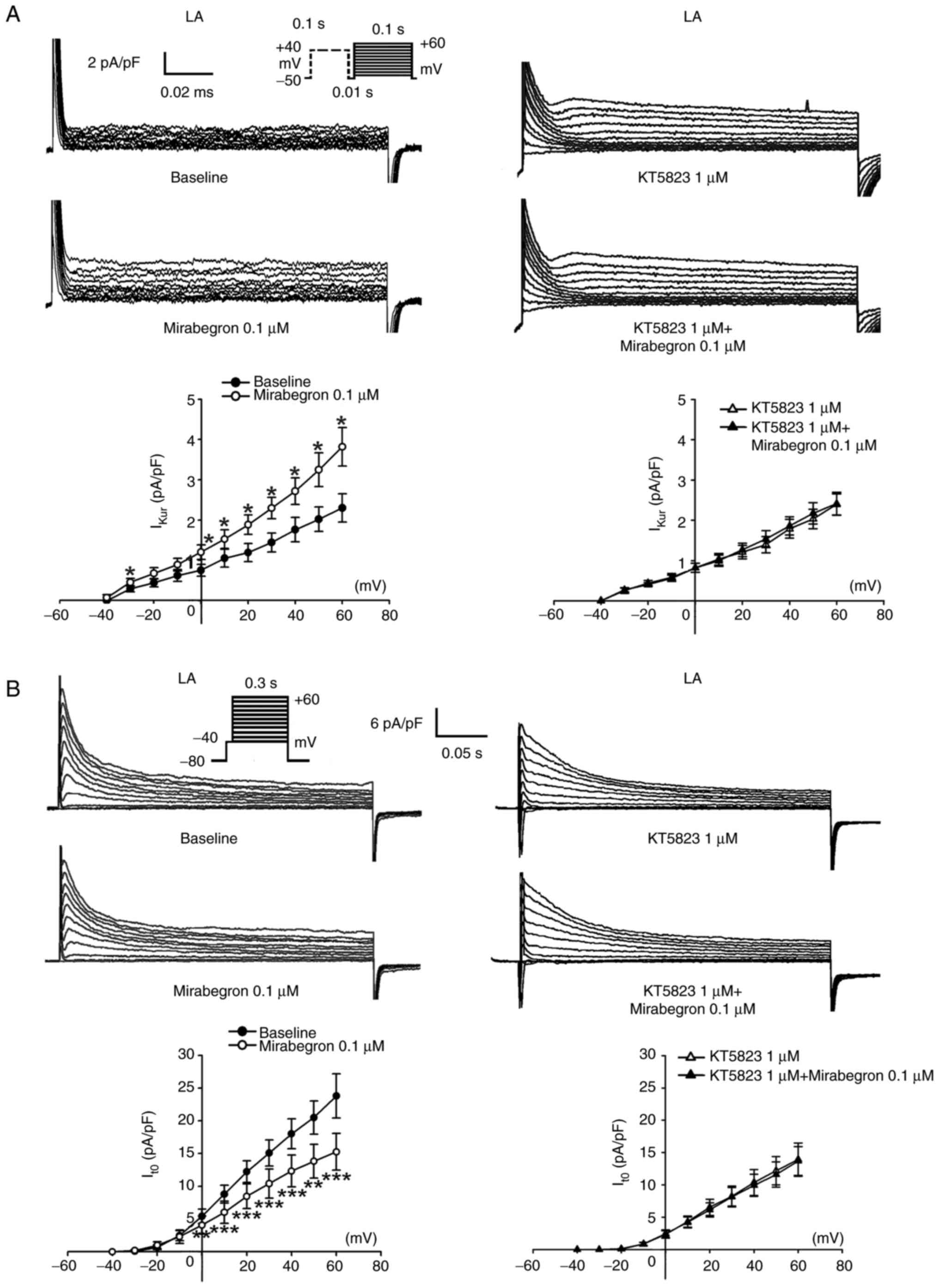
As shown in Fig. 5,
mirabegron (0.1 µM) significantly increased IKur but
reduced Ito. Similarly, KT5823 (1 µM) pretreatment
