Introduction
Microscopic polyangiitis (MPA) is a rare,
idiopathic, autoimmune, systemic disease, and it is classified as
antineutrophil cytoplasmic autoantibodies (ANCA) associated
vasculitis (AAV) (1). It is
defined as a necrotizing vasculitis, with few or no immune
deposits. It predominantly affects small blood vessels
(capillaries, venules, arterioles and small arteries) and medium
arteries, while it does not involve granulomatous inflammation
(1). This disease is associated
with the presence of ANCA, which are predominantly directed against
myeloperoxidase (MPO-ANCA), and in a minority of patients directed
against proteinase 3 (PR3-ANCA) (1). Other types of AAV include
granulomatosis with polyangiitis (GPA) and eosinophilic GPA (EGPA)
(1). The worldwide annual
cumulative incidence of MPA (new cases) is estimated to be 3-24 per
million inhabitants, with a prevalence of 25-94 per million
inhabitants (new and pre-existing cases). It affects all ethnical
groups with a predominance in Caucasian individuals (2-4).
Men seem to be slightly more frequently affected by this disease
than women (2-4).
The age at the onset of the symptoms is estimated to be around 50
years. MPA is more common in the south of Europe, while GPA is more
common in the north (2-4).
Although it is considered that MPA most frequently
involves the kidneys (5), the lung
involvement is another important feature of MPA. The current study
reports a case of MPA with PF.
Case report
The present study reports the case of a 48-year-old
woman who was admitted in Center of Rheumatic Diseases (Bucharest,
Romania) complaining of diffuse myalgia and arthralgia of both
hands and feet. The patient also described involuntary loss of
weight and occasional slight fever. The patient had family history
of colorectal cancer; however, she had no personal medical history
and was receiving non-steroidal anti-inflammatory drugs for joint
pain. The patient was a non-smoker and had no occupational history
of exposure to noxious substances.
On physical examination, the patient was clinically
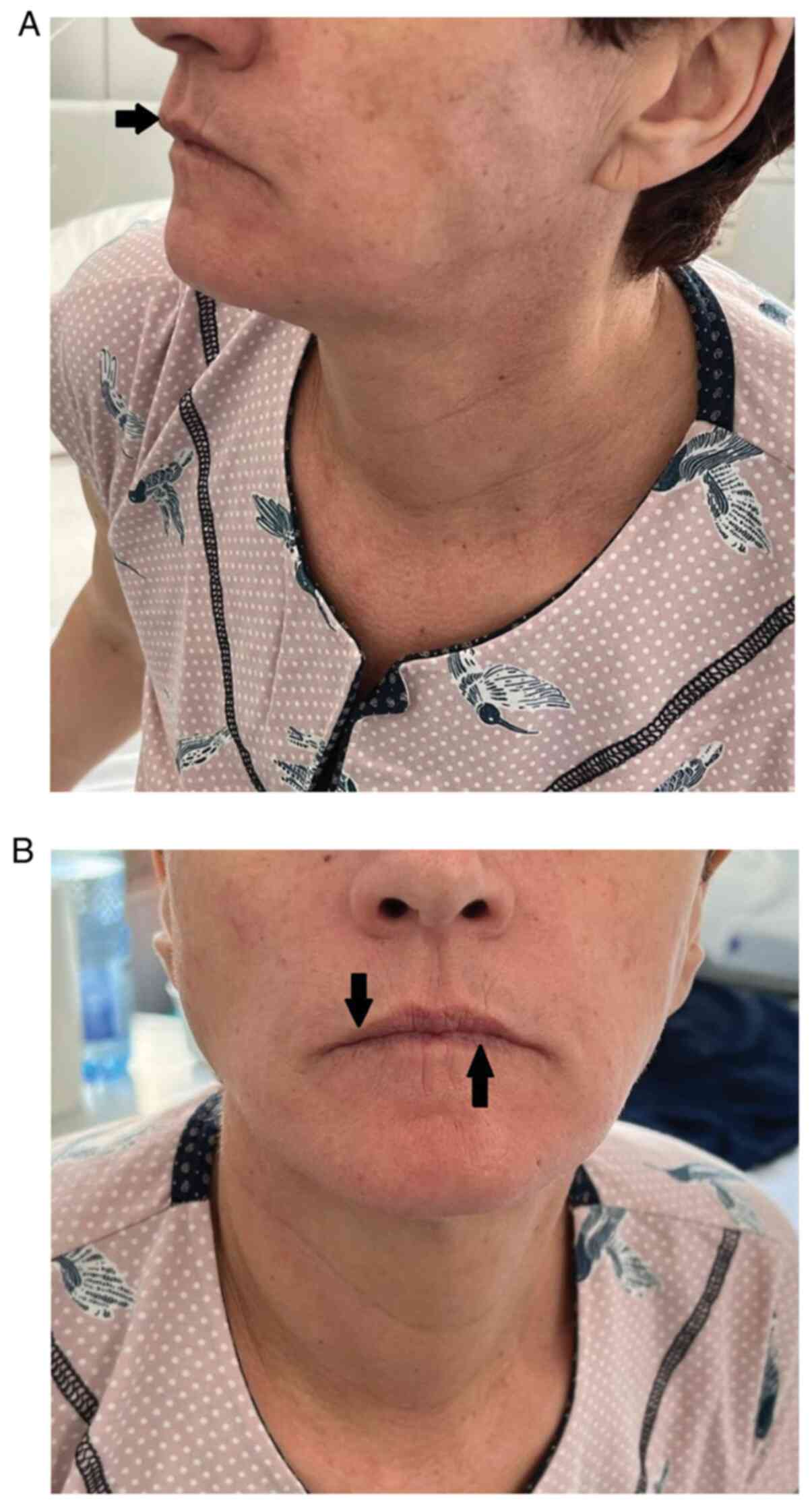
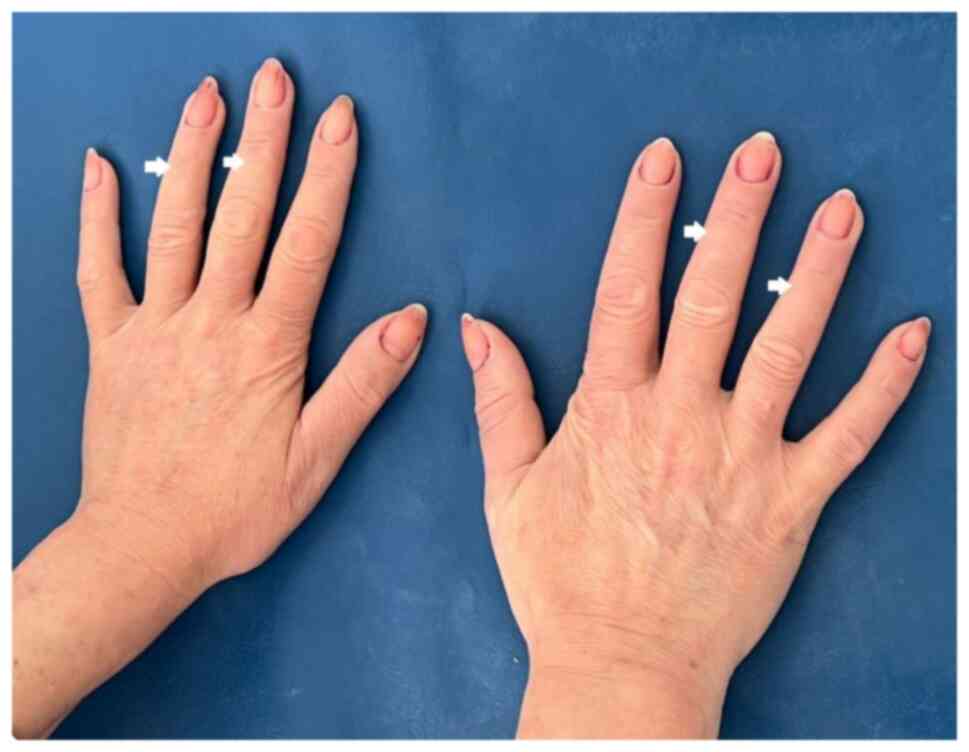
stable and had microstomia and perioral radial furrows (Fig. 1A and B), slight skin induration of the hands
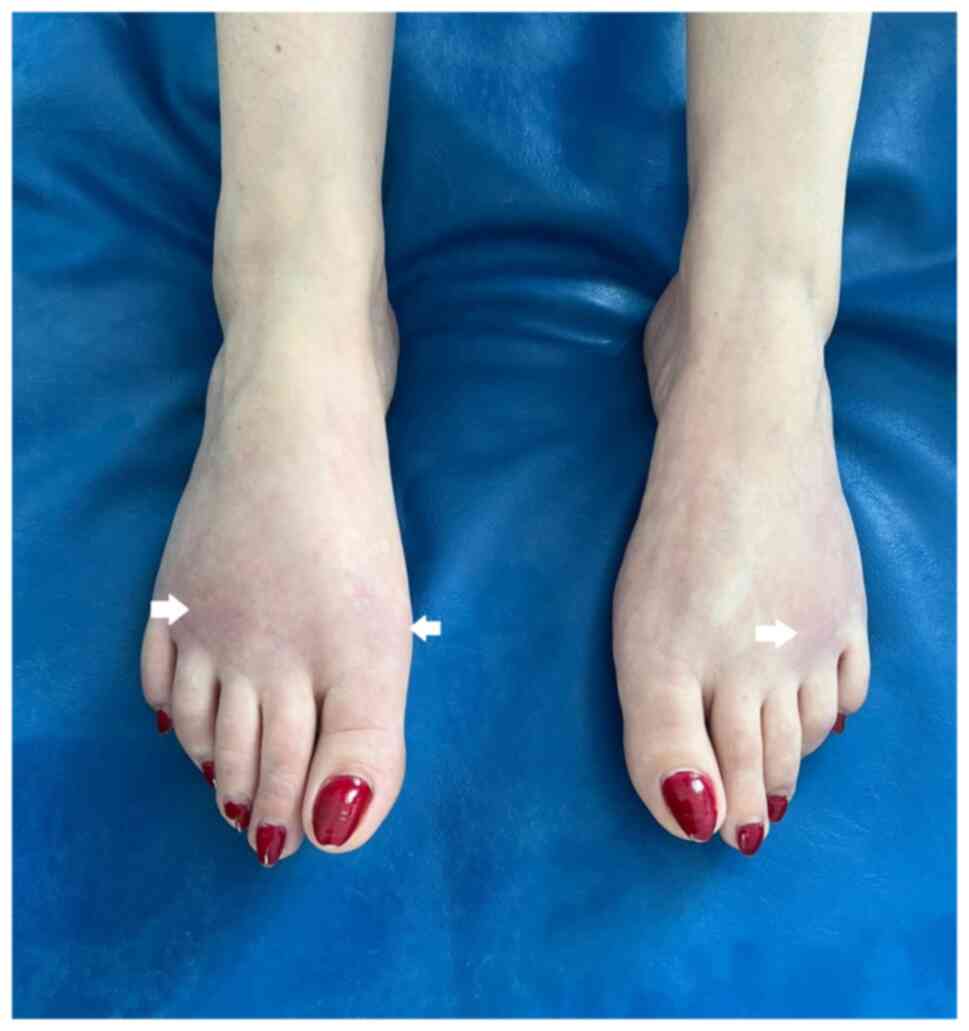
(Fig. 2) and discrete cyanotic
skin areas on the dorsal face of both feet (Fig. 3). The patient also had asymmetric
bilateral crepitant rales, bun normal vital signs (normal
temperature, oxygen saturation on pulse-oximetry of 98%, blood
pressure of 110/80 mmHg, heart rate of 100 beats/min and regular
cardiac rhythm).
