Introduction
Acute myocardial infarction (AMI) is acute ischemic
necrosis of myocardium occurring due to coronary artery disease,
which can lead to fatal complications, and even mortality in severe
cases (1). Since the beginning of
the 21st century, with the changes of the lifestyle and diet of the
public, the incidence of AMI has been increasing sharply (2). There are >3 million cases of MI in
China (3). The mortality rate of
cardiovascular diseases continues to increase. Annual number of
deaths owing to cardiovascular diseases increased from 2.51 million
to 3.97 million between 1990 and 2016 in China (4). Although the timely implementation of
thrombolytic drugs and interventional surgery permits coronary
artery recanalization, which is the only effective clinical
treatment method at present (5,6),
coronary artery recanalization itself can lead to a severe
inflammatory response, and continue to expand infarction lesions by
>50% (compared with original infarct size), leading to
irreversible injury, namely myocardial ischemia/reperfusion injury
(MIRI) (7,8). Therefore, it is of importance to find
a solution to alleviate heart injury and dysfunction caused by
I/R.
Roflumilast is a phosphodiesterase-4 (PDE-4)
inhibitor. The US Food and Drug Administration has approved
roflumilast for the treatment of severe chronic obstructive
pulmonary disease because of its strong anti-inflammatory and
immunomodulatory properties (9).
Studies have shown that roflumilast prevents ischemic
stroke-induced neuronal damage (10) and alleviates sepsis-induced acute
kidney injury (11). In addition,
roflumilast can protect myocardial cells from nitric oxide-induced
apoptosis (12) and decrease
cadmium-induced cardiotoxicity by inhibiting oxidative stress
(13) and doxorubicin-induced
cardiotoxicity by decreasing inflammation (14). These results indicate that
roflumilast has cardioprotective functions; to the best of our
knowledge, however, its role in MIRI has not been reported.
Mitochondrial dysfunction is a key cause of MIRI and
the main mechanisms include decreased mitochondrial ATP production
(15), excessive reactive oxygen
species (ROS) production and continuous mitochondrial permeability
transition pore (mPTP) opening (16). Roflumilast can reduce mitochondrial
dysfunction in smog-induced pulmonary bronchial epithelial cells by
downregulating phosphorylated (p)-dynamin-related protein 1 (DRP1)
and PTEN-induced kinase 1 (PINK1) (17). Therefore, it may be hypothesized
that roflumilast may also alleviate mitochondrial dysfunction
caused by MIRI. In addition, AMP-activated protein kinase (AMPK)
signaling is an intracellular energy sensor and the activation of
the AMPK signaling pathway can decrease mitochondrial damage.
Activation of AMPK inactivates DRP1 to inhibit mitochondrial
fission, thus preventing the opening of mPTP and contributing to
cell survival (18). Activation of
AMPK decreases oxidative stress induced by I/R (19) and roflumilast prevents diabetic
nephropathy by activating AMPK/sirtuin 1 (SIRT1) (20).
Therefore, the aim of the present study was to
investigate the effect of roflumilast on MIRI as well as to discuss
the underlying mechanisms. It was hypothesized that roflumilast
could alleviate MIRI by improving mitochondrial dysfunction by
activating the AMPK signaling pathway.
Materials and methods
PubChem
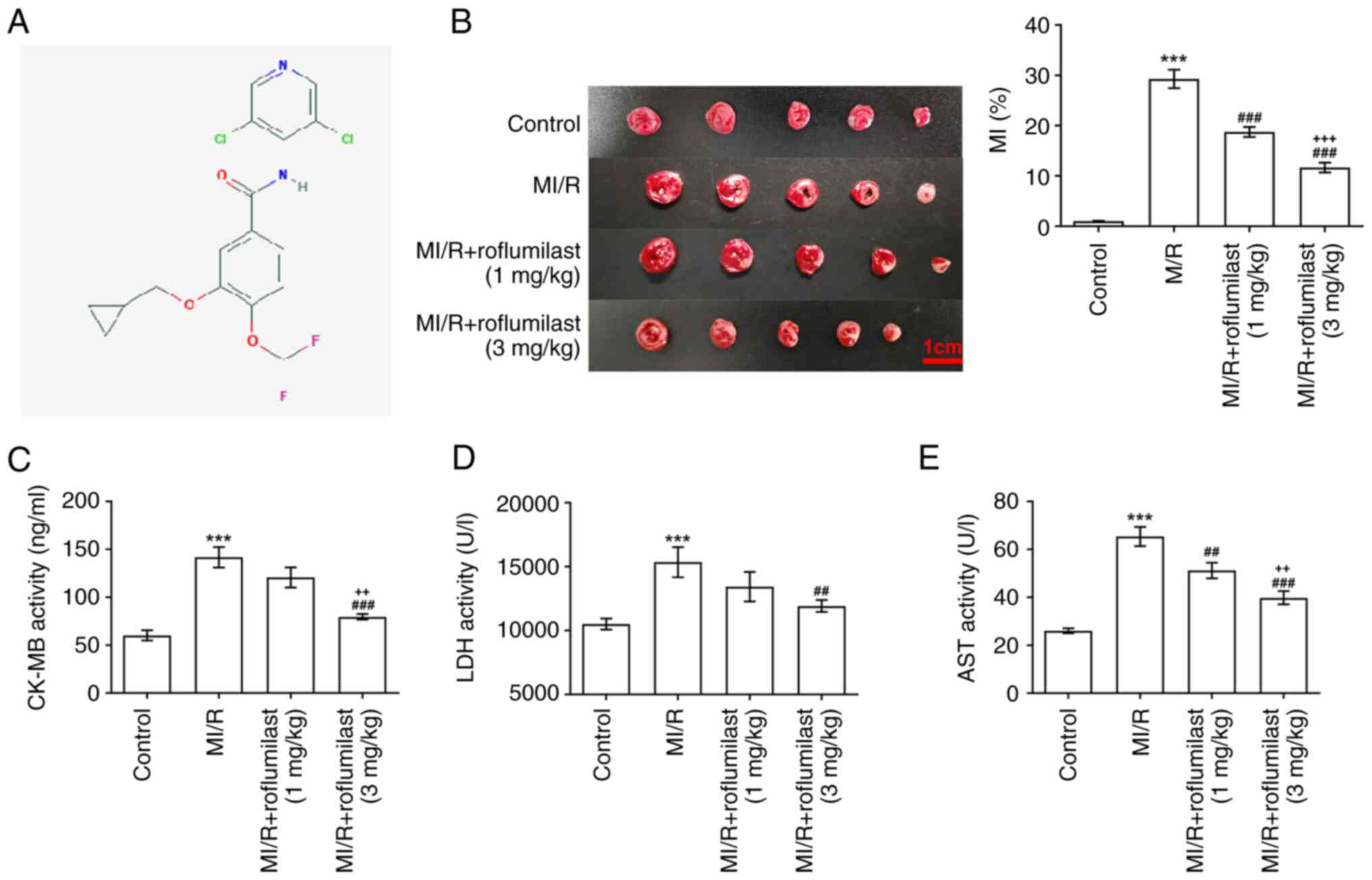
The chemical structure of roflumilast was determined
by PubChem (pubchem.ncbi.nlm.nih.gov/; Fig. 1A).