attenuated the effects of mirabegron on IKur and
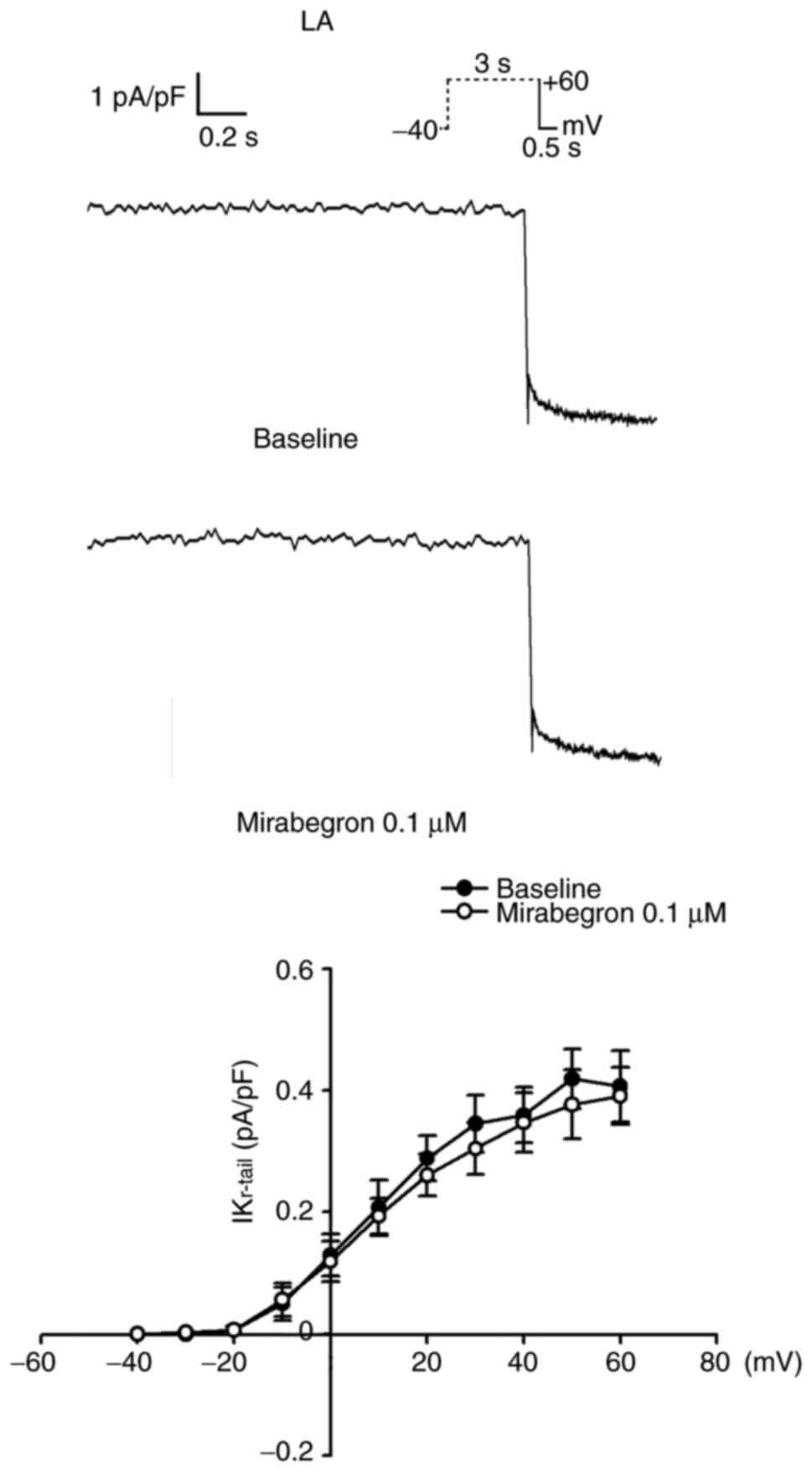
Ito. By contrast, IKr-tail in the LA myocytes
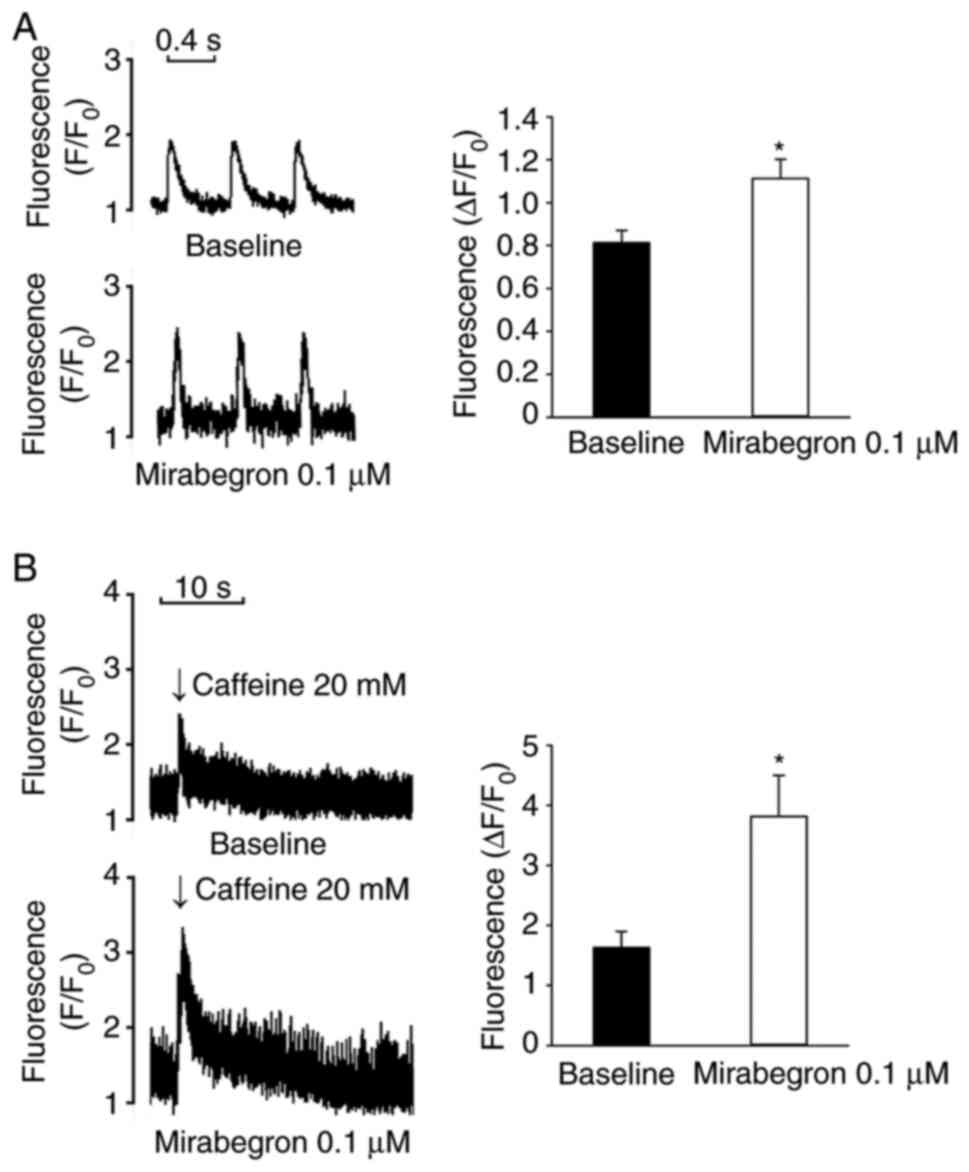
remained unchanged after 0.1 µM mirabegron treatment (Fig. 6). Moreover, mirabegron (0.1 µM)
increased Ca2+i and SR Ca2+ contents in the
LA myocytes (Fig. 7).