Laboratory findings at admission revealed normal
blood count with non-specific biological inflammatory syndrome,
consisting of high erythrocyte sedimentation rate (74 mm/h; normal,
<20 mm/h) and high C-reactive protein (111.15 mg/l; normal,
<5 mg/l), mild cytolysis with high alanine aminotransferase (82
U/l; normal, <55 U/l), high aspartate aminotransferase (53 U/l;
normal, <34 U/l) and high gamma-glutamyl transferase (158 U/l;
normal, <34 U/l). Urine analysis revealed no microscopic
hematuria or proteinuria.
The patient was initially suspected of systemic
sclerosis due to the appearance of microstomia and slight skin
induration of the hands with diffuse arthralgia and myalgia, but
with negative anti-SCL70 and anti-centromere B antibodies (Table I). Also, nailfold capillaroscopy
showed a normal aspect of the capillary bed. Cryoglobulins were
absent and the patient was seronegative for hepatitis B surface
antigen and antibodies to hepatitis C. Antiphospholipid syndrome
tests (anticardiolipin screening and anti-β2-glicoprotein I
screening) were negative, but high titers (>100 U/ml) of
MPO-ANCA were detected (Table I).
Since the patient did not exhibit vasculitis, the diagnosis
required further investigation to exclude other connective tissue
diseases. In this sense, rheumatoid factor, antinuclear antibodies
[indirect immunofluorescence (IIF); titer of 1:320 with homogeneous
nuclear appearance] and anti-double-stranded DNA antibodies were
positive (Table I). During
hospitalization, the patient experienced an episode of swelling,
forefoot erythema and local heat (predominantly in the fourth and
fifth right fingers), associated with severe local pain.
Simultaneously, the patient developed febrile episode (38.6˚C). The
patient's rapid COVID-19 antigen test was negative. Musculoskeletal
ultrasound examination of the forefoot revealed subcutaneous edema,
negative power Doppler signal and no synovitis or
tenosynovitis.
 | Table IPatient's serology (routine laboratory
diagnostic tests). |
Table I
Patient's serology (routine laboratory
diagnostic tests).
|
Test/antibodya | Value | Normal range |
|---|
| Rheumatoid
factor | 60.1 U/l | 0.0-30.0 U/l |
| Anti-citrullinated
protein antibody | 10.8 U/l | <20.0 U/l |
| Systemic sclerosis
panel (serum, Line Blot) | | |
|
Anti-SCL70 | Negative | Negative |
|
Anti-centromere
A | Negative | Negative |
|
anti-centromere
B | Negative | Negative |
|
Anti-RNA
polymerase III 11 kDa | Negative | Negative |
|
Anti-RNA
polymerase III 155 kDa | Negative | Negative |
|
Anti-fibrillarin | Negative | Negative |
|
Anti-NOR90 | Negative | Negative |
|
Anti
Th/To | Negative | Negative |
|
Anti-PM-SCL100 | Negative | Negative |
|
Anti-PM-SCL75 | Negative | Negative |
|
Anti-Ku | Negative | Negative |
|