MI/R rat model
The experimental protocol for animal studies was
reviewed and approved by the Committee for the Ethics of Animal
Experiments, Shenzhen Peking University-The Hong Kong University of
Science and Technology Medical Center (approval no. 2021-807;
Shenzhen, China). The MI/R rat model was established as previously
described (21). A total of 20
male Sprague-Dawley rats (age, 12 weeks; weight, 180-250 g) were
purchased from Beijing Vital River Laboratory Animal Technology
Co., Ltd. The rats were raised in an environmentally controlled
room (22±2˚C, humidity of 55±5%, 12/12-h light/dark cycle) with
free access to standard animal feed and filtered tap water for 7
days. Rats were anesthetized by intraperitoneal injection of 10%
chloral hydrate (0.3 ml/100 g) and the intercostal space was opened
under mechanical ventilation. The animals exhibited no signs of
peritonitis, pain or discomfort. To establish the MI/R rat model,
the left anterior descending coronary artery was ligated for 45
min, followed by reperfusion for 2 h. Rats in the control group
were not ligated. The rats were randomly divided into four groups
(n=5/group): Control; MI/R, MI/R + roflumilast (1 mg/kg; Adooq
Bioscience) and MI/R + roflumilast (3 mg/kg) group. Prior to MI/R
operation, roflumilast was administered to rats by gavage at 1 or 3
mg/kg once daily for 7 consecutive days. Rats were humanely
sacrificed under anesthesia by intraperitoneal injection of 1%
pentobarbital sodium (150 mg/kg). The blood samples (~6 ml)
collected from the hearts and cardiac tissues were stored at -80˚C
until further analysis.
Hypoxia/reoxygenation (H/R) injury
induction in vitro and roflumilast treatment
H9C2 cells were obtained from the American Tissue
Culture Collection (ATCC; cat. no. CRL-1446). H9C2 cells were
cultured in ATCC-formulated Dulbecco's Modified Eagle's Medium
(cat. no. ATCC 30-2002) supplemented with 10% fetal bovine serum
(cat. no. ATCC 30-2020) in 95% air and 5% CO2 at
37˚C.
For H/R stimulation, H9C2 cells were grown in an
anoxic chamber with 5% CO2 and 95% N2 for 6 h
and then in a normal chamber with 95% air and 5% CO2 for
12 h at 37˚C.
For roflumilast treatment, cultured cells were
pre-incubated with roflumilast (1.0, 2.5 and 5.0 µM; Adooq
Bioscience) or compound C (10 µM; an inhibitor of the AMPK
signaling pathway; MedChemExpress, United States) for 30 min at
37˚C before H/R treatment.
2,3,5-triphenyltetrazolium chloride
(TTC) staining
The MI area in each group was observed by TTC
staining. Following storage at -18˚C for 15 min, the heart tissue
perpendicular to the coronary sulcus was cut into five equal
pieces. Following incubation with 1% TTC solution (Sigma-Aldrich;
Merck KGaA) for 10 min at 37˚C and 10% neutral buffered formalin
(Thermo Fisher Scientific, Waltham, MA) for 90 min at 37˚C in the
dark, all slices (1 mm) were imaged using a digital camera. The MI
areas were determined by Image-Pro Plus image analysis software
(version 4.1; Media Cybernetics, Inc.). Finally, calculation of MI
area was conducted according to the following formula: Myocardial
infarct size (%)=(infarct area/whole heart area) x100%.
Hematoxylin and eosin (H&E)
staining
The cardiac tissue samples collected from the left
ventricle were fixed in 4% paraformaldehyde overnight at 4˚C,
dehydrated in ascending ethanol gradient and embedded in paraffin
for 50-60 min at room temperature. Subsequently, the embedded
cardiac tissues were cut into 4 µm slices, followed by staining
with 0.5% hematoxylin for 5 min and eosin for 2 min at room
temperature. Then, the sections were mounted and observed under a
light microscope (magnification, x100/400; Olympus Corporation
BX53).
Mitochondrial membrane potential
detection
The mitochondrial membrane potential in cardiac
tissue and H9C2 cells was detected using the JC-1 staining kit
(cat. no. C2006; Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. In brief, the isolated
cardiomyocyte suspension and H9C2 cells were stained with 2.5 mg/ml
JC-1 solution for 20 min at 37˚C. Subsequently, the cells were
washed with JC-1 staining buffer twice and observed using a
fluorescence microscope (magnification, x200; Olympus Corporation
BX63).
Detection of myocardial enzyme levels
in serum
The collected peripheral blood (15 ml) was
centrifuged at 2,072 x g at 4˚C for 10 min to separate the serum.
Heart muscle damage indicators, including aspartate transaminase
(AST), creatine kinase-myocardial band (CK-MB) and lactate
dehydrogenase (LDH) in serum were detected by AST (cat. no.
C010-2-1), CK-MB (cat. no. A032-1-1) and LDH (cat. no. A020-2-2)
assay kits (all Nanjing Jiancheng Bioengineering Institute)
according to the manufacturer's instructions, respectively.
Detection of inflammatory
cytokines
The concentrations of IL-1β and IFN-γ in myocardial
tissue and in cell supernatant were measured using ELISA kits for
IL-1β (cat. no. E-EL-R0012c; Elabscience Biotechnology, Inc.) and
IFN-γ (cat. no. E-EL-R0009c; Elabscience Biotechnology, Inc.)
according to the manufacturer's instructions.
Detection of oxidative stress
The activity of malondialdehyde (MDA) and superoxide
dismutase (SOD) in myocardial tissue and H9C2 cells were measured
using MDA (cat. no. S0131S; Beyotime Institute of Biotechnology)
and SOD assay kits (cat. no. S0109; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions.
Detection of ATP levels
The detection of ATP concentration in H9C2 cells was
conducted using ATP assay kit (cat. no. S0026; Beyotime Institute
of Biotechnology) according to the manufacturer's instructions.
Briefly, H9C2 cells were collected and mixed with cell lysis buffer
for 10 min at 4˚C, followed by centrifugation at 12,000 x g at 4˚C
for 5 min. Subsequently, cell supernatant was incubated with 100 µl
kit solution at room temperature for 5 min and the ATP levels in
the cell supernatant were detected using a LuminMax-C luminometer
(Maxwell Sensors Inc.).
Western blotting
Protein from cardiac tissues and H9C2 cells was
obtained using RIPA lysis buffer (Beyotime Institute of
Biotechnology) and qualified with a BCA detection kit (Beyotime
Institute of Biotechnology). A total of 25 µg/lane protein was
separated by 10% SDS-PAGE and transferred to a PVDF membrane.
Following blocking with 5% BSA (Beyotime Institute of
Biotechnology) at room temperature for 2 h, the membrane was
incubated with primary antibodies targeting AMP-activated protein
kinase alpha (AMPKα; cat. no. 5831; 1:1,000; Cell Signaling
Technology, Inc.), phosphorylated (p-)AMPKα (cat. no. 50081;
1:1,000; Cell Signaling Technology, Inc.), SIRT1 (cat. no.
ab189494; 1:1,000; Abcam), Bcl-2 (cat. no. ab196495; 1:1,000;
Abcam), Bax (cat. no. ab32503; 1:1,000; Abcam), PINK1 (cat. no.
ab186303; 1:1,000; Abcam), DRP1 (cat. no. 8570; 1:1,000; Cell
Signaling Technology, Inc.), p-DRP1 (cat. no. 4867; 1:1,000; Cell
Signaling Technology, Inc.) and β-actin (cat. no. 93473; 1:1,000;
Cell Signaling Technology, Inc.) overnight at 4˚C. Then, membranes
were incubated with horseradish peroxidase-conjugated goat
anti-rabbit IgG (cat. no. ab6721; 1:2,000; Abcam) or goat
anti-mouse IgG (cat. no. ab6789; 1:2,000; Abcam) for 1 h at room
temperature. Finally, the bands were examined with ECL reagent
(Beyotime Institute of Biotechnology) and band density was
quantified using ImageJ Software (version 1.46; National Institutes
of Health).
