1. Introduction
Sepsis-related acute kidney injury (S-AKI) is a
severe complication of sepsis in critically ill patients, which is
associated with a high morbidity and mortality rate and treatment
cost (1,2). A previous meta-analysis showed that
one in five adults and one in three children develop AKI during
hospitalization globally (3), and
a review reported that >50% of patients in intensive care units
(ICUs) suffer from AKI (4). The
mortality of patients with S-AKI is significantly higher than that
of patients with AKI without sepsis (5). It is estimated that >60% of
patients with sepsis or septic shock will develop S-AKI (5). Even if patients with S-AKI survive
the acute phase, the prevalence of chronic kidney disease in these
patients increases. Consequently, S-AKI is a major global challenge
to the human population (6).
The main therapy for S-AKI is antibiotics combined
with symptomatic treatment. When the complications cannot be
treated by drug therapy alone, renal replacement therapy should be
performed. Notably, there is still a lack of effective new drugs.
Unfortunately, the precise pathogenetic mechanism underlying S-AKI
is unclear; therefore, exploring the pathophysiology of S-AKI may
provide novel options for its diagnosis and treatment. Previous
studies have reported that heparanase (HPA) plays an important role
in the pathogenesis of S-AKI (7,8).
2. S-AKI and its underlying molecular
mechanisms
S-AKI is a syndrome that meets the Kidney Disease
Improving Global Outcomes' criteria in patients with sepsis
(9,10). In the criteria, AKI is defined as
any of the following: Increase in serum creatinine (SCr) by 0.3
mg/dl (26.5 µmol/l) within 48 h; or increase in SCr to 1.5 times
baseline, which is known or presumed to have occurred within the
prior 7 days; or urine volume <0.5 ml/kg/hour for 6 h. Previous
studies have reported that the pathogenesis of S-AKI may be
attributed to renal ischemia, hypoxia, toxic injury and sepsis in
the clinic (11,12). In addition, an epidemiological
study showed that the occurrence of S-AKI is associated with
decreased renal perfusion and subsequent tubular necrosis (13). Furthermore, certain researchers
have proposed alternative mechanisms for the pathogenesis of S-AKI.
In a sheep model of Escherichia coli-induced septic shock,
Maiden et al (14) showed
that early AKI was not related to changes in renal blood flow,
oxygen delivery or histological appearance, such as glomerular
mesangial expansion or podocyte disappearance. Lerolle et al
(15) found that the renal damage
caused by septic shock was not only a simple acute tubular injury,
but was also associated with capillary leukocyte infiltration and
apoptosis. Currently, microvascular dysfunction, inflammatory
response and metabolic reprogramming are considered the three basic
molecular mechanisms of S-AKI (16-18).
Microvascular dysfunction is characterized by an
increased number of capillaries, cessation of blood flow and an
uneven distribution of microvascular blood flow; it is considered
the main cause of the pathophysiology of sepsis (19). Microvessels are the primary site
for gas, nutrient and waste product exchange between blood and
tissues. Sepsis leads to the dysfunction of the main cell types and
fragments in microvessels, including endothelial cells (ECs),
smooth muscle cells, red blood cells, white blood cells and
platelets (20,21). ECs are vascular and non-traditional
immune cells that play a major role in the systemic response to
bacterial infection. Sepsis causes a series of impairments in ECs,
leading to the dysregulation of microcirculation, leukocyte
adhesion and migration, vasodilation, impaired transport of
nutrients and increased capillary permeability, amongst other
effects (22). During sepsis,
hypoxia weakens the regulatory effects of red blood cells on
microvascular relaxation and the deformability of red blood cells,
causing the aggregation of red blood cells and ECs, and impairing
microcirculatory blood flow (19,23).
It has been reported that inducible nitric oxide synthase (iNOS) is
heterogeneously expressed in different organic vascular beds,
leading to pathological shunting of microvascular blood flow
(24). Therefore, the increased
activity of iNOS and the subsequent increase in the levels of NO
may cause kidney (i.e., mitochondria and renal tubule) damage in
septic shock (24,25). Furthermore, degradation of the
endothelial glycocalyx, which is a major component of the vascular
barrier, and plays an important role in microvascular homeostasis
and organ perfusion, causes vascular leak, which also contributes
to the occurrence of S-AKI (26).
Thus, it may be inferred that sepsis causes microvascular
dysfunction in AKI, including impairments of the main cell types in
microvessels and the endothelial glycocalyx. As a result, the
impairment of nutrient transport, leukocyte adhesion and migration,
vasodilation and increased capillary permeability appear to be
responsible for S-AKI.
An inflammatory response is the main defense
mechanism of a host against pathogenic invasion. According to the
Third International Consensus Definitions for Sepsis and Septic
Shock (10), an imbalanced
inflammatory response in the host may be the cause of organ
dysfunction and adverse outcomes. Sepsis can induce the release of
inflammatory mediators, such as pathogens and injury-related
molecular patterns, into the intravascular lumen. These molecules
bind to pattern recognition receptors, such as T-lymphoid receptors
on the surface of immune cells, and initiate downstream cascade
signals, leading to the synthesis and release of pro-inflammatory
molecules (16). The dysregulated
inflammatory response during sepsis can promote the release of
cytokines and chemokines, commence leukocyte/platelet activation,
increase the production of oxygen free radicals and arachidonic
acid metabolites, and activate and recruit T cells, neutrophils,
macrophages, platelets and ECs (27). Studies have shown that Toll-like
receptors (TLRs) also play an important role in S-AKI (28,29).
TLRs are the main pattern recognition receptors and can mediate
cellular signaling cascades through a variety of
pathogen-associated molecular patterns (PAMPs) (30,31).
TLRs play a central role in the innate immune initiation process
against invasive microbial pathogens. Renal tubular epithelial
cells (TECs) also express TLRs, especially TLR2 and TLR4. Leemans
et al (32) demonstrated
that TLR2 expressed in the renal parenchyma plays a vital role in
inducing inflammation and injury. The expression of the TLR4/NF-κB
signaling pathway is enhanced in renal ischemia-reperfusion (I/R)
injury and septic kidney injury (28). Another study showed that TLR4
expression in renal TECs regulates S-AKI and inflammation (33).
Currently, increasing attention is being paid to the
interaction between inflammation, immunological mechanisms and
coagulation cascades in triggering adaptive immune responses in
renal TECs. These interactions amplify and enhance microvascular
dysfunction and endothelial injury (34). During sepsis, the expression of
adhesion molecules on ECs and immune cells is increased, resulting
in decreased EC deformability, and increased ability to aggregate
and activate neutrophils (22).
During S-AKI, ECs, red blood cells, monocytes and platelets produce
microvesicles (MVs), and endothelial microvascular damage may lead
to increased concentrations of MVs (35). When the kidney is exposed to
damage, PAMPs filter through glomeruli or adjacent peritubular
capillaries, and proximal renal TECs exhibit increased oxidative
stress, reactive oxygen species production and mitochondrial damage
(16). In systemic infection and
subsequent sepsis, dysregulation of immune thrombosis leads to
systemic coagulopathy and multiorgan failure, due to microvascular
obstruction depriving tissues of a blood supply. The main cellular
drivers of this process are platelets and innate immune cells, such
as neutrophils, eosinophils and macrophages (36). The activating interactions between
platelets and immune cells is composed of coagulation and
complement systems, which cause a coagulation cascade, leading to
thrombosis and microcirculation dysfunction. Uncontrolled
inflammation, activation of coagulation and complement cascades are
considered to be involved in the pathogenesis of S-AKI (37). Metabolic reprogramming in response
to injury is an evolutionarily conserved mechanism for cell
survival (38). Cells convert
nutrients to ATP through two key metabolic pathways: Oxidative
phosphorylation (OXPHOS) and glycolysis. OXPHOS is the metabolic
phenotype of TECs. The ability of OXPHOS to synthesize ATP depends
on functional mitochondria (39).
Sepsis is known to cause severe mitochondrial damage that may
hinder the ability of TECs to restore OXPHOS. Persistence of
glycolytic metabolism in renal TECs is associated with persistent
local inflammation and increased injury. During sepsis, glycolysis
may shift the TECs into ‘off’ mode, thus allowing the cell to
re-prioritize energy expenditure for survival at the expense of
organ function (38). As a result,
renal tubular ion transport is reduced, resulting in an increase in
chloride concentration in the renal tubular fluid. This decreases
the glomerular filtration rate by activating tubuloglomerular
feedback and causes AKI. An analysis of renal biopsies obtained 8 h
after the induction of sepsis by cecal ligation and puncture (CLP)
suggested a shift in the renal metabolic phenotype to glycolysis
(40).