Anti-PDGFR | Negative | Negative |
|
Anti-Ro52 | Negative | Negative |
| ANCAb | 1:80 | Negative |
| MPO-ANCA | 100 U/ml | <5 U/ml |
| PR3-ANCA | 2.4 U/ml | <5 U/ml |
| Antinuclear
antibodiesb | 1:320 | <1:160 |
| Anti-BPI | 1.9 U/ml | <10.0 U/ml |
| Anti-cathepsin G | 3.2 U/ml | <10.0 U/ml |
| Anti-lactoferin | 0.9 U/ml | <10.0 U/ml |
| Anti-lyzozyme | 1.3 U/ml | <10.0 U/ml |
| Anti-Sm | 1.3 U/ml | <15.0 U/ml |
| Anti-U1RNP | 2.4 U/ml | <25.0 U/ml |
| Anti-SSA
(anti-Ro) | 4.8 U/ml | <15.0 U/ml |
| Anti-SSB
(anti-La) | 2.2 U/ml | <15.0 U/ml |
| C3 serum
complement | 1.8 g/l | <1.8.0 g/l |
| C4 serum
complement | 0.3 g/l | <0.4.0 g/l |
| Anti-cardiolipin
screening | 5.5 U/ml | <10.0 U/ml |
|
Anti-β2-glicoprotein I screening | 2.3 U/ml | <10.0 U/ml |
| Anti-C1q IgG | 1.8 U/ml | <10.0 U/ml |
| Cryoglobulins | Negative | Negative |
| Anti-Jo 1 | 1.6 U/ml | <15.0 U/ml |
| Hepatitis B surface
antigen | Negative | Negative |
| Anti-hepatitis C
virus | Negative | Negative |
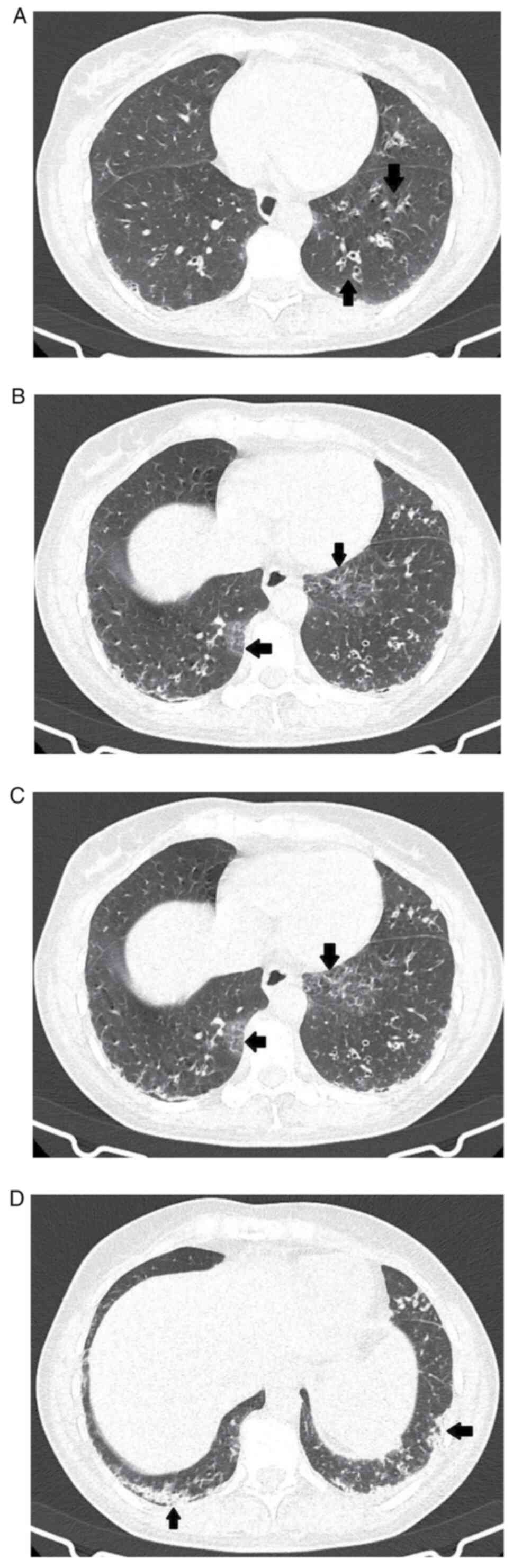
High resolution computerized tomography (CT) scan
revealed interlobular septal thickening, reticulations, tubular
bronchiectasis with thickened walls, some of which were with free
lumen and others occupied by mucus (Fig. 4A), accompanied by small areas of
pulmonary consolidation and ground glass alveolar opacities, which
were located predominantly in both lower lobes (posterior segments)
with peripheral topography (Fig.
4B and C). This imaging aspect
raised the suspicion of early-stage interstitial lung disease
(ILD). Moreover, isolated subpleural interstitial lung micronodules
were detected. The biggest micronodules measured 4 mm (in the
anterior segment of the right upper lobe) and 5 mm (in the lateral
segment of the middle lobe, subpleural in the lower segment of the
lingula), probably with an inflammatory substrate (Fig. 4D). The esophagus was presented as
having increased caliber in the whole trajectory (distended by air)
(data not shown).
Therefore, the diagnosis of MPA was formulated
considering the symptoms, the clinical findings and the high tither
of anti-MPO antibodies. Other vasculitides were excluded [the
patient had negative cytoplasmic (c)-ANCA, no pulmonary hemorrhage
and normal serum eosinophile count], as well as systemic lupus
erythematosus (SLE; normal serum complement level, absence of
proteinuria, normal blood count and absence of skin involvement)
and systemic sclerosis (negative anti-SCL70 and anti-centromere B
antibodies, normal nailfold capillaroscopy and absence of
proteinuria).
For the febrile episode, the patient was treated
with intravenous acetaminophen (1 g/day), intramuscular
dexamethasone (4 mg/day) and oral colchicine (1 mg/day) for 7 days,
with complete resolution of signs and symptoms.