Cell Counting Kit-8 (CCK-8) assay
H9C2 cells were inoculated into a 96-well plate at a
density of 1x103 cells/well. H9C2 cells were treated
with roflumilast (1.0, 2.5 and 5.0 µM) for 24 h at 37˚C and induced
by H/R. H9C2 cells in each well were mixed with 10 µl CCK-8
solution (Beyotime Institute of Biotechnology) and incubated at
37˚C for 1 h. The optical density at 450 nm was detected by a
spectrophotometer (Bio-Rad Laboratories, Inc.).
TUNEL assay
The apoptosis of H9C2 cells was determined by In
Situ Cell Death Detection kit (cat. no. 11684795910; Roche) for
the TUNEL assay. Briefly, H9C2 cells in a 24-well plate
(1x105 cells/well) were fixed in 4% paraformaldehyde at
room temperature for 15 min, then 0.1% Triton-X-100 at room
temperature for 10 min. Then, cells on the slides were stained with
TUNEL reaction mixture (50 µl terminal deoxynucleotidyl-transferase
and 450 µl fluorescein-labeled deoxyuridine triphosphate) at 37˚C
for 1 h in the dark and nuclei were stained with 10 µg/ml DAPI at
room temperature for 5 min. The cells were mounted with PBS and
glycerol (ratio, 1:2) and observed by fluorescence microscopy in
five randomly selected high-power microscope fields (magnification,
x200). The formula used to calculate the percentage of cell
apoptosis was as follows: Cell apoptosis (%)=(number of apoptotic
H9C2 cells/total number of H9c2 cells) x100%.
mPTP opening assay
The opening of the mPTP was detected using a
calcein-loading/cobalt chloride (CoCl2)-quenching
system. Briefly, 2x105 H9C2 cells seeded in a 6-well
plate were treated with 1 µM calcein and 2 mM CoCl2 for
20 min at 37˚C in the dark. After washing with PBS, H9C2 cells were
observed and imaged using a confocal laser scanning microscope
(model no. LSM 880; Carl Zeiss AG; magnification, x100). The mean
green fluorescence intensities in the mitochondria were quantified
using ImageJ Software (version 1.46; National Institutes of
Health). The changes of green fluorescence intensity in the
mitochondria were the index of mPTP opening.
Statistical analysis
GraphPad (version 8.0.1; GraphPad Software, Inc.;
Dotmatics) was used to analyze the experimental data. Data are
shown as the mean ± the standard deviation from three independent
experiments. The comparisons between multiple groups were conducted
by one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Roflumilast decreases MI/R-induced
MI
The chemical structure of roflumilast is shown in
Fig. 1A. TTC staining results
showed that the infarct area in the MI/R group was significantly
increased compared with that in the control group, indicating that
the rat model of MI/R was successfully constructed. Moreover,
roflumilast (1 and 3 mg/kg) significantly decreased the increased
infarct size in rats subjected to MIRI in a dose-dependent manner
(Fig. 1B). Furthermore, the
increased levels of CK-MB (Fig.
1C), LDH (Fig. 1D) and AST
(Fig. 1E) in the MI/R group were
decreased by treatment of roflumilast.
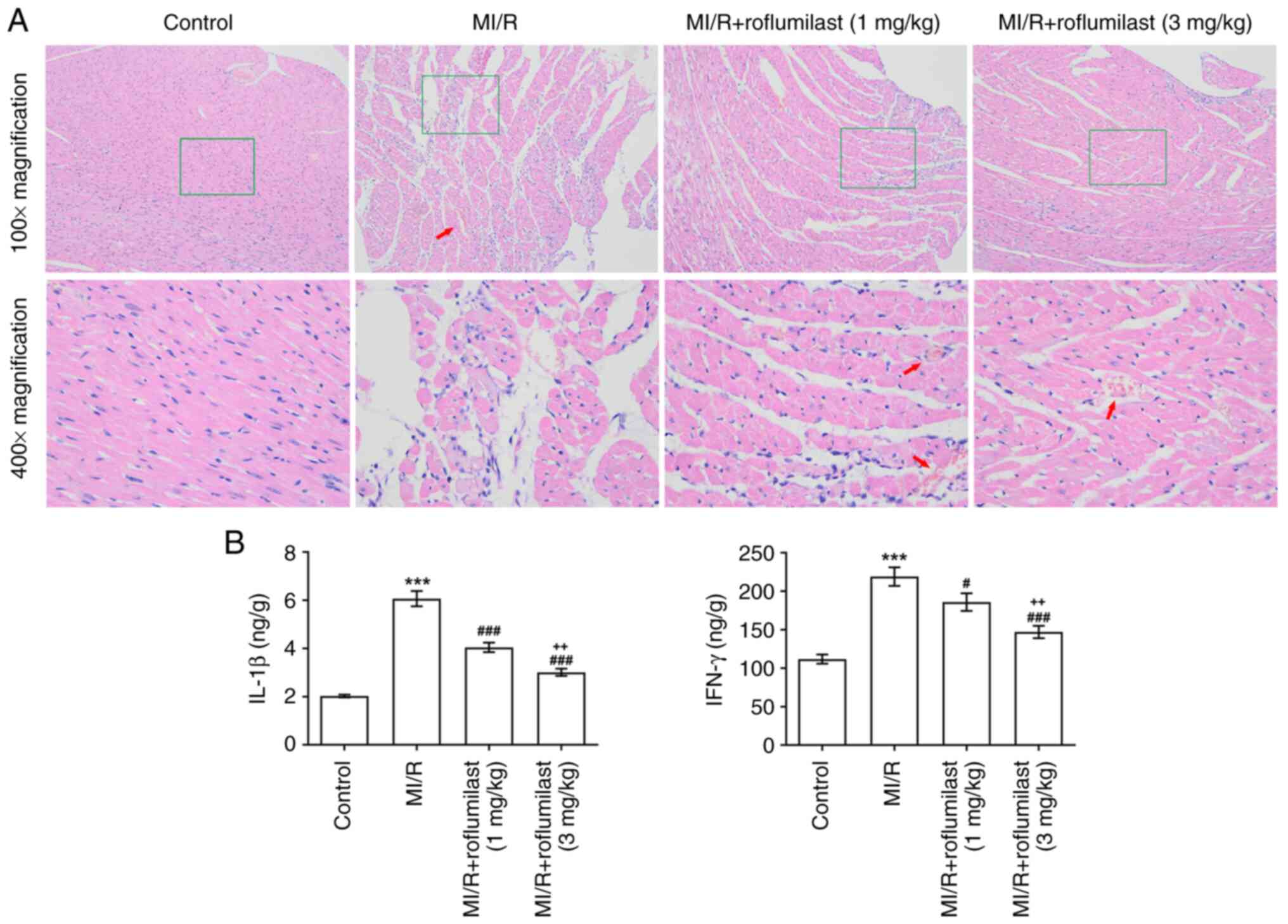
Roflumilast attenuates MI/R-induced
myocardial injury
The H&E staining results revealed that the
myocardial fiber structure was damaged, vascular walls were broken
and hemocyte infiltration was apparent in the MI/R group. However,
MI/R rats pre-injected with roflumilast exhibited mild tissue
damage (Fig. 2A). The inflammatory
factors (IL-1β and IFN-γ) in cardiac tissues were significantly
increased in the MI/R group but significantly reduced by
roflumilast treatment (Fig.
2B).