Although the mechanisms of S-AKI are widely studied,
it is still necessary to explore the complex mechanisms of S-AKI
pathogenesis to identify biomarkers and therapeutic targets.
Previous studies have shown that HPA plays an important role in the
S-AKI process (7,41).
3. Role of HPA in the pathogenesis of
S-AKI
HPA is an endoglycosidase
HPA is also named HPA-1, which differs from HPA-2.
The HPA gene, located on chromosome 4q21.2, is the sole known
mammalian endoglycosidase that cleaves the heparan sulfate (HS)
side chains of HS proteoglycans (HSPGs) intra- and extracellularly.
In vivo, HPA exists within lysosomes as a precursor with a
molecular weight of 65 kDa (42).
The active form of HPA is generated by lysosomal cathepsin-induced
cleavage of the pro-enzyme yielding 50- and 8-kDa fragments, both
of which are needed for its activity. Active HPA has enzymatic and
non-enzymatic activities that participate in multiple processes
(42). HPA serves an important
role in promoting pathological processes such as tumor growth,
metastasis, angiogenesis, thrombosis, fibrosis, inflammation,
autoimmunity and renal dysfunction (43-49).
Upregulation of HPA expression is associated with tumor size and
progression, enhanced metastasis and a poor prognosis (50).
When HPA is secreted outside the cell, it cleaves
the HS side chain and contributes to the activation of integrin,
epidermal growth factor receptor and other signaling pathways. HPA
not only trims HS that is bound to glycocalyx proteoglycan core
proteins, but also degrades proteoglycans that are attached to the
extracellular matrix (ECM), thus remodeling the ECM. HS is a
high-sulfur polysaccharide that is widely found in the ECM and
plasma membrane, and within the cell (51). Therefore, it plays an important
role in maintaining ECM integrity, barrier function and cell-ECM
interactions (52). HPA is crucial
for the normal turnover of HS (53). HPA affects the function of HSPGs by
degrading HS. Several linear HS chains covalently bind to a core
protein to form HSPGs, which participate in cell-cell and
cell-matrix adhesion (54). HSPGs
not only provide a repository for heparin-binding molecules, such
as growth factors, chemokines and enzymes in the tissue
microenvironment, but also regulate their accessibility, function
and mode of action. Degradation of HS by HPA not only begins to
remove physical barriers that prevent cell invasion, but also
releases various proteins that bind to HS, promoting activation of
cellular signaling pathways and responses (55). However, it has been reported that
the exact role of HPA in inflammation is difficult to determine, as
HPA may act in other ways, either enhancing or inhibiting the
inflammatory response (47).
Intracellular HPA binds to autophagosomes to enable
autophagy, binds to exosomes to promote their release from cells
and enters the nucleus to regulate gene expression (56). In addition, HPA has an important
role in the normal physiology of lysosomes (57). HPA regulates gene expression,
activates innate immune cells, promotes the formation of exosomes
and autophagosomes, and promotes signal transduction through
enzymatic and non-enzymatic activities (58). HPA promotes the secretion of
exosomes that interact with tumor and host cells, and drives their
transition to an aggressive tumor phenotype (57). Exosomes are mediators of
intercellular communication that initiate tumor progression by
regulating tumor and host cell behavior locally and distally
throughout the body in the tumor microenvironment (59). HPA enhances tumor growth and
chemotherapy resistance by enhancing autophagy (60).
HPA affects S-AKI by degrading the
endothelial glycocalyx
HPA promotes degradation of the endothelial
glycocalyx in glomeruli and renal tubules during sepsis (Fig. 1). The glycocalyx covers the surface
of the vascular endothelium and its degradation destroys the
integrity and permeability of the vascular system. Lygizos et
al (7) showed that glomerular
HPA was activated during sepsis and contributed to the occurrence
of S-AKI. HPA destroys the integrity and permeability of renal TECs
by degrading HS, resulting in renal inflammation, and urinary HS
primarily reflects the degradation of the renal glycocalyx
(7). Schmidt et al
(61) compared 30 patients with
septic shock in the medical ICU with 25 patients with severe trauma
in the surgical ICU that acted as controls. The study revealed that
compared with that in the control group, the level of HS in the
urine of septic patients was significantly higher. The degradation
function of HPA on HS suggests that HPA may contribute to
regulation of the renal glycocalyx during sepsis and lead to
S-AKI.
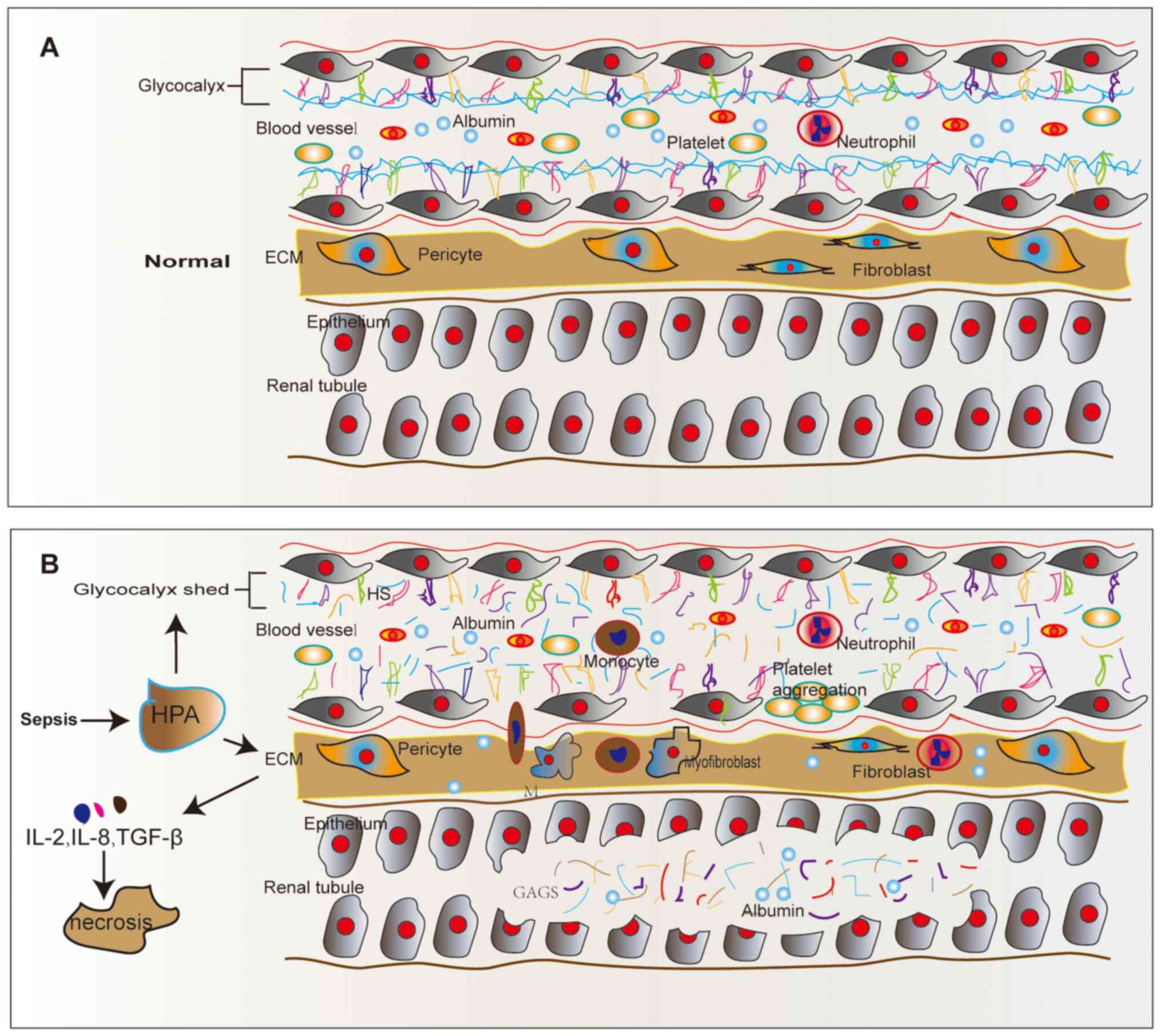 | Figure 1Structure of renal tubular epithelial
cells in normal and septic conditions. (A) Normal renal tubular
epithelial cells. (B) HPA acts on renal tubular epithelial cells
during sepsis. HPA engages in glycocalyx degradation in sepsis. HPA
also functions on ECM, causing the release of its pro-inflammatory
factors, such as IL-2, IL-8, bFGF and TGF-β, and stimulating
leukocyte recruitment, migration and extravasation by regulating
the interaction between leukocytes and endothelial cell surface.