After this initial treatment, when infection became
unlikely and immunology results indicated an autoimmune etiology,
the patient received intravenous dexamethasone (8 mg/day) and oral
treatment with pantoprazole (20 mg/day), potassium aspartate (39
mg/day) and magnesium aspartate tetrahydrate (12 mg/day) for 10
days. After the diagnosis of MPA was formulated, the patient
received a 3-day course of intravenous pulse-therapy with
methylprednisolone (500 mg/day), followed by oral
methylprednisolone (40 mg/day) and subcutaneous methotrexate (10
mg). After following this treatment, at discharge, the patient's
general condition improved with the remission of myalgia and
arthralgia.
At discharge, the patient was prescribed treatment
with subcutaneous methotrexate (15 mg/week, increased after 1 week
to 20 mg/week), combined with oral methylprednisolone (40 mg/day,
with progressive dose reduction and received together with
pantoprazole 20 mg/day) and oral diet supplements (folic acid 5
mg/day, potassium aspartate 39 mg/day, magnesium aspartate
tetrahydrate 12 mg/day and vitamin D 2000 IU/day).
After 5 months of treatment with progressive
decreasing methylprednisolone doses namely a decrease of 0.1-0.2
mg/kg every 2 weeks (for example, after taking 32 mg/day for 2
weeks, the dose was reduced to 24 mg/day for the following 2 weeks)
combined with methotrexate 20 mg/week, the patient's general
condition improved with absence of febrile state, arthralgia or
myalgia and remission of subcutaneous edema. Laboratory findings
after 5 months of follow-up showed the absence of biological
inflammatory syndrome and absence of anti-SCL70 and anti-centromere
antibodies. In addition, no renal damage was apparent, the patient
having normal serum creatinine levels and absence of proteinuria or
hematuria.
The absence of systemic sclerosis-specific
antibodies, both at the first evaluation and the subsequent
re-evaluations, and the normal aspect of the capillary bed at
nailfold capillaroscopy along with favorable clinical and
biological response to cortisone treatment, rendered the diagnosis
of systemic sclerosis unlikely.
Discussion
MPA is a rare disease that was initially considered
to be a microscopic form of polyarteritis nodosa (PAN) due to
similar clinical manifestations (6). In 1985, with the discovery of ANCA
antibodies, which are present in three types of vasculitis that
involve small vessels (MPA, GPA and EGPA) and are absent in PAN, a
differentiation between the two was possible (6). Thus, in 1994 at the International
Chapel Hill Consensus Conference (CHCC), MPA was defined as a
separate entity associated with MPO-ANCA, with absent immune
complex deposition and with the presence of pulmonary capillaritis
and glomerulonephritis (6-7). Subsequently, in 2012, the CHCC
revised the nomenclature of systemic vasculitis, and MPA was
defined as a form of necrotizing vasculitis, with minimal or
without immune deposits, which predominantly involves small vessels
(capillaries, venules and arterioles), but may also involve medium
vessels with absent granulomatous inflammation (1).
IIF can identify two major types of ANCAs: C-ANCA
and the perinuclear (p)-ANCA. Using enzyme-linked immunosorbent
assay (ELISA), c-ANCA was shown to be specific for PR3 (PR3-ANCA)
and p-ANCA to be specific for MPO (MPO-ANCA) (8). ANCAs are biomarkers used in the
diagnosis of small-vessel vasculitis (MPA, GPA and EGPA) that
should be detected using IIF, according to the 1999 international
consensus on ANCA testing (9). In
addition, in case of a positive result with IIF, a distinction
using ELISA should be made between the two types of ANCA,
anti-MPO-ANCA and anti-PR3-ANCA, due to the important clinical and
pathogenic implications (9).
The revised 2017 international consensus proposed by
a group of international experts (from the United States of
America, Europe, Asia and Australia) emphasizes the importance of
ANCA in diagnosis, but not as a follow-up tool for patients with
AAV (10). The same international
consensus recommends high-quality immunoassays for PR3-ANCAs and
MPO-ANCAs as the preferred method for diagnosing AAV. Moreover, it
does not consider IIF necessary. The recommendation applies to AAV
(particularly GPA and MPA) but does not apply to the diagnosis of
inflammatory bowel disease (IBD), immune hepatitis and drug-induced
autoimmunity (10).
Patients with AAV are usually seropositive for
PR3-ANCA or MPO-ANCA, but do not have both positive autoantibodies.
In MPA, it was found that 90% of patients are seropositive for ANCA
at diagnosis, ~55% being anti-MPO-ANCA positive. In GPA, at the
time of diagnosis, ANCA are present in 95% of patients, who are
mostly anti-PR3-ANCA positive (~65%). In EGPA, 40% of patients are
positive for ANCA, usually anti-MPO-ANCA (11,12).
ANCAs can also be found in other chronic inflammatory conditions,
such as IBD (seropositivity for p-ANCA in 50-67% of patients with
ulcerative colitis and 6-15% of those with Crohn's disease) and
liver disease [primary sclerosing cholangitis (88%), primary
biliary cirrhosis, autoimmune hepatitis (81%) and chronic viral
hepatitis]. In these diseases, p-ANCA is atypical and not anti-MPO
(13-17).