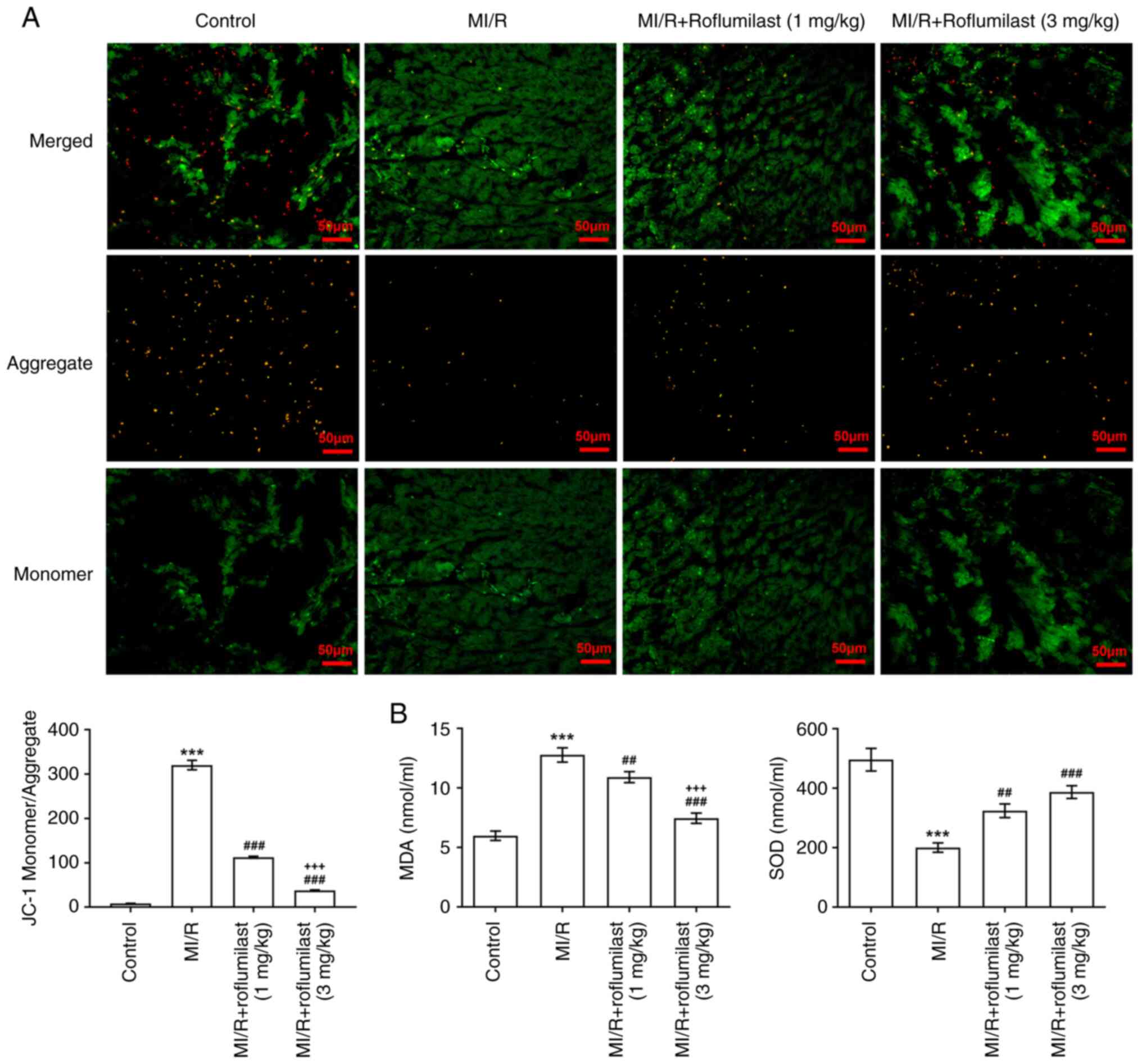
Roflumilast attenuates myocardial
mitochondrial damage induced by MI/R
The ratio of green and red fluorescence intensity
was significantly increased in MI/R rats compared with control
rats; this was reversed by roflumilast treatment, indicating that
roflumilast decreased depolarization of the mitochondrial membrane
(Fig. 3A). In addition, MDA was
significantly increased and SOD was significantly decreased in the
cardiac tissues of rats subjected to MI/R; these effects were
subsequently reversed by roflumilast (Fig. 3B).
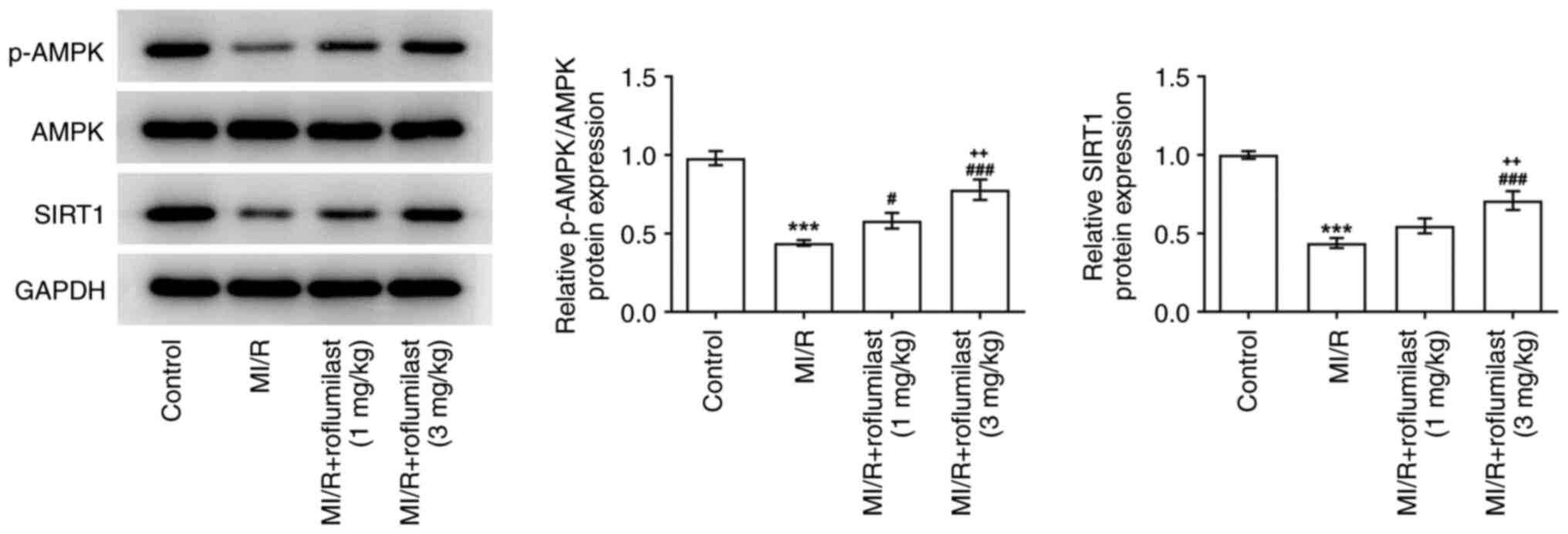
Roflumilast activates the AMPK
signaling pathway in MIRI
The expression of p-AMPK and SIRT1 in the cardiac
tissue of rats subjected to MI/R were significantly decreased
compared with the control group, while roflumilast treatment
promoted the expression of p-AMPK and SIRT1 in a dose-dependent
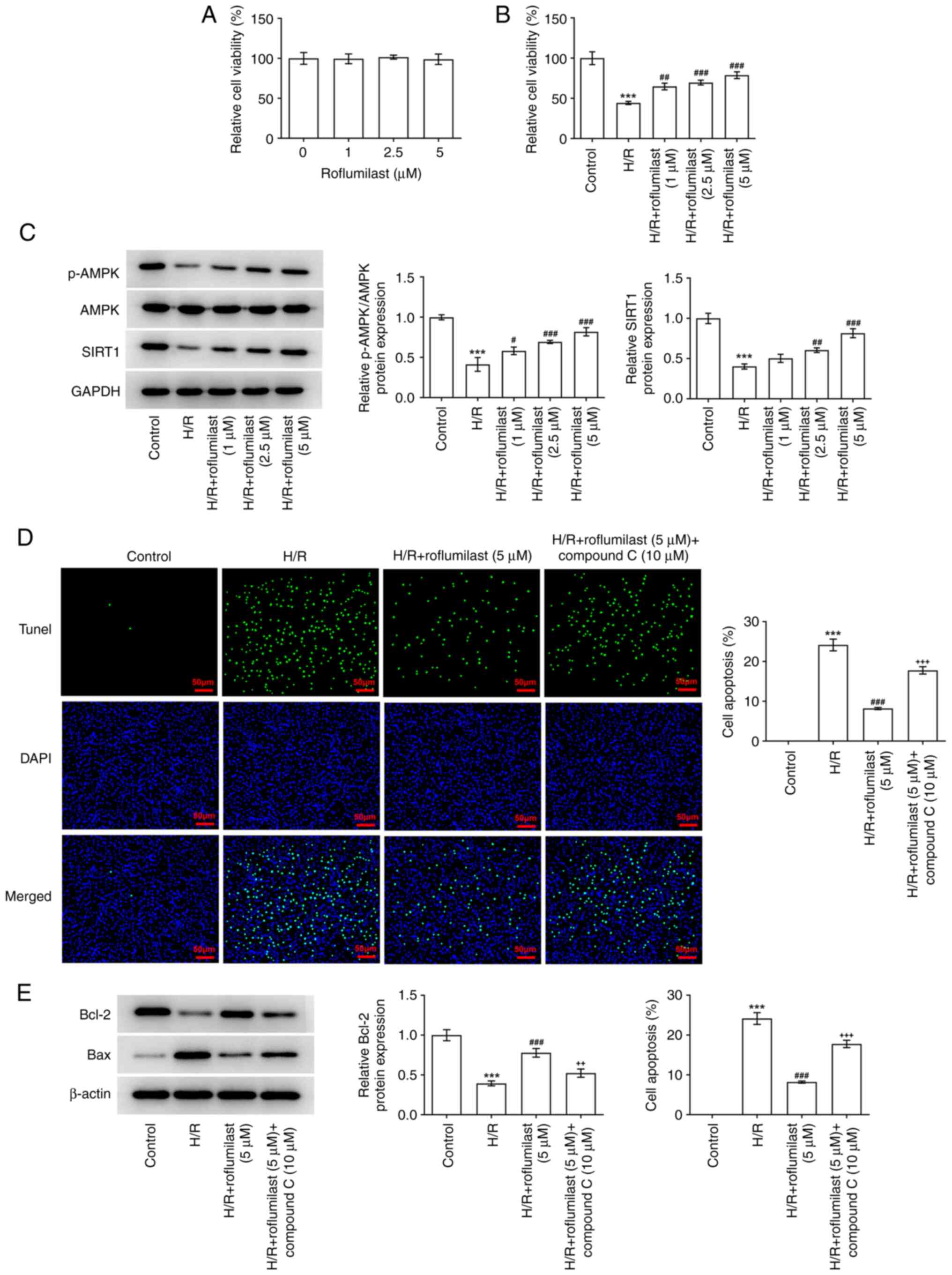
manner (Fig. 4).
Roflumilast mitigates damage of H/R to
H9C2 cell viability by activating the AMPK signaling pathway
After H9C2 cells were treated with roflumilast,
their viability was unchanged, indicating that roflumilast has no
significant effect on H9C2 cells at these concentrations (Fig. 5A). H/R-induced H9C2 cells showed
significantly decreased viability, which was reversed by