ECM, extracellular matrix; HS, heparan sulfate; HPA, heparanase;
GAGs, glycosaminoglycans; M, monocyte. |
HPA functions in I/R AKI and toxic
AKI
HPA not only promotes the progression of S-AKI, but
also plays an important role in other types of AKI, such as toxic
AKI. I/R is a complication of AKI and HPA is part of the biological
network triggered by I/R injury (62). Masola et al (62) demonstrated through in vivo
and in vitro experiments that HPA can regulate macrophage
polarization, and renal damage and repair following I/R, and that
HPA inhibitors can partially restore renal function and reduce
apoptosis. In addition, another study showed that HPA promotes the
onset of I/R-induced AKI in an I/R mouse model (63). This previous study found that HPA
is upregulated in I/R mouse models, particularly in
HPA-overexpressing transgenic mice, whereas pretreatment with
PG545, an HPA inhibitor, can eliminate I/R-induced renal
dysfunction. Another study demonstrated that HPA is a key factor
involved in the occurrence and development of I/R-induced
epithelial-mesenchymal transition (EMT) (64). In a podocyte model of
Adriamycin-induced toxic kidney injury, HPA overexpression was
shown to preserve glomerular structure and renal function, and
elevated HPA levels could promote cell protection against apoptosis
(65), thus exhibiting a
protective effect. This previous study also reported that exposure
to toxic damage resulted in a significant increase in autophagic
flux in the podocytes of HPA-overexpressing mice, which could be
reversed by the HPA inhibitor, Roneparstat.
Therefore, previous studies have suggested that HPA
plays an important role in pathological processes, such as tumor
growth, migration/invasion of ECs, infiltration of immune cells,
metastasis, angiogenesis, thrombosis, fibrosis, autoimmunity,
autophagy, exosome release, promotion of pro-inflammatory cell
adhesion, signal transduction and renal dysfunction (42-44,46,57,66-71).
HPA mediates its effects on S-AKI
through inflammatory and immune responses
HPA plays an important role in inflammation, sepsis
and AKI (72-74).
Furthermore, HPA has effects on the endothelial glycocalyx and ECM
in the kidney. HPA is activated under inflammatory conditions and
can promote the degradation and shedding of the glycocalyx, leading
to increased cell permeability, thus causing vascular leakage and
hypovolemia. HPA also mediates its enzymatic activity on the ECM,
particularly the basement membrane, and promotes the release of
pro-inflammatory factors (i.e., IL-2, IL-8, basic fibroblast growth
factor and TGF-β) and heparin-binding molecules in the ECM. These
inflammatory factors stimulate the recruitment, rolling process and
extravasation of leukocytes by regulating the interaction between
leukocytes and the ECM surface (75-77)
(Fig. 1).
There is additional evidence to support the role of
HPA in S-AKI through the immune response during sepsis. In S-AKI,
HPA increases the presence of adhesion molecules, and enhances
vascular permeability, tissue edema, leukocyte adhesion, platelet
aggregation and vasodilation-related dysfunction by promoting
glycocalyx degradation (78,79).
The glycocalyx plays a vital role in maintaining the integrity and
permeability of the vascular system, providing vascular tension and
regulating leukocyte adhesion (80). HPA induces the apoptosis of tubular
cells and induces the production of damage-associated molecular
patterns. HS fragments released by HPA can also activate TLRs on
macrophages and tubular cells (62). Tubular cells produce
proinflammatory cytokines in response to direct hypoxic stimulation
and TLR activation, resulting in the attraction and activation of
macrophages. In addition, high levels of HPA can promote M1
polarization of infiltrating macrophages and aggravate parenchymal
injury (62). HPA also induces a
procoagulant effect and renal fibrosis by binding to cellular HS
(48), and interacts with tissue
factor pathway inhibitor (TFPI) on the surface of ECs, resulting in
TFPI dissociation and cell coagulation activity promotion (81). In addition, during S-AKI, HPA leads
to platelet aggregation and vasodilation-related dysfunction by
promoting glycocalyx degradation. Furthermore, it can lead to
increased microvascular dysfunction and endothelial injury, thus
affecting microcirculatory blood flow and aggravating coagulation
(74). HPA is also involved in
hemostasis through non-enzymatic mechanisms (48). For example, HPA reduces
unfractionated heparin to low-molecular weight heparin, which
functions as a co-factor for factor Xa inhibition by antithrombase
(48). Bayam et al
(82) confirmed that elevated
heparin levels may promote thrombosis. Therefore, HPA may also
enhance procoagulant activity in S-AKI. In addition, HPA can
promote renal fibrosis. Abassi et al (63) confirmed that PG545 suppressed renal
dysfunction and the upregulation of HPA, as well as
pro-inflammatory and pro-fibrotic factors that were induced by I/R
in AKI. HPA promotes renal fibrosis by participating in fibroblast
growth factor-2-dependent EMT of renal TECs (83).
In conclusion, HPA plays a major role in S-AKI,
mainly through the degradation of the endothelial glycocalyx, the
remodeling of the ECM, and the release of pro-inflammatory
cytokines and heparin-binding molecules in the ECM, thus
aggravating the immune response and inducing a procoagulant effect
and renal fibrosis. However, HPA may also influence S-AKI through
other mechanisms.
HPA mediates its effects on S-AKI
through autophagy
Autophagy is a highly conserved lysosome degradation
pathway in mammals, which removes protein aggregates and damaged
organelles to maintain cellular homeostasis (84,85).
It is reported that autophagy occurs at low levels under
physiological conditions to maintain the homeostasis of the
intracellular environment, but is upregulated upon exposure to
stress conditions, such as hunger, hypoxia, ischemia and oxidative
stress (86). In healthy and
diseased states, autophagy plays a key role in maintaining the
morphology, activity and dynamic balance between various cell types
in the kidney (87). It has been
reported that autophagy induced by unilateral ureteral obstruction
(UUO) has renoprotective effects, and treatment with the autophagy
inhibitor, 3-methyladenine, enhances tubular cell apoptosis and
tubulointerstitial fibrosis in the obstructed kidney following UUO
(88). However, uncontrolled
autophagy leads to excessive degradation of cellular proteins and
organelles, eventually resulting in the death of autophagic cells
(89).
A previous study has shown that HPA regulates
autophagy (90). Shteingauz et
al (60) and White (91) first described the role of lysosomal
HPA in regulating autophagy, which presents intracellular proteins,
lipids and other molecules or organelles for degradation and
recycling. The PI3K/AKT/mTOR signaling pathway is known to inhibit
autophagy (92). Certain studies
(60,93) have reported that overexpression of
HPA downregulates mTOR activity, and the inhibition of HPA by PG545
reverses this. mTOR is usually scattered throughout the cytoplasm
of normal cells; however, when HPA expression increases, the
majority of mTOR and HPA is concentrated around the nucleus, thus
reducing the activity of mTOR and increasing autophagy (94). Therefore, it was speculated that
HPA may cause renal autophagy through the PI3K/Akt/mTOR signaling
pathway and promote the progression of S-AKI (Table I; the protective role in S-AKI).
HPA may also cause renal autophagy through the AMPK/mTOR signaling
pathway, which is another autophagy signaling pathway. Further
in-depth investigations are needed to validate these
hypotheses.
 | Table IRole of heparanase in sepsis-related
acute kidney injury. |
Table I
Role of heparanase in sepsis-related
acute kidney injury.
| A, Adverse
role |
|---|
| Activity | Action | Area | Outcomes | (Refs.) |
|---|
| Enzymatic
activity | Degrade HS side
chain | Glomerular ECs and
glomerular basement membrane | Damage to
glomerular filtration barrier; proteinuria; renal
insufficiency | (62,69) |
| | | Renal interstitial
vascular ECs | Renal interstitial
inflammatory infiltration | (34) |
| | | Renal TECs | Destruction of
integrity and permeability; enhancement of renal inflammation | (69) |
| | | Glycocalyx | Enhancement of
vascular permeability, tissue edema, leukocyte adhesion and
platelet aggregation; vasodilation disorder | (78-80) |
| | | ECM | Destruction of ECM
integrity and barrier function; release of its pro-inflammatory
factors; stimulation of leukocyte recruitment, rolling process and
extravasation | (52,75-78) |
| Non-enzymatic
activity | Procoag ulant
activity | Extracellular | Promotion of
thrombosis | (82) |
| | Bind to autopha
gosome | Intracellular | Promotion of
autophagy to break the cellular homeostasis | (60,91,92) |
| | Bind to
exosome | Intracellular | Entering of the
nucleus to affect gene transcription | (58,108) |
| B, Protective
role |
| Activity | Action | Area | Outcomes | (Refs.) |
| Non-enzymatic
activity | Bind to autopha
gosome | Intracellular | Promotion of
autophagy to maintain cellular homeostasis | (89,90) |
| | Bind to
exosome | Intracellular | Entering of the
nucleus to affect gene transcription through the substances carried
by exosomes | (58) |
| | Procoag ulant
activity | Extracellular | Involvement in
hemostasis and relieve bleeding | (48) |
HPA mediates its action through
exosomes during S-AKI
Exosomes are membranous vesicles with a diameter of
40-100 mm. All living cells secrete exosomes, which exist in the
blood, urine, saliva, lymph and other body fluids (95,96).