Certain studies confirm the simultaneous existence of AAV and IBD,
but the association is rare. In a previously reported case, IBD
occurred for a few years before the onset of AAV (18). ANCAs may also be positive in
rheumatoid arthritis, SLE, malignant hematological diseases
(19), as well as in infectious
endocarditis and tuberculosis (20-22).
In the current patient, the presence of p-ANCA was detected using
ELISA and IIF, followed by confirming the intense positivity of
MPO-ANCA through ELISA.
In 2022, an international group of researchers
formulated and validated several criteria for classifying and
differentiating the three types of AAV (23). These criteria have been approved by
the American College of Rheumatology and the European Alliance
Associations for Rheumatology. The study included 149 patients with
MPA and 408 healthy comparators. Out of 10 items identified by
regression analysis, the authors retained the following six
criteria: i) P-ANCA/MPO-ANCA positivity (+6); ii) pauci-immune
glomerulonephritis (+3); iii) PF or ILD (+3); iv) sino-nasal
symptoms or signs (-3); v) c-ANCA or PR3-ANCA positivity (-1); and
vi) eosinophil count ≥109/l (-4) (Table II). These criteria have a
sensitivity of 91% and a specificity of 94%. At a cumulative score
of ≥5 the patient is classified as having MPA. An important point
to note is that these criteria should be used after the diagnosis
of vasculitis of small or medium vessels and after other conditions
that mimic vasculitis have been excluded (23). Applying these criteria, the present
patient accumulated a total of nine points due to anti-MPO-ANCA
positivity, the presence of ILD and the absence of sino-nasal
symptoms or signs, renal damage and anti-PR3 antibodies, as well as
normal eosinophil count. An important observation of these criteria
reveals an equal score between renal and pulmonary involvement.
Thus, MPA can be classified as vasculitis of small/medium vessels
with PF or ILD and without renal impairment if there is positivity
for anti-MPO-ANCA, normal eosinophil count and the patient has no
sino-nasal symptoms or signs (Table
II).
| Table IIAmerican College of
Rheumatology/European Alliance Associations for Rheumatology 2022
classification criteria for microscopic polyangiitisa. |
Table II
American College of
Rheumatology/European Alliance Associations for Rheumatology 2022
classification criteria for microscopic polyangiitisa.
| Criteria | Score | Present
patient |
|---|
| ANCA/MPO-ANCA
positivity | +6 | +6 |
| Pauci-immune
glomerulonephritis | +3 | 0 |
| Lung
fibrosis/ILD | +3 | +3 |
| Sino-nasal
symptoms/signs | -3 | 0 |
| Cytoplasmic
ANCA/PR3-ANCA positivity | -1 | 0 |
| Eosinophil count
≥109/l | -4 | 0 |
| Total score | ≥+5 | +9 |
It is known that rapidly progressive
glomerulonephritis is a common manifestation of MPA. Renal
involvement has been indicated to occur in almost all cases in the
first series of reported cases of MPA, but according to the third
edition of the European Alliance Associations for Rheumatology
Textbook on Rheumatic diseases, this can be attributed to the fact
that the first cases were reported by nephrologists (24).
The results of a 2018 study on 97 patients diagnosed
with MPA meeting the CHCC 2012 criteria, which were followed up for
a median period of 47.6 months, showed the following: The median
age at the onset of symptoms was 50.7 years, 66% of patients were
positive for MPO-ANCA, 24.7% for PR3-ANCA and the remaining 9.3%
were undifferentiated ANCA. A total of 79.4% of patients had
pulmonary involvement, this being present in 55.8% of patients at
the time of diagnosis, the remaining 44.2% developing symptoms and
signs during follow-up. The most common identified CT patterns were
pulmonary infiltrates (50.5%) and ground glass opacities (40.2%).
Diffuse alveolar hemorrhage had been present since the onset of the
disease in 15.5% of patients, and it was developed by 30% of all
patients. PF was the most common involvement at the end of
follow-up, being present in 53.6% of patients. It was noted that,
at the end of follow-up, interstitial changes from the onset of the
disease were associated with the development of PF and
bronchiectasis. This study concluded that in patients with MPA the
signs of ILD were usually reversible, and predicted a higher
incidence of PF changes and bronchiectasis at the end of follow-up
(25).
A study conducted by a group of researchers from
Greece, published in 2010, included a group of 33 patients
diagnosed with MPA who were followed up for a mean of 38 months.
The authors reported that the most common manifestations were
nonspecific symptoms, such as fever, fatigue and weight loss. This
study demonstrated and emphasized that PF is a common manifestation
(39%) and a leading cause of death in patients with MPA. They also
concluded that PF may be manifested at the time of diagnosis (36%)
or may occur before other manifestations (3%) of MPA (26). Similarly, in the present case
report, the patient presented weight loss and episodes of fever.
The diagnosis of PF in the early stages was concomitant with the
diagnosis of MPA.
In a retrospective study performed in China, out of
67 MPA cases, 19 patients (28%) presented with PF with a median age
of 63.6 years (27). All patients
had non-specific biological inflammatory syndrome and were positive
for MPO-ANCA. The following were the most common manifestations:
Fever (89.5%), cough (84.2%), dyspnea (78.9%) and velcro rales
(84.2%). This study found that 36.8% of cases presented fever
before the diagnosis of PF and 63.2% after the diagnosis. In
addition, the remission of febrile episodes after the
administration of glucocorticoids was noticed in most cases, with
no benefit from antibiotic therapy. The patient of the present case
report presented two of the symptoms identified as the most common
in the aforementioned study, such as fever and velcro rales.