roflumilast in a dose-dependent manner (Fig. 5B). H/R induction significantly
decreased the expression of p-AMPK and SIRT1, while roflumilast
promoted the expression of p-AMPK and SIRT1 in H/R-induced H9C2
cells (Fig. 5C). H9C2 cells
subjected to H/R induction demonstrated a significant increase in
the number of TUNEL-positive/apoptotic cells; this was
significantly decreased following treatment with roflumilast, while
compound C weakened the effect of roflumilast (Fig. 5D). Roflumilast resulted in the
increased expression of Bcl-2 and decreased expression of Bax in
H/R induced H9C2 cells, which was then reversed by compound C
(Fig. 5E).
Roflumilast activates the AMPK
signaling pathway to alleviate oxidative stress and the
inflammatory response of H9C2 cells induced by H/R
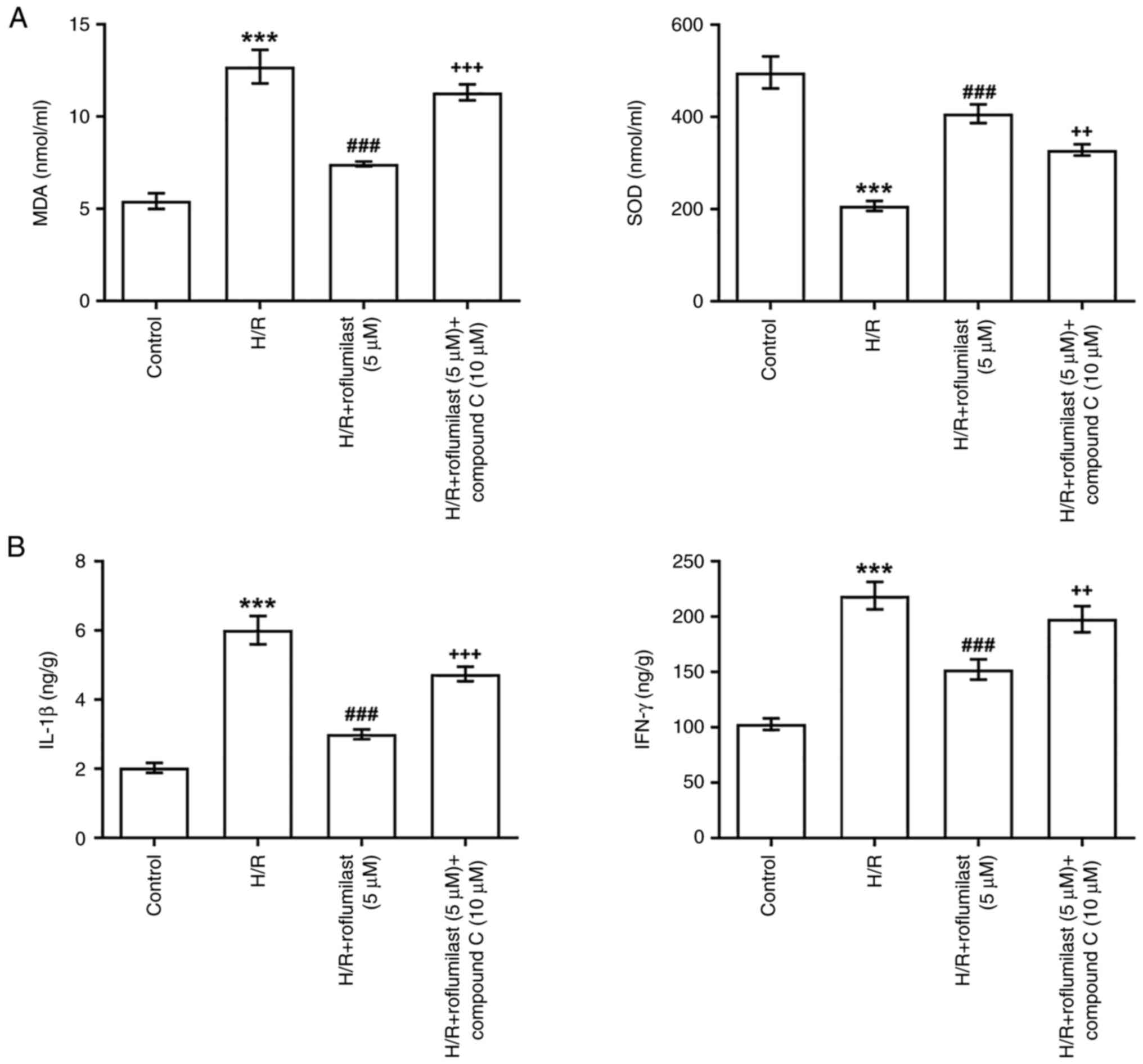
Roflumilast significantly suppressed MDA and
significantly upregulated SOD in H/R-induced H9C2 cells, while
compound C significantly impaired the function of roflumilast
(Fig. 6A). ELISA revealed a
significant increase in levels of IL-1β and IFN-γ in the H/R group
compared with the control group, which were significantly decreased
by roflumilast treatment. However, the decreased levels of IL-1β
and IFN-γ in the H/R + roflumilast (5 µM) group were partially
elevated by compound C (Fig.