Exosomes do not contain nuclei and cannot self-replicate. Exosomes
are, reportedly, relatively good drug delivery systems due to their
stability. Recipient cells endocytose exosomes and release exosome
contents, and thus play a crucial role in cell-to-cell
communication and information transmission, as exosomes are
involved in transporting proteins, lipids, microRNA (miRNA/miR),
mRNA and other bioactive substances. Exosomes also participate in
the regulation of the inflammatory response, immune response,
tumorigenesis, infection and other pathophysiological processes
(97,98). Additionally, exosomes have a
significant role in sepsis. The overall contribution of exosomes to
sepsis was previously studied using GW4869, which was shown to
inhibit the production of exosomes. Essandoh et al (99) showed that GW4869 significantly
improved the survival rate of mice injected with lipopolysaccharide
(LPS) or the septic mouse model of CLP.
Exosomes affect S-AKI by carrying
relevant cargo, such as miRNAs
miRNAs are released into body fluids, such as serum
and urine, and their high specificity and sensitivity render them
suitable for use as potential biomarkers for monitoring the
progression of AKI (100). Viñas
et al (101) reported that
the levels of miR-486-5p expressed in the proximal tubules and ECs
were increased upon injection with miR-486-5p mimics into AKI mice.
Furthermore, the levels of plasma creatinine, tissue damage, and
neutrophil infiltration and apoptosis improved. Urinary exosomes
are excreted by all nephron segments. Exosome miR-27b from bone
marrow mesenchymal stem cells has been shown to inhibit the
development of sepsis by downregulating Jumonji domain-containing
protein-3 and inactivating the NF-κB signaling pathway (102). Zhang et al (103) showed that human umbilical cord
mesenchymal stem cell-derived exosomes downregulated the expression
of the miR-146b target gene, interleukin-1 receptor-associated
kinase, by upregulating the expression of miR-146b. Furthermore, a
previous study showed that exosome-mediated pyroptosis of
miR-93-TXNIP-NLRP3 creates functional differences between M1 and M2
macrophages in S-AKI (104).
miR-19b-3p, derived from LPS-induced AKI mouse TECs, has been
reported to promote macrophage infiltration and tubulointerstitial
inflammation by inhibiting the suppressor of cytokine signaling-1
in vitro, leading to the activation of NF-κB, and
upregulation of monocyte chemoattractant protein-1 (MCP-1), IL-1β,
IL-6, TNF-α and iNOS (105)
(Fig. 2).
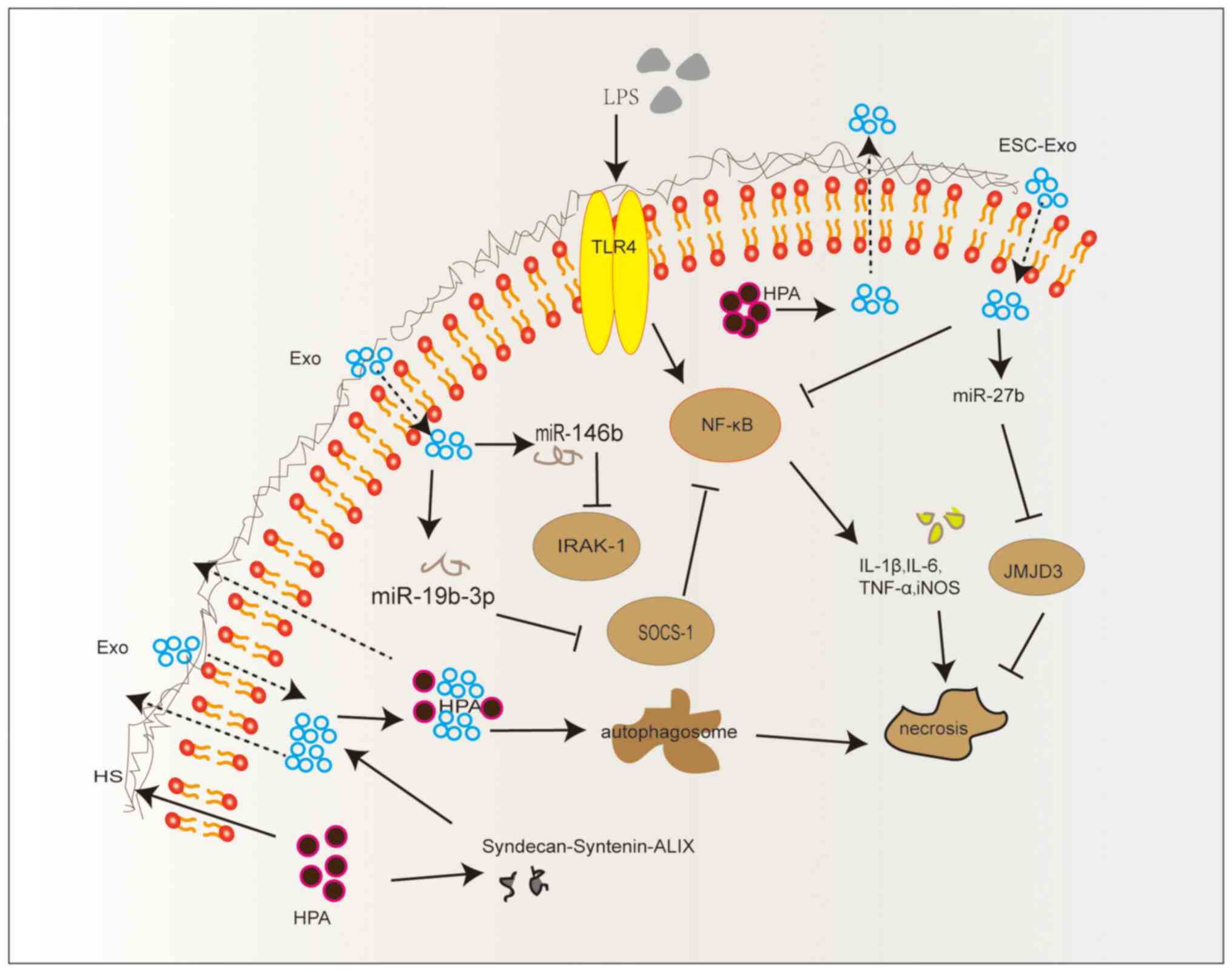 | Figure 2HPA activates the
syndecan-syntenin-ALIX exosome pathway. Exo, exosomes; HS, heparan
sulfate; HPA, heparanase; LPS, lipopolysaccharide; TLR4, Toll-like
receptor 4; ESC-Exo, mesenchymal stem cell-derived exosomes;
SOCS-1, suppressor of cytokine signaling-1; JMJD3, Jumonji domain
containing 3; iNOS, inducible nitric oxide synthase; IRAK-1,
interleukin-1 receptor associated kinase 1; miR, microRNA. |
HPA promotes the formation of
exosomes
HPA-induced enhancement of exosome biogenesis was
initially identified in human myeloma cells transfected with HPA
cDNA (106). The formation of
extracellular vesicles is dependent on syntenin and its interaction
with syndecans. Roucourt et al (107) reported that HPA activated the
syndecan-syntenin-ALIX exosome pathway, and that it also played a
role in regulating this pathway. Syndecans, a family of membrane
proteoglycans, are composed of chondroitin sulfate and HS chains
linked to a 31-kDa integral membrane protein (108). Syndecans and syntenin are
involved in the regulation of exosome biogenesis through their
interaction with ALIX (109).
However, HPA alters syndecan and syntenin composition, and
biological function through promoting exosome secretion (106). In human cancer cells, exosome
secretion is significantly increased when HPA expression is
enhanced or when tumor cells are exposed to exogenous HPA.
Furthermore, HPA activity is necessary to enhance exosome secretion
(106). In conclusion, HPA is
involved in exosome biogenesis and activates the
syndecan-syntenin-ALIX pathway (Fig.
2).