Regarding the febrile episode in the present case report, the
patient recovered after glucocorticoids without the use of
antibiotic therapy. In addition, apart from fever, the patient
showed no other clinical or biological signs of infection.
A retrospective review from Argentina of 28 patients
with MPA who were divided into two subgroups, with PF (MPA-PF) and
without PF (MPA-non-PF), revealed that 9 patients (32%) were
classified as MPA-PF. This subgroup had more respiratory symptoms
and higher mortality than the MPA-non-PF subgroup. In 5 patients
(17%) PF preceded other manifestations of vasculitis (28).
In a study on a cohort of 85 patients (47 men and 38
women) who met the CHCC 2012 criteria for MPA, it was found that in
addition to renal manifestations (78.8%), weight loss (72.9%), skin
changes (62.4%) and fever (55.3%), more than half of the patients
had joint pain (50.6%). Myalgias were present in 48.2% of patients
(29).
Following analysis of these studies, it can be
concluded that general symptoms, such as fever, fatigue or weight
loss, are common manifestations of MPA. The prevalence of ILD in
patients with MPA is quite prevalent worldwide, being common at the
time of diagnosis of MPA. Most patients are seropositive for
MPO-ANCA, but they may also be PR3-ANCA positive or have an
undifferentiated-ANCA disease.
In the present case report, although the initial
clinical presentation was not suggestive of a vasculitis-type
pathology, this diagnosis was later highlighted during
hospitalization by the cyanotic skin changes, the subcutaneous
edema associated with the febrile episode and the paraclinical
investigations. Unfortunately, lung biopsy was not performed
considering that the early diffuse interstitial lung lesions were
minimal and difficult to approach. In addition, the patient
required urgent treatment because of deteriorating condition.
Obtaining the result of a lung biopsy would have taken too long,
jeopardizing the clinical evolution. Thus, the main guiding
significance of the present case is that even in the absence of
renal damage the diagnosis of MPA should not be excluded by the
clinician.
In conclusion, MPA is a necrotizing systemic
vasculitis that affects small and medium-sized vessels and has long
been thought to affect the kidneys most frequently. The current
case report presented a patient with MPA, with constitutional and
musculoskeletal symptoms, atypical skin changes, intense positivity
for MPO-ANCA, in whom renal dysfunction was absent, while ILD was
present.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
DIM, AM and AR participated in the design of the
case report and wrote the manuscript. DIM, ALC and CP were
responsible for the patient evaluation and management. VG, MD and
DGM were responsible for analysis and interpretation of data and
critically reviewed the manuscript. All authors confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient prior to publication at the time of admission.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jennette JC, Falk RJ, Bacon PA, Basu N,
Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen
EC, et al: 2012 revised international Chapel Hill consensus
conference nomenclature of vasculitides. Arthritis Rheum. 65:1–11.
2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mohammad AJ, Jacobsson LT, Mahr AD,
Sturfelt G and Segelmark M: Prevalence of Wegener's granulomatosis,
microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss
syndrome within a defined population in southern Sweden.
Rheumatology. 46:1329–1337. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Mohammad AJ, Jacobsson LT, Westman KW,
Sturfelt G and Segelmark M: Incidence and survival rates in
Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss
syndrome and polyarteritis nodosa. Rheumatology (Oxford).
48:1560–1565. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Watts RA, Lane SE, Scott DG, Koldingsnes
W, Nossent H, Gonzalez-Gay MA, Garcia-Porrua C and Bentham GA:
Epidemiology of vasculitis in Europe. Ann Rheum Dis. 60:1156–1157.
2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Greco A, De Virgilio A, Rizzo MI, Gallo A,
Magliulo G, Fusconi M, Ruoppolo G, Tombolini M, Turchetta R and de
Vincentiis M: Microscopic polyangiitis: Advances in diagnostic and
therapeutic approaches. Autoimmun Rev. 14:837–844. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
van der Woude FJ, Rasmussen N, Lobatto S,
Wiik A, Permin H, van Es LA, van der Giessen M, van der Hem GK and
The TH: Autoantibodies against neutrophils and monocytes: tool for
diagnosis and marker of disease activity in Wegener's
granulomatosis. Lancet. 1:425–429. 1985.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Jennette JC, Falk RJ, Andrassy K, Bacon
PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg
CG, et al: Nomenclature of systemic vasculitides. Proposal of an
international consensus conference. Arthritis Rheum. 37:187–192.
1994.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Jennette JC and Falk RJ: Antineutrophil
cytoplasmic autoantibodies and associated diseases: A review. Am J
Kidney Dis. 15:517–529. 1990.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Savige J, Gillis D, Benson E, Davies D,
Esnault V, Falk RJ, Hagen EC, Jayne D, Jennette JC, Paspaliaris B,
et al: International consensus statement on testing and reporting
of antineutrophil cytoplasmic antibodies (ANCA). Am J Clin Pathol.