6B).
Roflumilast activates the AMPK
signaling pathway to decrease mitochondrial damage of H9C2 cells
induced by H/R
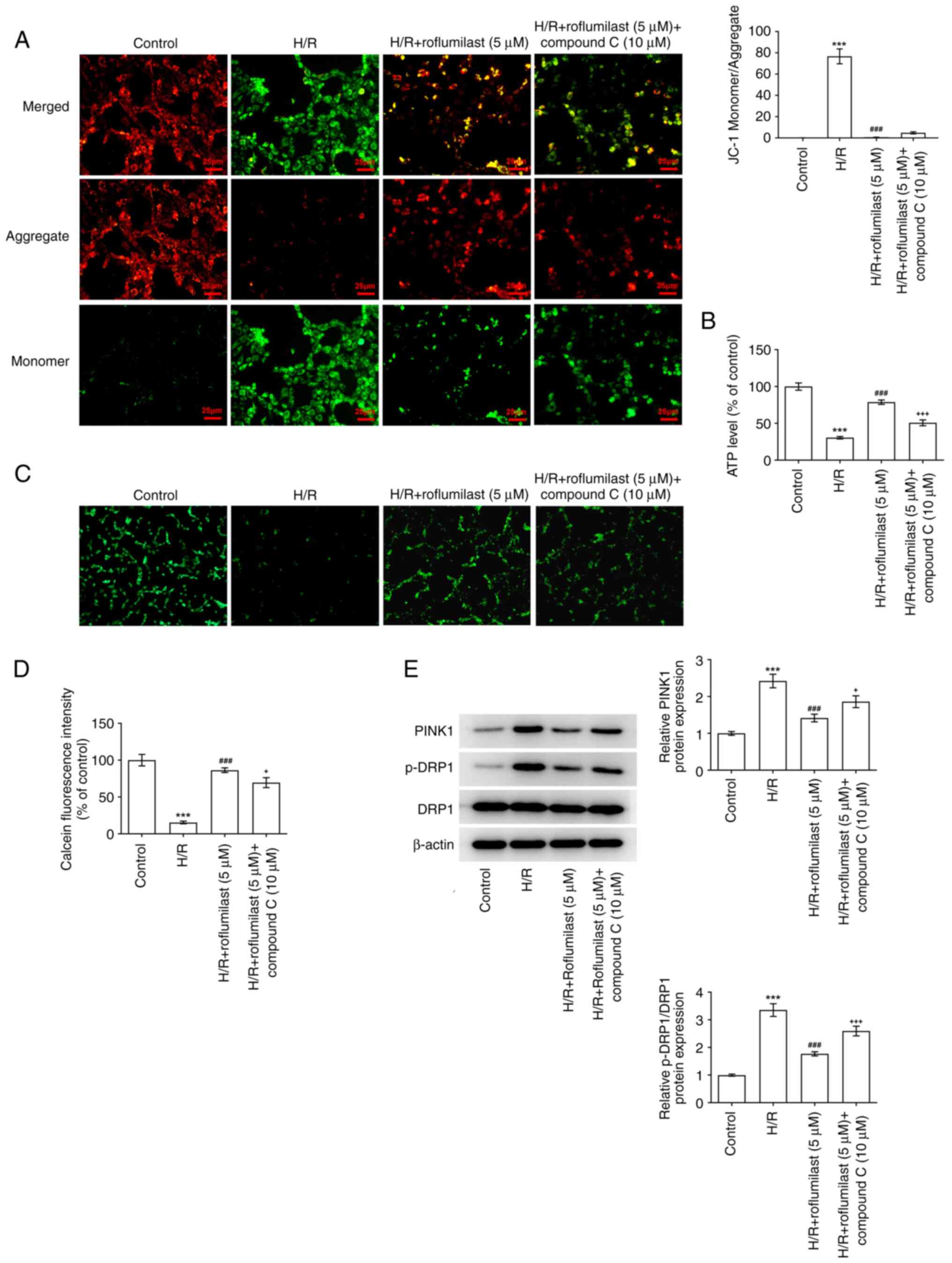
Roflumilast treatment significantly decreased the
ratio of green and red fluorescence intensity in H/R-induced H9C2
cells compared with that in the H/R group; this was increased
following the administration of compound C (Fig. 7A). H/R induction significantly
decreased the ATP levels, which were promoted by roflumilast.
Compound C decreased the ATP levels in H/R + roflumilast + compound
C group compared with the H/R + roflumilast group (Fig. 7B). Compared with the control, the
fluorescence intensity of mitochondrial calcein was significantly
decreased in the H/R group, indicating that the extent of mPTP
opening was enhanced following H/R. Moreover, roflumilast
significantly increased fluorescence intensity compared with the
H/R group, indicating that roflumilast inhibited H/R-induced mPTP
opening in H9C2 cardiomyocytes, which was reversed by compound C
(Fig. 7C and D). The enhanced expression of PINK1 and
p-DRP1 in H9C2 cells due to H/R induction were suppressed by
roflumilast treatment; these effects were then reversed by compound
C (Fig. 7E).
Discussion
The present study demonstrated that roflumilast
treatment decreased MIRI in vivo and in vitro by
activating the AMPK signaling pathway. Furthermore, mechanistic
investigations demonstrated that compound C, an inhibitor of the
AMPK signaling pathway, reversed the protective effects of
roflumilast on MIRI in vivo and in vitro.
The rhythmic contraction of cardiomyocytes consumes
a lot of energy, and 90% of ATP is produced by mitochondria.
Therefore, maintaining good mitochondrial morphology and function
is crucial for the survival and normal function of cardiomyocytes
(22). Mitochondria are also
involved in calcium homeostasis, which regulates cell division and
initiates signal transduction pathways (23-25).