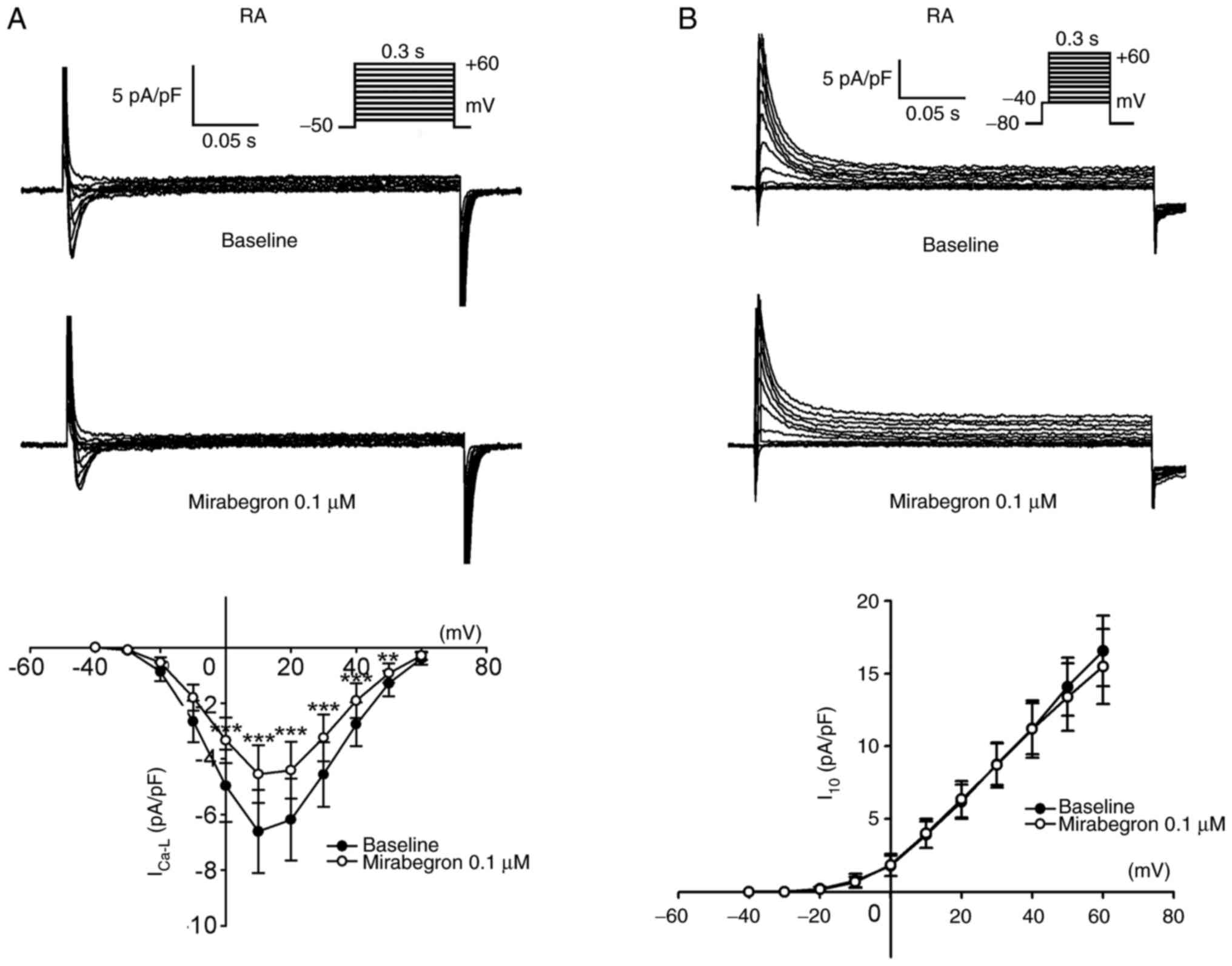
To clarify whether mirabegron may have different
ionic effects between RA and LA myocytes, the present study
investigated the effects of mirabegron on ICa-L and
Ito in the RA myocytes. In contrast to the effects of
mirabegron on ICa-L and Ito in LA myocytes,
mirabegron (0.1 µM) reduced ICa-L and did not alter
Ito in RA myocytes (Fig.