HPA mediates its action through
exosomes during S-AKI
HPA promotes exosome formation, which act on the
kidneys through their biologically relevant cargo. It has been
reported that the upregulation of miR-21-5p improves renal function
through several mechanisms; it has been shown to reduce the
pathological damage to renal tissue, reduce serum inflammatory
response, reduce renal tissue apoptosis, reduce oxidative stress
responses and regulate the expression of endothelial glycocalyx
injury markers, such as syndecan-1 and HPA in CLP rats (8). HPA also enters the nucleus and
regulates gene transcription through exosomes. The role of HPA in
exosome activity has been determined by its regulation of HS
cleavage (55). HPA localizes to
the exosome surface, where it is activated and degrades HS within
the ECM (110). HPA can regulate
the transcription of various genes involved in neovascularization,
such as matrix metalloproteinase 9 (MMP-9), coagulation and
inflammatory responses. HPA has been shown to increase the
expression of MMP-9 and to increase cleavage of syndecan-1. The
released syndecan-1 can be transported into the nucleus, where the
bound HS chain and HPA transported into the nucleus can affect a
number of mechanisms, including promotion of mitotic spindle
formation and subsequent chromosome stability, inhibition of DNA
topoisomerase I activity and regulation of cell proliferation
(55). In conclusion, HPA
functions on S-AKI through exosomes and the release of bioactive
substances, and certain miRNAs derived from exosomes can improve
renal function in S-AKI (Table I;
the protective roles of HPA in S-AKI).
In conclusion, HPA contributes to the pathogenesis
of S-AKI by inducing the degradation of HS and the destruction of
ECM, thus promoting inflammation, macrophage polarization,
fibrosis, dysregulation of inflammatory response, excessive
activation of autophagy and exosome biogenesis (Table I, the adverse roles of HPA in
S-AKI.). However, the precise molecular mechanisms underlying the
pathogenesis of S-AKI require further investigation.
4. HPA as a diagnostic biomarker and
therapeutic target for S-AKI
Currently, several molecules have been identified as
potential biomarkers for the early detection of renal damage prior
to increased serum creatinine levels (4). For example, tissue inhibitor of
metalloproteinase 2, insulin-like growth factor binding protein 7,
liver fatty acid binding protein, neutrophil gelatinase-associated
lipocalin and cystatin C (111-114),
amongst others, have been reported as potential biomarkers for
renal damage. However, due to their limited specificity and
sensitivity, injury markers are mainly only used for research
purposes at this time (4).
Notably, in a septic model of CLP, the level of HPA was moderately
increased at 4 h and further increased after 24 h (115). HPA is activated in AKI and plays
a key role in endothelial glycan shedding (74,116). HPA may also serve as a potential
biomarker for S-AKI. In a CLP-induced mouse model of S-AKI, renal
function was damaged after 4 h (7). The study showed that CLP-treated mice
exhibited early activation of glomerular HPA and loss of glomerular
filtration, as indicated by a greater than two-fold increase in
blood urea nitrogen and a >50% decrease in inulin clearance.
However, administration of HPA inhibitors 2 h prior to CLP was
revealed to attenuate sepsis-induced glomerular filtration rate
loss.
The inhibition of HPA activity alleviates the
degradation of HS, thereby protecting the glycocalyx and preserving
the vascular barrier. HPA inhibitors, such as PG545, PI-88,
SST0001, other HS analogues and heparin-derived drugs, have been
reported to inhibit the activity of HPA during inflammation
(42). Endothelial progenitor
cell-derived exosomes were reported to increase the level of
syndecan-1 and decrease the level of HPA in the renal tissues of
CLP rats (8). In vivo,
phillyrin, the main pharmacological component of the traditional
Chinese medicine Forsythia suspensa, has an effective
protective effect on glycocalyx HS degradation in LPS-induced AKI
mice (41).
Based on the effects of HPA in S-AKI, it is expected
to serve as a novel biomarker for the detection and treatment of
S-AKI. However, HPA inhibitors have not yet been tested in the
clinic. The identification of a specific, safe and efficacious HPA
inhibitor may pave the way for its application in the clinic for
the treatment of patients with S-AKI, perhaps reducing the
mortality rate.
5. Limitations
HPA-2 is a protein located on chromosome 10q23-24.
HPA-2 has 40% sequence homology with the coding region of another
protein, HPA-1, but despite this, they have different functions and
effects (117). The total length
of HPA-2 is 592 amino acids, with the two proteins, HPA-2 and
HPA-1, sharing 47% of their amino acids. HPA-2 can bind to HS, but
has no enzymatic activity and lacks the ability to degrade HS
(118). Despite its lack of
endoglycosidase activity, HPA-2 has a higher affinity for HS than
HPA-1; therefore, by competing for HS, it inhibits the enzymatic
activity of HPA-1(119). HPA-2
plays an important role in embryonic development, but its
mechanisms and biological functions remain unclear. HPA-2 appears
to be upregulated in benign and less aggressive tumors, and may
function as a tumor suppressor (118). At present, there are no studies
on the association between HPA-2, sepsis and S-AKI. Therefore, the
HPA mentioned in this review only refers to HPA-1 without much
focus on HPA-2.
6. Summary and future perspectives
HPA plays a major role in the pathogenesis of S-AKI,
but the precise molecular mechanism requires further exploration.
HPA may serve as a potential biomarker for the early diagnosis and
treatment of S-AKI. HPA mediates its action through exosomes in
S-AKI, which may transport potential biomarkers of S-AKI.
Therefore, such biomarkers could potentially offer a novel approach
to the diagnosis of S-AKI.
Early intervention with targeted therapy for
patients with S-AKI may improve their prognosis. HPA inhibitors,
exosomes and autophagy regulators could serve as potential
therapeutic strategies for S-AKI. Further studies exploring the
crosstalk between HPA, autophagy and exosomes are required to
determine the underlying molecular mechanisms.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the Science and
Technology Department of Gansu (grant no. 20JR5RA35), the Talent
Innovation and Entrepreneurship Project of Science and Technology
Bureau of Chengguan, Lanzhou (grant no. 2020RCCX0030), the Lanzhou
Science and Technology Development Guiding Plan Project (grant no.
2019-ZD-37), the Fund of The First Hospital of Lanzhou University
(grant no. Ldyyyn2018-48), The Open Topics of the Key Laboratory of
Biological Treatment and Regenerative Medicine in Gansu (grant no.
zdsyskfkt-201702), and the Science and Technology Program of Gansu
(grant no. 21JR7RA418).
Availability of data and materials
Not applicable.
Authors' contributions
LPL proposed the current study and was responsible
for its design and review. JCL designed and wrote the manuscript.
LJW analyzed the feasibility of the manuscript, researched the
literature and was responsible for reviewing the manuscript. FF and
TTC researched and reviewed the literature and the manuscript. WGS
corrected and revised the manuscript. Data authentication is not
applicable. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lameire NH, Bagga A, Cruz D, De Maeseneer
J, Endre Z, Kellum JA, Liu KD, Mehta RL, Pannu N, Van Biesen W and
Vanholder R: .: Acute kidney injury: An increasing global concern.
Lancet. 382:170–179. 2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hoste EA and Schurgers M: Epidemiology of
acute kidney injury: How big is the problem? Crit Care Med. 36 (4
Suppl):S146–S151. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Susantitaphong P, Cruz DN, Cerda J,
Abulfaraj M, Alqahtani F, Koulouridis I and Jaber BL: Acute Kidney
Injury Advisory Group of the American Society of Nephrology. World
incidence of AKI: A meta-analysis. Clin J Am Soc Nephrol.
8:1482–1493. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ronco C, Bellomo R and Kellum JA: Acute
kidney injury. Lancet. 394:1949–1964. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kellum JA, Chawla LS, Keener C, Singbartl
K, Palevsky PM, Pike FL, Yealy DM, Huang DT and Angus DC: ProCESS
and ProGReSS-AKI Investigators. The effects of alternative
resuscitation strategies on acute kidney injury in patients with
septic shock. Am J Respir Crit Care Med. 193:281–287.
2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mayeux PR and MacMillan-Crow LA:
Pharmacological targets in the renal peritubular microenvironment:
Implications for therapy for sepsis-induced acute kidney injury.
Pharmacol Ther. 134:139–155. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lygizos MI, Yang Y, Altmann CJ, Okamura K,
Hernando AA, Perez MJ, Smith LP, Koyanagi DE, Gandjeva A, Bhargava
R, et al: Heparanase mediates renal dysfunction during early sepsis
in mice. Physiol Rep. 1(e00153)2013.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Zhang Y, Huang H, Liu W, Liu S, Wang XY,
Diao ZL, Zhang AH, Guo W, Han X, Dong X and Katilov O: Endothelial
progenitor cells-derived exosomal microRNA-21-5p alleviates
sepsis-induced acute kidney injury by inhibiting RUNX1 expression.