Mitochondrial dysfunction plays a key role in H/R injury, including
decreased ATP synthesis, excessive production of ROS,
Ca2+ overload and continuous opening of mPTP. Abnormal
mitochondrial function can lead to systolic-diastolic dysfunction
of cardiac myocytes, and apoptosis (22,26-28).
mPTP opening reduces mitochondrial membrane potential, which is an
important factor for the decrease of ATP production, resulting in
the death of myocardial cells (29). The present study indicated that
abnormal mitochondrial function occurred in the MI/R rat model. The
enhanced mPTP opening occurred in H/R injury, which resulted in a
decreased mitochondrial membrane potential and ATP levels and
increased apoptosis.
When the energy crisis of the body is caused by
stress conditions (ischemia, hypoxia, oxidative stress and other
factors), the ATP levels in the body decrease or the newly
generated ATP cannot rapidly replace its consumption by tissues and
organs, resulting in the insufficient energy supply in cells and
the activation of AMPK (30).
Notably, previous study found that mitochondrial dysfunction could
activate AMPK signaling to promote cell survival (31). At the initial stage of reperfusion,
oxidative stress injury caused by oxygen free radical explosion is
one of the main pathogenic mechanisms of IRI. Activation of the
AMPK signaling pathway increases cell viability and alleviates
cardiomyocyte apoptosis induced by oxidative stress (32-34).
Activated AMPK can inhibit myocardial cell apoptosis (35,36).
Numerous studies suggested that pharmacological agonists of AMPK
had anti-apoptotic activity in MI/R by activating the AMPK
signaling pathway (37,38). In addition, AMPK decreases
production of proinflammatory factors IL-1β and TNF-α, increases
the content of anti-inflammatory factor IL-10 and alleviates
myocardial injury from MI in rats (39). To confirm the effects of AMPK on
inflammation, oxidative stress, apoptosis and mitochondrial
function in I/R injury, compound C was administered to cells. As
expected, compound C promoted inflammation, oxidative stress and
apoptosis and aggravated mitochondrial injury, in H/R-induced H9C2
cells.
A previous study indicated that roflumilast could
mitigate inflammation, oxidative stress and apoptosis in the acute
lung injury of rabbits (40). Xu
et al (10) demonstrated
that roflumilast inhibits oxidative stress caused by ischemic
stroke. Additionally, it was also discovered that roflumilast
protected against cardiotoxicity by reducing inflammation and
oxidative stress (13,14). In the present study, it was also
found that roflumilast could suppress inflammation, oxidative
stress and apoptosis in H/R-induced H9C2 cells and inhibit
inflammation in rats subjected to MI/R. In addition, roflumilast
could inhibit weight gain, promote insulin sensitivity and suppress
hepatic steatosis in mice by increasing mitochondrial
chondrogenesis (41). A PDE-4
inhibitor (rolipram) protected against malathion-induced toxic
damage in rat blood and brain mitochondria (42). Based on aforementioned findings, it
was hypothesized that a PDE-4 inhibitor might be related to the
regulation of mitochondrial function. In the present study,
roflumilast improved mitochondrial function in MI/R rats and
H/R-induced H9C2 cells. Xu et al (43) found that inhibition of AMPKα by
compound C almost abolished the promotive effects of roflumilast on
proliferator-activated Receptor-gamma and CCAAT enhancer-binding
protein alpha: When AMPKα was inhibited, roflumilast treatment was
almost abated. The protective effect of roflumilast on injury in
MI/R rats or H/R-induced H9C2 cells was observed in the present
study; this was weakened by compound C via the inhibition of the
AMPK signaling pathway.
However, there were certain limitations to the
current study. Firstly, cardiac functional studies, such as imaging
and cardiac echo, were not performed. Secondly, female rats were
not included in the MIRI model. Finally, the target of roflumilast
in the MIRI model was not determined. These factors should be
considered in further studies.
In conclusion, the present study demonstrated that
roflumilast could alleviate MI in rats subjected to MI/R and
attenuate H/R-induced oxidative stress, inflammatory response and
mitochondrial damage in H9C2 cells by activating the AMPK signaling
pathway.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZH designed and conceived the study. BL conducted
the experiments, analyzed the data and drafted the manuscript. ZH
and BL confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocol for the animal studies was
reviewed and approved by the Committee for the Ethics of Animal
Experiments, Shenzhen Peking University-The Hong Kong University of
Science and Technology Medical Center (approval no. 2021-807;
Shenzhen, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Weinberger T and Schulz C: Myocardial
infarction: A critical role of macrophages in cardiac remodeling.
Front Physiol. 6(107)2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Anderson JL, Adams CD, Antman EM, Bridges
CR, Califf RM, Casey DE Jr, Chavey WE II, Fesmire FM, Hochman JS,
Levin TN, et al: ACC/AHA 2007 guidelines for the management of
patients with unstable angina/non-ST-Elevation myocardial
infarction: A report of the American college of cardiology/american
heart association task force on practice guidelines (Writing
Committee to Revise the 2002 Guidelines for the Management of
Patients With Unstable Angina/Non-ST-Elevation Myocardial
Infarction) developed in collaboration with the American college of
emergency physicians, the society for cardiovascular angiography
and interventions, and the society of Thoracic surgeons endorsed by
the American association of cardiovascular and pulmonary
rehabilitation and the society for academic emergency medicine. J
Am Coll Cardiol. 50:e1–e157. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hu S, Gao R, Liu L, Zhu M, Wang W, Wang Y,
Wu Z, Li H, Gu D, Yang Y, et al: Summary of the 2018 report on
cardiovascular diseases in China. Chin Circulat J. 34:209–220.
2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
4
|
Liu S, Li Y, Zeng X, Wang H, Yin P, Wang
L, Liu Y, Liu J, Qi J, Ran S, et al: Burden of cardiovascular
diseases in China, 1990-2016: Findings from the 2016 global burden
of disease study. JAMA Cardiol. 4:342–352. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wu Y, Li S, Patel A, Li X, Du X, Wu T,
Zhao Y, Feng L, Billot L, Peterson ED, et al: Effect of a quality
of care improvement initiative in patients with acute coronary
syndrome in resource-constrained hospitals in China: A randomized
clinical trial. JAMA Cardiol. 4:418–427. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Farah A and Barbagelata A: Unmet goals in
the treatment of acute myocardial infarction: Review. F1000Res 6:
Faculty Rev-1243, 2017.
|
|
7
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Janjua S, Fortescue R and Poole P:
Phosphodiesterase-4 inhibitors for chronic obstructive pulmonary
disease. Cochrane Database Syst Rev. 5(CD002309)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Xu B, Xu J, Cai N, Li M, Liu L, Qin Y, Li
X and Wang H: Roflumilast prevents ischemic stroke-induced neuronal
damage by restricting GSK3β-mediated oxidative stress and
IRE1α/TRAF2/JNK pathway. Free Radic Biol Med. 163:281–296.
2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xu X, Liao L, Hu B, Jiang H and Tan M:
Roflumilast, a phosphodiesterases-4 (PDE4) inhibitor, alleviates
sepsis-induced acute kidney injury. Med Sci Monit.
26(e921319)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kwak HJ, Park KM, Choi HE, Chung KS, Lim
HJ and Park HY: PDE4 inhibitor, roflumilast protects cardiomyocytes
against NO-induced apoptosis via activation of PKA and Epac dual
pathways. Cell Signal. 20:803–814. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ansari MN, Ganaie MA, Rehman NU, Alharthy
KM, Khan TH, Imam F, Ansari MA, Al-Harbi NO, Jan BL, Sheikh IA and
Hamad AM: Protective role of Roflumilast against cadmium-induced
cardiotoxicity through inhibition of oxidative stress and NF-κB
signaling in rats. Saudi Pharm J. 27:673–681. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang S, Wu P, Liu J, Du Y and Yang Z:
Roflumilast attenuates doxorubicin-induced cardiotoxicity by
targeting inflammation and cellular senescence in cardiomyocytes
mediated by SIRT1. Drug Des Devel Ther. 15:87–97. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Perrelli MG, Pagliaro P and Penna C:
Ischemia/reperfusion injury and cardioprotective mechanisms: Role
of mitochondria and reactive oxygen species. World J Cardiol.