8).
Discussion
The present study, for the first time to the best of
the authors' knowledge, found that mirabegron significantly
increased the inducibility of burst firing through tachypacing in
the LA but not in the RA, suggesting that mirabegron differentially
induces atrial arrhythmogenesis in the LA but not the RA. Moreover,
the results indicated that KT5823, a cAMP-dependent protein kinase
inhibitor, suppressed burst firing induced by 20-Hz tachypacing and
mirabegron in the LA. These results suggested that the
cAMP-dependent protein kinase pathway may serve a role in
mirabegron-induced atrial arrhythmogenesis.
cAMP-dependent protein kinase phosphorylates several
key intracellular Ca2+ regulatory proteins such as
Ca2+ channels, sarco/endoplasmic reticulum
Ca2+-ATPase (SERCA), phospholamban (PLB), ryanodine
receptor channel (RyR) and Na+/Ca2+
exchangers (NCXs). In particular, cAMP-dependent protein kinase
activation increases PLB, RyR and NCX phosphorylation, resulting in
Ca2+ dysregulation and arrhythmogenesis (11-13,27).
In the whole-cell patch clamp experiments, mirabegron significantly
increased ICa-L in the LA myocytes. A prolonged increase
in ICa-L causes Ca2+ overload, which may
contribute to arrhythmogenesis (28). Moreover, KT5823 pretreatment
inhibited the effects of mirabegron on ICa-L in the LA
myocytes in the current study and mirabegron significantly
increased Ca2+i and SR Ca2+ content in these
myocytes. Therefore, the findings indicated that mirabegron may
change the electrophysiological characteristics of the LA and cause
LA arrhythmogenesis by activating the cAMP-dependent protein kinase
pathway and inducing Ca2+ dysregulation.
In the present study, mirabegron was found to
accelerate LA and RA repolarization, which possibly reduces the
effective refractory period, favoring the genesis of reentry
(29). The results of the present
study revealed that mirabegron significantly shortened the
APD20 and APD50 but not APD90 of
the LA and RA. In theory, APD20 and APD50 are
affected by not only Ca2+ channels but also other ion
channels, such as voltage-gated K+ channels
(Ito and IKur). The present study found that
mirabegron reduced ICa-L but did not alter
Ito in the RA myocytes. By contrast, mirabegron
significantly increased ICa-L but reduced Ito
in the LA myocytes. These findings suggested that mirabegron
affected the electrophysiological characteristics of the LA and RA
differentially, ultimately leading to atrial arrhythmogenesis. The
whole-cell patch clamp experiments demonstrated that mirabegron
increased ICa-L and IKur but reduced
Ito in the LA myocytes. These results suggested that the
shortening of the APD20 and APD50 of the LA
is mainly attributable to the composite effects of mirabegron on
ICa-L, IKur and Ito. The results
also indicated that mirabegron did not alter the APD90
of the LA and RA. Phase 3 repolarization occurs because of the
time-dependent inactivation of ICa-L and activation of
IKr-tail. The IKr-tail, rapid component of
IKr-tail and slow component of IKr-tail all
play major roles in phase 3 repolarization (30). In the current study,
IKr-tail in the LA myocytes remained unchanged after
mirabegron treatment and this unchanged IKr-tail was
responsible for APD90 expression caused by mirabegron in
the LA.