Cell Death Dis. 12(335)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kellum JA and Lameire N: KDIGO AKI
Guideline Work Group. Diagnosis, evaluation, and management of
acute kidney injury: A KDIGO summary (Part 1). Crit Care.
17(204)2013.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The Third International Consensus
Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA.
315:801–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bonventre JV and Yang L: Cellular
pathophysiology of ischemic acute kidney injury. J Clin Invest.
121:4210–4221. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Schrier RW, Wang W, Poole B and Mitra A:
Acute renal failure: Definitions, diagnosis, pathogenesis, and
therapy. J Clin Invest. 114:5–14. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Schmidt C, Steinke T, Moritz S, Graf BM
and Bucher M: Acute renal failure and sepsis: Just an organ
dysfunction due to septic multiorgan failure? Anaesthesist.
59:682–699. 2010.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
14
|
Maiden MJ, Otto S, Brealey JK, Finnis ME,
Chapman MJ, Kuchel TR, Nash CH, Edwards J and Bellomo R: Structure
and function of the kidney in septic shock. A prospective
controlled experimental study. Am J Respir Crit Care Med.
194:692–700. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lerolle N, Nochy D, Guérot E, Bruneval P,
Fagon JY, Diehl JL and Hill G: Histopathology of septic shock
induced acute kidney injury: Apoptosis and leukocytic infiltration.
Intensive Care Med. 36:471–478. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Peerapornratana S, Manrique-Caballero CL,
Gómez H and Kellum JA: Acute kidney injury from sepsis: Current
concepts, epidemiology, pathophysiology, prevention and treatment.
Kidney Int. 96:1083–1099. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Poston JT and Koyner JL: Sepsis associated
acute kidney injury. BMJ. 364(k4891)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gomez H, Ince C, De Backer D, Pickkers P,
Payen D, Hotchkiss J and Kellum JA: A unified theory of
sepsis-induced acute kidney injury: Inflammation, microcirculatory
dysfunction, bioenergetics, and the tubular cell adaptation to
injury. Shock. 41:3–11. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bateman RM, Sharpe MD, Jagger JE and Ellis
CG: Sepsis impairs microvascular autoregulation and delays
capillary response within hypoxic capillaries. Crit Care.
19(389)2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ye C, Kawasaki M, Nakano K, Ohnishi T,
Watanabe E, Oda S, Nakada TA and Haneishi H: Acquisition and
analysis of microcirculation image in septic model rats. Sensors
(Basel). 22(8471)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ince C: The microcirculation is the motor
of sepsis. Crit Care. 9 (Suppl 4):S13–S19. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Joffre J, Hellman J, Ince C and
Ait-Oufella H: Endothelial responses in sepsis. Am J Respir Crit
Care Med. 202:361–370. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Anniss AM and Sparrow RL: Variable
adhesion of different red blood cell products to activated vascular
endothelium under flow conditions. Am J Hematol. 82:439–445.
2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ishikawa K, Calzavacca P, Bellomo R,
Bailey M and May CN: Effect of selective inhibition of renal
inducible nitric oxide synthase on renal blood flow and function in
experimental hyperdynamic sepsis. Crit Care Med. 40:2368–2375.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Heemskerk S, Pickkers P, Bouw MP, Draisma
A, van der Hoeven JG, Peters WH, Smits P, Russel FG and Masereeuw
R: Upregulation of renal inducible nitric oxide synthase during
human endotoxemia and sepsis is associated with proximal tubule
injury. Clin J Am Soc Nephrol. 1:853–862. 2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Inkinen N, Pettilä V, Lakkisto P, Kuitunen
A, Jukarainen S, Bendel S, Inkinen O, Ala-Kokko T and Vaara ST:
FINNAKI Study Group. Association of endothelial and glycocalyx
injury biomarkers with fluid administration, development of acute
kidney injury, and 90-day mortality: Data from the FINNAKI
observational study. Ann Intensive Care. 9(103)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Gustot T: Multiple organ failure in
sepsis: Prognosis and role of systemic inflammatory response. Curr
Opin Crit Care. 17:153–159. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhu J, Zhang Y, Shi L, Xia Y, Zha H, Li H
and Song Z: RP105 protects against ischemic and septic acute kidney
injury via suppressing TLR4/NF-κB signaling pathways. Int
Immunopharmacol. 109(108904)2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Krivan S, Kapelouzou A, Vagios S,
Tsilimigras DI, Katsimpoulas M, Moris D, Aravanis CV, Demesticha
TD, Schizas D, Mavroidis M, et al: Increased expression of
Toll-like receptors 2, 3, 4 and 7 mRNA in the kidney and intestine
of a septic mouse model. Sci Rep. 9(4010)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kawai T and Akira S: Signaling to
NF-kappaB by Toll-like receptors. Trends Mol Med. 13:460–469.
2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kawai T and Akira S: TLR signaling. Semin
Immunol. 19:24–32. 2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Leemans JC, Stokman G, Claessen N,
Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T,
Weening JJ and Florquin S: Renal-associated TLR2 mediates
ischemia/reperfusion injury in the kidney. J Clin Invest.
115:2894–2903. 2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
El-Achkar TM, Huang X, Plotkin Z, Sandoval
RM, Rhodes GJ and Dagher PC: Sepsis induces changes in the
expression and distribution of Toll-like receptor 4 in the rat
kidney. Am J Physiol Renal Physiol. 290:F1034–F1043.
2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Fani F, Regolisti G, Delsante M,
Cantaluppi V, Castellano G, Gesualdo L, Villa G and Fiaccadori E:
Recent advances in the pathogenetic mechanisms of sepsis-associated
acute kidney injury. J Nephrol. 31:351–359. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zafrani L, Gerotziafas G, Byrnes C, Hu X,
Perez J, Lévi C, Placier S, Letavernier E, Leelahavanichkul A,
Haymann JP, et al: Calpastatin controls polymicrobial sepsis by
limiting procoagulant microparticle release. Am J Respir Crit Care
Med. 185:744–755. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Stark K and Massberg S: Interplay between
inflammation and thrombosis in cardiovascular pathology. Nat Rev
Cardiol. 18:666–682. 2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Benedetti C, Waldman M, Zaza G, Riella LV
and Cravedi P: COVID-19 and the Kidneys: An update. Front Med
(Lausanne). 7(423)2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Toro J, Manrique-Caballero CL and Gómez H:
Metabolic reprogramming and host tolerance: A novel concept to
understand sepsis-associated AKI. J Clin Med.
10(4184)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wilson DF: Oxidative phosphorylation:
Regulation and role in cellular and tissue metabolism. J Physiol.
595:7023–7038. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Waltz P, Carchman E, Gomez H and
Zuckerbraun B: Sepsis results in an altered renal metabolic and
osmolyte profile. J Surg Res. 202:8–12. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang D, Qi B, Li D, Feng J, Huang X, Ma
X, Huang L, Wang X and Liu X: Phillyrin relieves
lipopolysaccharide-induced AKI by protecting against glycocalyx
damage and inhibiting inflammatory responses. Inflammation.
43:540–551. 2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Vlodavsky I, Singh P, Boyango I,
Gutter-Kapon L, Elkin M, Sanderson RD and Ilan N: Heparanase: From
basic research to therapeutic applications in cancer and
inflammation. Drug Resist Updat. 29:54–75. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Goldberg R, Meirovitz A, Hirshoren N,
Bulvik R, Binder A, Rubinstein AM and Elkin M: Versatile role of
heparanase in inflammation. Matrix Biol. 32:234–240.
2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Goldberg R, Rubinstein AM, Gil N, Hermano
E, Li JP, van der Vlag J, Atzmon R, Meirovitz A and Elkin M: Role
of heparanase-driven inflammatory cascade in pathogenesis of
diabetic nephropathy. Diabetes. 63:4302–4313. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Goldshmidt O, Zcharia E, Abramovitch R,
Metzger S, Aingorn H, Friedmann Y, Schirrmacher V, Mitrani E and
Vlodavsky I: Cell surface expression and secretion of heparanase
markedly promote tumor angiogenesis and metastasis. Proc Natl Acad
Sci USA. 99:10031–10036. 2002.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Parish CR, Freeman C, Ziolkowski AF, He
YQ, Sutcliffe EL, Zafar A, Rao S and Simeonovic CJ: Unexpected new
roles for heparanase in type 1 diabetes and immune gene regulation.