3:186–200. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Penna C, Perrelli MG and Pagliaro P:
Mitochondrial pathways, permeability transition pore, and redox
signaling in cardioprotection: Therapeutic implications. Antioxid
Redox Signal. 18:556–599. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kyung SY, Kim YJ, Son ES, Jeong SH and
Park JW: The phosphodiesterase 4 inhibitor roflumilast protects
against cigarette smoke extract-induced mitophagy-dependent cell
death in epithelial cells. Tuberc Respir Dis (Seoul). 81:138–147.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63(e12413)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tian L, Cao W, Yue R, Yuan Y, Guo X, Qin
D, Xing J and Wang X: Pretreatment with Tilianin improves
mitochondrial energy metabolism and oxidative stress in rats with
myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha
signaling pathway. J Pharmacol Sci. 139:352–360. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tikoo K, Lodea S, Karpe PA and Kumar S:
Calorie restriction mimicking effects of roflumilast prevents
diabetic nephropathy. Biochem Biophys Res Commun. 450:1581–1586.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Qian L, Shi J, Zhang C, Lu J, Lu X, Wu K,
Yang C, Yan D, Zhang C, You Q, et al: Downregulation of RACK1 is
associated with cardiomyocyte apoptosis after myocardial
ischemia/reperfusion injury in adult rats. In Vitro Cell Dev Biol
Anim. 52:305–313. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tahrir FG, Langford D, Amini S, Ahooyi TM
and Khalili K: Mitochondrial quality control in cardiac cells:
Mechanisms and role in cardiac cell injury and disease. J Cell
Physiol. 234:8122–8133. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Seidlmayer LK, Juettner VV, Kettlewell S,
Pavlov EV, Blatter LA and Dedkova EN: Distinct mPTP activation
mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS,
pH, and inorganic polyphosphate. Cardiovasc Res. 106:237–248.
2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ong SB and Hausenloy DJ: Mitochondrial
dynamics as a therapeutic target for treating cardiac diseases.
Handb Exp Pharmacol. 240:251–279. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Vásquez-Trincado C, García-Carvajal I,
Pennanen C, Parra V, Hill JA, Rothermel BA and Lavandero S:
Mitochondrial dynamics, mitophagy and cardiovascular disease. J
Physiol. 594:509–525. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Shoshan-Barmatz V and De S: Mitochondrial
VDAC, the Na(+)/Ca(2+) exchanger, and the Ca(2+) uniporter in
Ca(2+) dynamics and signaling. Adv Exp Med Biol. 981:323–347.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li Y and Liu X: Novel insights into the
role of mitochondrial fusion and fission in cardiomyocyte apoptosis
induced by ischemia/reperfusion. J Cell Physiol. 233:5589–5597.
2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Maneechote C, Palee S, Chattipakorn SC and
Chattipakorn N: Roles of mitochondrial dynamics modulators in
cardiac ischaemia/reperfusion injury. J Cell Mol Med. 21:2643–2653.
2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Leucker TM, Bienengraeber M, Muravyeva M,
Baotic I, Weihrauch D, Brzezinska AK, Warltier DC, Kersten JR and
Pratt PF Jr: Endothelial-cardiomyocyte crosstalk enhances
pharmacological cardioprotection. J Mol Cell Cardiol. 51:803–811.
2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hardie DG: AMPK: Positive and negative
regulation, and its role in whole-body energy homeostasis. Curr
Opin Cell Biol. 33:1–7. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhao B, Qiang L, Joseph J, Kalyanaraman B,
Viollet B and He YY: Mitochondrial dysfunction activates the AMPK
signaling and autophagy to promote cell survival. Genes Dis.
3:82–87. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hwang JT, Kwon DY, Park OJ and Kim MS:
Resveratrol protects ROS-induced cell death by activating AMPK in
H9c2 cardiac muscle cells. Genes Nutr. 2:323–326. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sasaki H, Asanuma H, Fujita M, Takahama H,
Wakeno M, Ito S, Ogai A, Asakura M, Kim J, Minamino T, et al:
Metformin prevents progression of heart failure in dogs: Role of
AMP-activated protein kinase. Circulation. 119:2568–2577.
2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wang XF, Zhang JY, Li L and Zhao XY:
Beneficial effects of metformin on primary cardiomyocytes via
activation of adenosine monophosphate-activated protein kinase.
Chin Med J (Engl). 124:1876–1884. 2011.PubMed/NCBI
|
|
35
|
Yeh CH, Chen TP, Wang YC, Lin YM and Fang
SW: AMP-activated protein kinase activation during
cardioplegia-induced hypoxia/reoxygenation injury attenuates
cardiomyocytic apoptosis via reduction of endoplasmic reticulum
stress. Mediators Inflamm. 2010(130636)2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chen MB, Wu XY, Gu JH, Guo QT, Shen WX and
Lu PH: Activation of AMP-activated protein kinase contributes to
doxorubicin-induced cell death and apoptosis in cultured myocardial
H9c2 cells. Cell Biochem Biophys. 60:311–322. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chin JT, Troke JJ, Kimura N, Itoh S, Wang
X, Palmer OP, Robbins RC and Fischbein MP: A novel cardioprotective
agent in cardiac transplantation: Metformin activation of
AMP-activated protein kinase decreases acute ischemia-reperfusion
injury and chronic rejection. Yale J Biol Med. 84:423–432.
2011.PubMed/NCBI
|
|
38
|
Kim AS, Miller EJ, Wright TM, Li J, Qi D,
Atsina K, Zaha V, Sakamoto K and Young LH: A small molecule AMPK
activator protects the heart against ischemia-reperfusion injury. J
Mol Cell Cardiol. 51:24–32. 2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
McGaffin KR, Witham WG, Yester KA, Romano
LC, O'Doherty RM, McTiernan CF and O'Donnell CP: Cardiac-specific
leptin receptor deletion exacerbates ischaemic heart failure in
mice. Cardiovasc Res. 89:60–71. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kosutova P, Mikolka P, Kolomaznik M,
Balentova S, Adamkov M, Calkovska A and Mokra D: Reduction of lung
inflammation, oxidative stress and apoptosis by the PDE4 inhibitor
roflumilast in experimental model of acute lung injury. Physiol
Res. 67:S645–S654. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kahles F, Mllmann J, Bck C, Liberman A and
Lehrke M: The PDE-4 Inhibitor Roflumilast reduces weight gain,
enhances insulin sensitivity and prevents hepatic steatosis in mice
by increasing mitochondrogenesis. Diabetologie Und Stoffwechsel.
9:1993–2003. 2014.
|
|
42
|
Rezvanfar MA, Rezvanfar MA, Ranjbar A,
Baeeri M, Mohammadirad A and Abdollahi M: Biochemical evidence on
positive effects of rolipram a phosphodiesterase-4 inhibitor in
malathion-induced toxic stress in rat blood and brain mitochondria.
Pesticide Biochemistry & Physiology. 98:135–143. 2010.
|
|
43
|
Xu W, Zhang J and Xiao J: Roflumilast
suppresses adipogenic differentiation via AMPK mediated pathway.
Front Endocrinol (Lausanne). 12(662451)2021.PubMed/NCBI View Article : Google Scholar
|