The present study had some limitations. First, it
found that mirabegron has a direct electrophysiological effect on
the LA (an AF substrate), suggesting that mirabegron possibly
contributed to atrial arrhythmogenesis. However, simply
investigating the acute effects of mirabegron may not reveal the
complete mechanisms underlying the arrhythmogenesis induction
caused by mirabegron. Chronic mirabegron treatment in patients with
overactive bladder may result in the various electrophysiological
effects involved in arrhythmogenesis. Second, in the present study,
mirabegron increased ICa-L in the LA myocytes, whereas
KT5823 suppressed this effect as well as arrhythmogenesis in the
LA. It was also observed that mirabegron significantly increased
Ca2+i and SR Ca2+ content, which may result
in Ca2+ dysregulation. Therefore, mirabegron may induce
LA arrhythmogenesis through Ca2+ overload induction and
activation of cAMP-dependent protein kinase. Although acute
mirabegron administration may not alter the expression of RyR,
SERCA and PLB due to short experimental periods, it is not clear
whether mirabegron may change the phosphorylation of RyR, SERCA and
PLB, leading to Ca2+ dysregulation in the present study.
The precise signaling pathways underlying mirabegron-induced LA
arrhythmogenesis were not completely elucidated.
In conclusion, mirabegron differentially modulated
the electrophysiological and arrhythmogenic effects in the LA and
RA. Through the activation of the cAMP-dependent protein kinase
pathway and induction of Ca2+ dysregulation, mirabegron
may increase LA arrhythmogenesis, increasing AF risk in patients
receiving mirabegron treatment.
Supplementary Material
Comparison of baseline
ICa-L and Ito in the LA and RA myocytes. (A)
Tracings and current-voltage relationship of ICa-L in
the LA (n=11) and RA (n=12) myocytes. (B) Tracings and
current-voltage relationship of Ito in the LA (n=9) and
RA (n=10) myocytes. Insets in the current traces show various clamp
protocols. *P<0.05, **P<0.01,
***P<0.005 compared with LA group at same mV.
ICa-L, Ca2+ current; Ito,
transient outward K+ current; LA, left atrium; RA, right
atrium.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the Ministry of Science and
Technology of Taiwan (grant no. MOST 110-2314-B-038-126-MY2),
Taipei Medical University-Taipei Medical University Hospital (grant
no. 108TMU-TMUH-25), Taipei Medical University (grant no.
TMU109-AE1-B29) and the Foundation for the Development of Internal
Medicine in Okinawa (grant no. 03-009).
Availability of data and materials
The datasets used and/or analyzed during the
present study are available from the corresponding authors on
reasonable request.
Authors' contributions
CC, YL and CH contributed to analyzing the
experimental results and writing the manuscript. FL, CL and YCC
contributed to the in vitro experiments and provided
technical assistance. SH, SC and YJC contributed to conceiving and
designing the study and reviewing the manuscript. CC and YCC
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All the experimental procedures were approved by
the Institutional Animal Care and Use Committee of Taipei Veterans
General Hospital, Taipei, Taiwan (approval no. IACUC-2021-011).