Matrix Biol. 32:228–233. 2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Meirovitz A, Goldberg R, Binder A,
Rubinstein AM, Hermano E and Elkin M: Heparanase in inflammation
and inflammation-associated cancer. FEBS J. 280:2307–2319.
2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Levey AS and James MT: Acute kidney
injury. Ann Intern Med. 167:ITC66–ITC80. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Masola V, Zaza G, Onisto M, Lupo A and
Gambaro G: Impact of heparanase on renal fibrosis. J Transl Med.
13(181)2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Rosenfeldt MT and Ryan KM: The multiple
roles of autophagy in cancer. Carcinogenesis. 32:955–963.
2011.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Bishop JR, Schuksz M and Esko JD: Heparan
sulphate proteoglycans fine-tune mammalian physiology. Nature.
446:1030–1037. 2007.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Bernfield M, Götte M, Park PW, Reizes O,
Fitzgerald ML, Lincecum J and Zako M: Functions of cell surface
heparan sulfate proteoglycans. Annu Rev Biochem. 68:729–777.
1999.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Goldshmidt O, Nadav L, Aingorn H, Irit C,
Feinstein N, Ilan N, Zamir E, Geiger B, Vlodavsky I and Katz BZ:
Human heparanase is localized within lysosomes in a stable form.
Exp Cell Res. 281:50–62. 2002.PubMed/NCBI View Article : Google Scholar
|
|
54
|
van den Hoven MJ, Rops AL, Vlodavsky I,
Levidiotis V, Berden JH and van der Vlag J: Heparanase in
glomerular diseases. Kidney Int. 72:543–548. 2007.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Gaskin SM, Soares Da Costa TP and Hulett
MD: Heparanase: Cloning, function and regulation. Adv Exp Med Biol.
1221:189–229. 2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Masola V, Bellin G, Gambaro G and Onisto
M: Heparanase: A multitasking protein involved in extracellular
matrix (ECM) remodeling and intracellular events. Cells.
7(236)2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Sanderson RD, Elkin M, Rapraeger AC, Ilan
N and Vlodavsky I: Heparanase regulation of cancer, autophagy and
inflammation: new mechanisms and targets for therapy. FEBS J.
284:42–55. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
David G and Zimmermann P: Heparanase
involvement in exosome formation. Adv Exp Med Biol. 1221:285–307.
2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Simons M and Raposo G: Exosomes-vesicular
carriers for intercellular communication. Curr Opin Cell Biol.
21:575–581. 2009.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Shteingauz A, Boyango I, Naroditsky I,
Hammond E, Gruber M, Doweck I, Ilan N and Vlodavsky I: Heparanase
enhances tumor growth and chemoresistance by promoting autophagy.
Cancer Res. 75:3946–3957. 2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Schmidt EP, Overdier KH, Sun X, Lin L, Liu
X, Yang Y, Ammons LA, Hiller TD, Suflita MA, Yu Y, et al: Urinary
glycosaminoglycans predict outcomes in septic shock and acute
respiratory distress syndrome. Am J Respir Crit Care Med.
194:439–449. 2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Masola V, Zaza G, Bellin G, Dall'Olmo L,
Granata S, Vischini G, Secchi MF, Lupo A, Gambaro G and Onisto M:
Heparanase regulates the M1 polarization of renal macrophages and
their crosstalk with renal epithelial tubular cells after
ischemia/reperfusion injury. FASEB J. 32:742–756. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Abassi Z, Hamoud S, Hassan A, Khamaysi I,
Nativ O, Heyman SN, Muhammad RS, Ilan N, Singh P, Hammond E, et al:
Involvement of heparanase in the pathogenesis of acute kidney
injury: Nephroprotective effect of PG545. Oncotarget.
8:34191–34204. 2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Masola V, Zaza G, Gambaro G, Onisto M,
Bellin G, Vischini G, Khamaysi I, Hassan A, Hamoud S, Nativ O, et
al: Heparanase: A potential new factor involved in the renal
epithelial mesenchymal transition (EMT) induced by
ischemia/reperfusion (I/R) Injury. PLoS One.
11(e0160074)2016.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Abu-Tayeh Suleiman H, Said S, Ali Saleh H,
Gamliel-Lazarovich A, Haddad E, Minkov I, Zohar Y, Ilan N,
Vlodavsky I, Abassi Z and Assady S: Heparanase increases podocyte
survival and autophagic flux after adriamycin-induced injury. Int J
Mol Sci. 23(12691)2022.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Ilan N, Elkin M and Vlodavsky I:
Regulation, function and clinical significance of heparanase in
cancer metastasis and angiogenesis. Int J Biochem Cell Biol.
38:2018–2039. 2006.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Parish CR, Freeman C and Hulett MD:
Heparanase: A key enzyme involved in cell invasion. Biochim Biophys
Acta. 1471:M99–M108. 2001.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Secchi MF, Masola V, Zaza G, Lupo A,
Gambaro G and Onisto M: Recent data concerning heparanase: Focus on
fibrosis, inflammation and cancer. Biomol Concepts. 6:415–421.
2015.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Vlodavsky I, Beckhove P, Lerner I, Pisano
C, Meirovitz A, Ilan N and Elkin M: Significance of heparanase in
cancer and inflammation. Cancer Microenviron. 5:115–132.
2012.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Vreys V and David G: Mammalian heparanase:
What is the message? J Cell Mol Med. 11:427–452. 2007.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Sanderson RD, Bandari SK and Vlodavsky I:
Proteases and glycosidases on the surface of exosomes: Newly
discovered mechanisms for extracellular remodeling. Matrix Biol.
75-76:160–169. 2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Xavier RJ and Podolsky DK: Unravelling the
pathogenesis of inflammatory bowel disease. Nature. 448:427–434.
2007.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Belmiro CL, Souza HS, Elia CC,
Castelo-Branco MT, Silva FR, Machado RL and Pavão MS: Biochemical
and immunohistochemical analysis of glycosaminoglycans in inflamed
and non-inflamed intestinal mucosa of patients with Crohn's
disease. Int J Colorectal Dis. 20:295–304. 2005.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Abassi Z and Goligorsky MS: Heparanase in
acute kidney injury. Adv Exp Med Biol. 1221:685–702.
2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Axelsson J, Xu D, Kang BN, Nussbacher JK,
Handel TM, Ley K, Sriramarao P and Esko JD: Inactivation of heparan
sulfate 2-O-sulfotransferase accentuates neutrophil infiltration
during acute inflammation in mice. Blood. 120:1742–1751.
2012.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Götte M: Syndecans in inflammation. FASEB
J. 17:575–591. 2003.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Carter NM, Ali S and Kirby JA: Endothelial
inflammation: The role of differential expression of
N-deacetylase/N-sulphotransferase enzymes in alteration of the
immunological properties of heparan sulphate. J Cell Sci. 116(Pt
17):3591–3600. 2003.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Uchimido R, Schmidt EP and Shapiro NI: The
glycocalyx: A novel diagnostic and therapeutic target in sepsis.
Crit Care. 23(16)2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Becker BF, Jacob M, Leipert S, Salmon AH
and Chappell D: Degradation of the endothelial glycocalyx in
clinical settings: Searching for the sheddases. Br J Clin
Pharmacol. 80:389–402. 2015.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Lupu F, Kinasewitz G and Dormer K: The
role of endothelial shear stress on haemodynamics, inflammation,
coagulation and glycocalyx during sepsis. J Cell Mol Med.
24:12258–12271. 2020.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Ponticelli C: Ischaemia-reperfusion
injury: A major protagonist in kidney transplantation. Nephrol Dial
Transplant. 29:1134–1140. 2014.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Bayam E, Kalçık M, Gürbüz AS, Yesin M,
Güner A, Gündüz S, Gürsoy MO, Karakoyun S, Cerşit S, Kılıçgedik A,
et al: The relationship between heparanase levels, thrombus burden
and thromboembolism in patients receiving unfractionated heparin
treatment for prosthetic valve thrombosis. Thromb Res. 171:103–110.
2018.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Masola V, Gambaro G, Tibaldi E, Brunati
AM, Gastaldello A, D'Angelo A, Onisto M and Lupo A: Heparanase and
syndecan-1 interplay orchestrates fibroblast growth
factor-2-induced epithelial-mesenchymal transition in renal tubular
cells. J Biol Chem. 287:1478–1488. 2012.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Jiang P and Mizushima N: Autophagy and
human diseases. Cell Res. 24:69–79. 2014.PubMed/NCBI View Article : Google Scholar
|
|
85
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Singh R and Cuervo AM: Autophagy in the
cellular energetic balance. Cell Metab. 13:495–504. 2011.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Melk A, Baisantry A and Schmitt R: The yin
and yang of autophagy in acute kidney injury. Autophagy.