111:507–513. 1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bossuyt X, Cohen Tervaert JW, Arimura Y,
Blockmans D, Flores-Suárez LF, Guillevin L, Hellmich B, Jayne D,
Jennette JC, Kallenberg CGM, et al: Revised 2017 international
consensus on testing of ANCAs in granulomatosis with polyangiitis
and microscopic polyangiitis. Nat Rev Rheumatol. 13:683–692.
2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hagen EC, Daha MR, Hermans J, Andrassy K,
Csernok E, Gaskin G, Lesavre P, Lüdemann J, Rasmussen N, Sinico RA,
et al: Diagnostic value of standardized assays for anti-neutrophil
cytoplasmic antibodies in idiopathic systemic vasculitis for the
EC/BCR project for ANCA assay standardisation. Kidney Int.
53:743–753. 1998.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sablé-Fourtassou R, Cohen P, Mahr A,
Pagnoux C, Mouthon L, Jayne D, Blockmans D, Cordier JF, Delaval P,
Puechal X, et al: Antineutrophil cytoplasmic antibodies and the
Churg-Strauss syndrome. Ann Intern Med. 143:632–638.
2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bossuyt X: Serologic markers in
inflammatory bowel disease. Clin Chem. 52:171–181. 2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Terjung B: Anti-neutrophil antibodies in
primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol.
15:629–642. 2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Terjung B, Bogsch F, Klein R, Söhne J,
Reichel C, Wasmuth JC, Beuers U, Sauerbruch T and Spengler U:
Diagnostic accuracy of atypical p-ANCA in autoimmune hepatitis
using ROC-and multivariate regression analysis. Eur J Med Res.
9:439–448. 2004.PubMed/NCBI
|
|
16
|
Roozendaal C, de Jong MA, van den Berg AP,
van Wijk RT, Limburg PC and Kallenberg CG: Clinical significance of
anti-neutrophil cytoplasmic antibodies (ANCA) in autoimmune liver
diseases. J Hepatol. 32:734–741. 2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Terjung B, Worman HJ, Herzog V, Sauerbruch
T and Spengler U: Differentiation of antineutrophil nuclear
antibodies in inflammatory bowel and autoimmune liver diseases from
antineutrophil cytoplasmic antibodies (p-ANCA) using
immunofluorescence microscopy. Clin Exp Immunol. 126:37–46.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Humbert S, Guilpain P, Puéchal X, Terrier
B, Rivière S, Mahr A, Pagnoux C, Bagnères D, Cordier JF, Le Quellec
A, et al: Inflammatory bowel diseases in anti-neutrophil
cytoplasmic antibody-associated vasculitides: 11 retrospective
cases from the French vasculitis study group. Rheumatology
(Oxford). 54:1970–1975. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Philipponnet C, Garrouste C, Le Guenno G,
Cartery C, Guillevin L, Boffa JJ and Heng AE: Antineutrophilic
cytoplasmic antibody-associated vasculitis and malignant
hemopathies, a retrospective study of 16 cases. Joint Bone Spine.
84:51–57. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Weiner M and Segelmark M: The clinical
presentation and therapy of diseases related to anti-neutrophil
cytoplasmic antibodies (ANCA). Autoimmune Rev. 15:978–982.
2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Langlois V, Lesourd A, Girszyn N, Ménard
JF, Levesque H, Caron F and Marie I: Antineutrophil cytoplasmic
antibodies associated with infectious endocarditis. Medicine
(Baltimore). 95(e2564)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Flores-Suárez LF, Cabiedes J, Villa AR,
van der Woude FJ and Alcocer-Varela J: Prevalence of antineutrophil
cytoplasmic autoantibodies in patients with tuberculosis.
Rheumatology (Oxford). 42:223–229. 2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Suppiah R, Robson JC, Grayson PC, Ponte C,
Craven A, Khalid S, Judge A, Hutchings A, Merkel PA, Luqmani RA, et
al: 2022 American college of rheumatology/european alliance of
associations for rheumatology classification criteria for
microscopic polyangiitis. Ann Rheum Dis. 81:321–326.
2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
EULAR Textbook on Rheumatic diseases.
Third Edition. BMJ Publishing Group, London, 2018.
|
|
25
|
Shchegoleva E, Zykova A, Bulanov N,
Meshkov A, Novikov P and Moiseev S: Lung damage in patients with
microscopic polyangiitis. Ann Rheumatic Dis. 77(766)2018.
|
|
26
|
Tzelepis GE, Kokosi M, Tzioufas A, Toya
SP, Boki KA, Zormpala A and Moutsopoulos HM: Prevalence and outcome
of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J.
36:116–121. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Huang H, Wang YX, Jiang CG, Liu J, Li J,
Xu K and Xu ZJ: A retrospective study of microscopic polyangiitis
patients presenting with pulmonary fibrosis in China. BMC Pulm Med.
14(8)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fernandez Casares M, Gonzalez A, Fielli M,
Caputo F, Bottinelli Y and Zamboni M: Microscopic polyangiitis
associated with pulmonary fibrosis. Clin Rheumatol. 34:1273–1277.
2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Guillevin L, Durand-Gasselin B, Cevallos
R, Gayraud M, Lhote F, Callard P, Amouroux J, Casassus P and
Jarrousse B: Microscopic polyangiitis: Clinical and laboratory
findings in eighty-five patients. Arthritis Rheum. 42:421–430.
1999.PubMed/NCBI View Article : Google Scholar
|