Furthermore, the experimental protocols conform to the
institutional guideline for the care and use of laboratory animals
and the Guide for the Care and Use of Laboratory Animals
published by the United States National Institutes of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gauthier C, Tavernier G, Charpentier F,
Langin D and Le Marec H: Functional β3-adrenoceptor in the human
heart. J Clin Invest. 98:556–562. 1996.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Khullar V, Amarenco G, Angulo JC,
Cambronero J, Høye K, Milsom I, Radziszewski P, Rechberger T,
Boerrigter P, Drogendijk T, et al: Efficacy and tolerability of
mirabegron, a β3-adrenoceptor agonist, in patients with overactive
bladder: Results from a randomised European-Australian phase 3
trial. Eur Urol. 63:283–295. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wagg A, Cardozo L, Nitti VW, Castro-Diaz
D, Auerbach S, Blauwet MB and Siddiqui E: The efficacy and
tolerability of the β3-adrenoceptor agonist mirabegron for the
treatment of symptoms of overactive bladder in older patients. Age
Ageing. 43:666–675. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wagg A, Staskin D, Engel E, Herschorn S,
Kristy RM and Schermer CR: Efficacy, safety, and tolerability of
mirabegron in patients aged ≥ 65yr with overactive bladder wet: A
phase IV, double-blind, randomised, placebo-controlled study
(PILLAR). Eur Urol. 77:211–220. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gauthier C, Leblais V, Kobzik L, Trochu
JN, Khandoudi N, Bril A, Balligand JL and Marec HL: The negative
inotropic effect of beta3-adrenoceptor stimulation is mediated by
activation of a nitric oxide synthase pathway in human ventricle. J
Clin Invest. 102:1377–1384. 1998.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Moniotte S, Kobzik L, Feron O, Trochu JN,
Gauthier C and Balligand JL: Upregulation of beta3-adrenoceptors
and altered contractile response to inotropic amines in human
failing myocardium. Circulation. 103:1649–1655. 2001.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Skeberdis VA, Gendvilienė C, Zablockaitė
D, Treinys R, Macianskiene R, Bogdelis A, Jurevicius J and
Fischmeister R: β3-adrenergic receptor activation increases human
atrial tissue contractility and stimulates the L-type
Ca2+ current. J Clin Invest. 118:3219–3227.
2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nitti VW, Khullar V, Kerrebroeck P,
Herschorn S, Cambronero J, Angulo JC, Blauwet MB, Dorrepaal C,
Siddiqui E and Martin NE: Mirabegron for the treatment of
overactive bladder: A prespecified pooled efficacy analysis and
pooled safety analysis of three randomised, double-blind,
placebo-controlled, phase III studies. Int J Clin Pract.
67:619–632. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Batista JE, Kölbl H, Herschorn S,
Rechberger T, Cambronero J, Halaska M, Coppell A, Kaper M, Huang M
and Siddiqui E: The efficacy and safety of mirabegron compared with
solifenacin in overactive bladder patients dissatisfied with
previous antimuscarinic treatment due to lack of efficacy: results
of a noninferiority, randomized, phase IIIb trial. Ther Adv Urol.
7:167–179. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nitti VW, Chapple CR, Walters C, Blauwet
MB, Herschorn S, Milsom I, Auerbach S and Radziszewski P: Safety
and tolerability of the β3-adrenoceptor agonist mirabegron, for the
treatment of overactive bladder: Results of a prospective pooled
analysis of three 12-week randomised phase III trials and of a
1-year randomised phase III trial. Int J Clin Pract. 68:972–985.
2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lindemann JP, Jones LR, Hathaway DR, Henry
BG and Watanabe AM: β-Adrenergic stimulation of phospholamban
phosphorylation and Ca2+-ATPase activity in guinea pig
ventricles. J Biol Chem. 258:464–471. 1983.PubMed/NCBI
|
|
12
|
Takasago T, Imagawa T and Shigekawa M:
Phosphorylation of the cardiac ryanodine receptor by cAMP-dependent
protein kinase. J Biochem. 106:872–877. 1989.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Perchenet L, Hinde AK, Patel KC, Hancox JC
and Levi AJ: Stimulation of Na+/Ca2+ exchange
by the β-adrenergic/protein kinase A pathway in guinea-pig
ventricular myocytes at 37 degrees C. Pflugers Arch. 439:822–828.