12:596–597. 2016.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Kim WY, Nam SA, Song HC, Ko JS, Park SH,
Kim HL, Choi EJ, Kim YS, Kim J and Kim YK: The role of autophagy in
unilateral ureteral obstruction rat model. Nephrology (Carlton).
17:148–159. 2012.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Zhang M, Sui W, Xing Y, Cheng J, Cheng C,
Xue F, Zhang J, Wang X, Zhang C, Hao P and Zhang Y: Angiotensin IV
attenuates diabetic cardiomyopathy via suppressing FoxO1-induced
excessive autophagy, apoptosis and fibrosis. Theranostics.
11:8624–8639. 2021.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Jin H and Zhou S: The functions of
heparanase in human diseases. Mini Rev Med Chem. 17:541–548.
2017.PubMed/NCBI View Article : Google Scholar
|
|
91
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri
S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K,
et al: Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187.
2011.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Ferro V, Dredge K, Liu L, Hammond E,
Bytheway I, Li C, Johnstone K, Karoli T, Davis K, Copeman E and
Gautam A: PI-88 and novel heparan sulfate mimetics inhibit
angiogenesis. Semin Thromb Hemost. 33:557–568. 2007.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Rabelink TJ, van den Berg BM, Garsen M,
Wang G, Elkin M and van der Vlag J: Heparanase: Roles in cell
survival, extracellular matrix remodelling and the development of
kidney disease. Nat Rev Nephrol. 13:201–212. 2017.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Suchorska WM and Lach MS: The role of
exosomes in tumor progression and metastasis (Review). Oncol Rep.
35:1237–1244. 2016.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Oosthuyzen W, Sime NE, Ivy JR, Turtle EJ,
Street JM, Pound J, Bath LE, Webb DJ, Gregory CD, Bailey MA and
Dear JW: Quantification of human urinary exosomes by nanoparticle
tracking analysis. J Physiol. 591:5833–5842. 2013.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Petrik J and Seghatchian J: Big things
from small packages: The multifaceted roles of extracellular
vesicles in the components quality, therapy and infection. Transfus
Apher Sci. 55:4–8. 2016.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Conlan RS, Pisano S, Oliveira MI, Ferrari
M and Mendes Pinto I: Exosomes as reconfigurable therapeutic
systems. Trends Mol Med. 23:636–650. 2017.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Essandoh K, Yang L, Wang X, Huang W, Qin
D, Hao J, Wang Y, Zingarelli B, Peng T and Fan GC: Blockade of
exosome generation with GW4869 dampens the sepsis-induced
inflammation and cardiac dysfunction. Biochim Biophys Acta.
1852:2362–2371. 2015.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Kanki M, Moriguchi A, Sasaki D, Mitori H,
Yamada A, Unami A and Miyamae Y: Identification of urinary miRNA
biomarkers for detecting cisplatin-induced proximal tubular injury
in rats. Toxicology. 324:158–168. 2014.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Viñas JL, Spence M, Porter CJ, Douvris A,
Gutsol A, Zimpelmann JA, Campbell PA and Burns KD: micro-RNA-486-5p
protects against kidney ischemic injury and modifies the apoptotic
transcriptome in proximal tubules. Kidney Int. 100:597–612.
2021.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Sun J, Sun X, Chen J, Liao X, He Y, Wang
J, Chen R, Hu S and Qiu C: microRNA-27b shuttled by mesenchymal
stem cell-derived exosomes prevents sepsis by targeting JMJD3 and
downregulating NF-κB signaling pathway. Stem Cell Res Ther.
12(14)2021.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Zhang R, Zhu Y, Li Y, Liu W, Yin L, Yin S,
Ji C, Hu Y, Wang Q, Zhou X, et al: Human umbilical cord mesenchymal
stem cell exosomes alleviate sepsis-associated acute kidney injury
via regulating microRNA-146b expression. Biotechnol Lett.
42:669–679. 2020.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Juan CX, Mao Y, Cao Q, Chen Y, Zhou LB, Li
S, Chen H, Chen JH, Zhou GP and Jin R: Exosome-mediated pyroptosis
of miR-93-TXNIP-NLRP3 leads to functional difference between M1 and
M2 macrophages in sepsis-induced acute kidney injury. J Cell Mol
Med. 25:4786–4799. 2021.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong
X, Wu WJ, Chen J, Ni HF, Tang TT, et al: Exosomal miRNA-19b-3p of
tubular epithelial cells promotes M1 macrophage activation in
kidney injury. Cell Death Differ. 27:210–226. 2020.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Thompson CA, Purushothaman A, Ramani VC,
Vlodavsky I and Sanderson RD: Heparanase regulates secretion,
composition, and function of tumor cell-derived exosomes. J Biol
Chem. 288:10093–10099. 2013.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Roucourt B, Meeussen S, Bao J, Zimmermann
P and David G: Heparanase activates the syndecan-syntenin-ALIX
exosome pathway. Cell Res. 25:412–428. 2015.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Bernfield M and Sanderson RD: Syndecan, a
developmentally regulated cell surface proteoglycan that binds
extracellular matrix and growth factors. Philos Trans R Soc Lond B
Biol Sci. 327:171–186. 1990.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Baietti MF, Zhang Z, Mortier E, Melchior
A, Degeest G, Geeraerts A, Ivarsson Y, Depoortere F, Coomans C,
Vermeiren E, et al: Syndecan-syntenin-ALIX regulates the biogenesis
of exosomes. Nat Cell Biol. 14:677–685. 2012.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Bandari SK, Purushothaman A, Ramani VC,
Brinkley GJ, Chandrashekar DS, Varambally S, Mobley JA, Zhang Y,
Brown EE, Vlodavsky I and Sanderson RD: Chemotherapy induces
secretion of exosomes loaded with heparanase that degrades
extracellular matrix and impacts tumor and host cell behavior.
Matrix Biol. 65:104–118. 2018.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Cummings JJ, Shaw AD, Shi J, Lopez MG,
O'Neal JB and Billings FT IV: Intraoperative prediction of cardiac
surgery-associated acute kidney injury using urinary biomarkers of
cell cycle arrest. J Thorac Cardiovasc Surg. 157:1545–1553.e5.
2019.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Parikh CR, Thiessen-Philbrook H, Garg AX,
Kadiyala D, Shlipak MG, Koyner JL, Edelstein CL, Devarajan P, Patel
UD, Zappitelli M, et al: Performance of kidney injury molecule-1
and liver fatty acid-binding protein and combined biomarkers of AKI
after cardiac surgery. Clin J Am Soc Nephrol. 8:1079–1088.
2013.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Nakamura T, Sugaya T, Node K, Ueda Y and
Koide H: Urinary excretion of liver-type fatty acid-binding protein
in contrast medium-induced nephropathy. Am J Kidney Dis.
47:439–444. 2006.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Mori K, Lee HT, Rapoport D, Drexler IR,
Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, et al:
Endocytic delivery of lipocalin-siderophore-iron complex rescues
the kidney from ischemia-reperfusion injury. J Clin Invest.
115:610–621. 2005.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Chen S, He Y, Hu Z, Lu S, Yin X, Ma X, Lv
C and Jin G: Heparanase mediates intestinal inflammation and injury
in a mouse model of sepsis. J Histochem Cytochem. 65:241–249.
2017.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Kiyan Y, Tkachuk S, Kurselis K, Shushakova
N, Stahl K, Dawodu D, Kiyan R, Chichkov B and Haller H:
Heparanase-2 protects from LPS-mediated endothelial injury by
inhibiting TLR4 signalling. Sci Rep. 9(13591)2019.PubMed/NCBI View Article : Google Scholar
|
|
117
|
McKenzie E, Tyson K, Stamps A, Smith P,
Turner P, Barry R, Hircock M, Patel S, Barry E, Stubberfield C, et
al: Cloning and expression profiling of Hpa2, a novel mammalian
heparanase family member. Biochem Biophys Res Commun.
276:1170–1177. 2000.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Pinhal MAS, Melo CM and Nader HB: The good
and bad sides of heparanase-1 and heparanase-2. Adv Exp Med Biol.
1221:821–845. 2020.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Bashkin P, Doctrow S, Klagsbrun M, Svahn
CM, Folkman J and Vlodavsky I: Basic fibroblast growth factor binds
to subendothelial extracellular matrix and is released by
heparitinase and heparin-like molecules. Biochemistry.
28:1737–1743. 1989.PubMed/NCBI View Article : Google Scholar
|