2000.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lou Q, Janardhan A and Efimov IR:
Remodeling of calcium handling in human heart failure. Adv Exp Med
Biol. 740:1145–1174. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Heijman J, Voigt N, Wehrens XH and Dobrev
D: Calcium dysregulation in atrial fibrillation: the role of
CaMKII. Front Pharmacol. 5(30)2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pappone C, Rosanio S, Oreto G, Tocchi M,
Gugliotta F, Vicedomini G, Salvati A, Dicandia C, Mazzone P,
Santinelli V, et al: Circumferential radiofrequency ablation of
pulmonary vein ostia: A new anatomic approach for curing atrial
fibrillation. Circulation. 102:2619–2628. 2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Roithinger FX, Steiner PR, Goseki Y,
Sparks PB and Lesh MD: Electrophysiologic effects of selective
right versus left atrial linear lesions in a canine model of
chronic atrial fibrillation. J Cardiovasc Electrophysiol.
10:1564–1574. 1999.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lin YK, Lai MS, Chen YC, Cheng CC, Huang
JH, Chen SA, Chen YJ and Lin CI: Hypoxia and reoxygenation modulate
the arrhythmogenic activity of the pulmonary vein and atrium. Clin
Sci. 122:121–132. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chan CS, Lin YK, Chen YC, Kao YH, Chen SA
and Chen YJ: Hydrogen sulphide increases pulmonary veins and atrial
arrhythmogenesis with activation of protein kinase C. J Cell Mol
Med. 22:3503–3513. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chan CS, Lin YS, Lin YK, Chen YC, Kao YH,
Hsu CC, Chen SA and Chen YJ: Atrial arrhythmogenesis in a rabbit
model of chronic obstructive pulmonary disease. Transl Res.
223:25–39. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the care and use of laboratory animals. 8th
edition. National Academies Press (US), Washington, DC, 2011.
|
|
22
|
Chan CS, Chen YC, Chang SL, Lin YK, Kao
YH, Chen SA and Chen YJ: Heart failure differentially modulates the
effects of ivabradine on the electrical activity of the sinoatrial
node and pulmonary veins. J Card Fail. 24:763–772. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Calvo-Guirado JL, Satorres M, Negri B,
Ramirez-Fernandez P, Maté-Sánchez JE, Delgado-Ruiz R, Gomez-Moreno
G, Abboud M and Romanos GE: Biomechanical and histological
evaluation of four different titanium implant surface
modifications: An experimental study in the rabbit tibia. Clin Oral
Invest. 18:1495–1505. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen YJ, Chen YC, Wongcharoen W, Lin CI
and Chen SA: Effect of K201, a novel antiarrhythmic drug on calcium
handling and arrhythmogenic activity of pulmonary vein
cardiomyocytes. Br J Pharmacol. 153:915–925. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lu YY, Chung FB, Chen YC, Tsai CF, Kao YH,
Chao TF, Huang JH, Chen SA and Chen YJ: Distinctive
electrophysiological characteristics of right ventricular outflow
tract cardiomyocytes. J Cell Mol Med. 18:1540–1548. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Huang SY, Lu YY, Lin YK, Chen YC, Chen YA,
Chung CC, Lin WS, Chen SA and Chen YJ: Ceramide modulates
electrophysiological characteristics and oxidative stress of
pulmonary vein cardiomyocytes. Eur J Clin Invest.
52(e13690)2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
McDonald TF, Pelzer S, Trautwein W and
Pelzer DJ: Regulation and modulation of calcium channels in
cardiac, skeletal, and smooth muscle cells. Physiol Rev.
74:365–507. 1994.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Landstrom AP, Dobrev D and Wehrens XHT:
Calcium signaling and cardiac arrhythmias. Circ Res. 120:1969–1993.
2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li D, Zhang L, Kneller J and Nattel S:
Potential ionic mechanism for repolarization differences between
canine right and left atrium. Circ Res. 88:1168–1175.
2001.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Grand AO: Cardiac ion channels. Circ
Arrhythm Electrophysiol. 2:185–194. 2009.PubMed/NCBI View Article : Google Scholar
|