Introduction
β-Amyloid peptide (Aβ) deposition in the brain is
one of the most important pathological changes that occur in
Alzheimer's disease (AD) (1).
Previous studies have suggested that Aβ is able to cause the
phosphorylation of the tubulin associated unit protein, which leads
to the formation of neurofibrillary tangles (2), in addition to exerting a role in
synaptic dysfunction (3), neuron
loss (4), microglia activation and
neuroinflammation (5), calcium
deregulation (6), oxidative stress
(7), mitochondrial dysfunction
(8), and cholinergic dysfunction
(9), all of which ultimately
affect cognitive function and lead to the development of AD.
Insulin-degrading enzyme (IDE) is a target gene of a nuclear
transcription factor called peroxisome proliferator-activated
receptor (PPAR) γ (10). IDE is
able to effectively proteolyze Aβ in the brain (11). At present, targeted IDE therapy has
become one of the hotspots of research for AD treatment (12).
Ginseng is the dried root of Panax ginseng,
which has been used as a natural medicine for thousands of years in
Asia, especially in China, due its effects of reinforcing vitality,
increasing bodily resistance and enhancing cognitive ability
(13,14). Ginsenoside Rg1 is one of the active
ingredients of ginseng, and it has been demonstrated to exert
numerous neuroprotective effects on AD (15), including alleviating oxidative
stress damage (16), improving the
bioenergetics and morphology of mitochondria (17), and altering the gut microbiota
(18). A recent study (19) indicated that ginsenoside Rg1 exerts
a scavenging effect on Aβ, which is able to inhibit the
phosphorylation of PPARγ at Ser273 through inhibition of the
cyclin-dependent kinase 5 expression pathway, subsequently reducing
Aβ production and exerting neuroprotective effects on AD by
affecting the expression levels of IDE and β-amyloid
cleavage enzyme 1 (BACE1), which are targeted by PPARγ. The
MAPK family comprises a series of serine-threonine protein kinases
that form a common signaling pathway through which extracellular
signals elicit nuclear responses such as gene expression, cell
proliferation and apoptosis (20,21).
Upon activation by extracellular stimulation, MAPK is translocated
from the cytoplasm to the nucleus, thereby causing the
phosphorylation of nuclear transcription factors, and subsequently
regulates the transcriptional levels of the corresponding genes
(22). It has been demonstrated
that ERK, a member of the MAPK family, is able to mediate the
phosphorylation of the A/B structural domain of PPARγ at Ser112,
thereby inhibiting its transcriptional activity (23).
The present study aimed to investigate whether
ginsenoside Rg1 could affect the regulation of PPARγ based on the
expression of its target gene, IDE, and whether it could
promote Aβ degradation via inhibition of the ERK/PPARγ
phosphorylation pathway.
Materials and methods
Reagents
Ginsenoside Rg1 was purchased from Baoji Chenguang
Biotechnology Co., Ltd.; it has the molecular formula
C42H72O14, a molecular weight of
801.01 and high-performance liquid chromatography was performed by
Baoji Chenguang Biotechnology Co., Ltd. to determine that the
purity was ~98%. Dulbecco's modified Eagle's medium, neurobasal
medium and B27 were purchased from Gibco; Thermo Fisher Scientific,
Inc., fetal bovine serum was purchased from Biological Industries,
glutamine and cytarabine were purchased from Aladdin, goat serum
was purchased from Beijing Solarbio, Aβ1-42 was
purchased from MilliporeSigma and PD98059 was purchased from Abcam.
Streptomycin, penicillin and rabbit anti-rat phosphorylated
(p-)PPARγ-Ser112 (cat. no. LM-3737R) polyclonal antibody were
obtained from LMAI Bio Co., Ltd. Mouse anti-rat ERK monoclonal
antibody (cat. no. bsm-33232M), and rabbit anti-rat ERK (cat. no.
bs-0022R), IDE (cat. no. bs-0018R), PPARγ (cat. no. bs-4590R),
Histone H3 (cat. no. bs-17422R), GAPDH (cat. no. bs-41373R) and
β-actin (cat. no. bs-0061R) polyclonal antibodies were purchased
from BIOSS. Horseradish peroxidase-labeled goat anti-rabbit
secondary antibody (cat. no. WLA023), and the TUNEL assay, BCA
protein concentration assay (cat. no. WLA004), whole cell lysis
assay (cat. no. WLA019) and nuclear and cytoplasmic protein
extraction (cat. no. WLA020) kits, and ECL luminescent solution
were purchased from Wanleibio Co., Ltd. Aβ1-42 ELISA kit
(cat. no. CEA946Ra) was purchased from Wuhan USCN Business Co.,
Ltd. Trypsin, RIPA, DAPI, Cy3-labeled goat anti-rabbit IgG (cat.
no. A0516; for red fluorescence) and FITC-labeled goat anti-mouse
IgG (cat. no. A0568; for green fluorescence) were obtained from
Beyotime Institute of Biotechnology.
Rat hippocampal neuron isolation and
culture
A total of 130 2-day-old Sprague-Dawley rats (weight
range, 8-10 g; males, 65; females, 65) were purchased from the
Experimental Animal Center of Xi'an Jiaotong University Health
Science Center [License no. SCXK (Shaan) 2018-001]. These animals
were kept in a specific pathogen-free facility with a 12/12-h
light/dark cycle at a temperature of 25˚C and a humidity of 50-65%
and were allowed free access to breastmilk from their mothers
before the study began. After being obtained, these animals were
temporarily housed in a cage covered with soft bedding at a
temperature of 25˚C, and then moved to a specialized room where
euthanasia via decapitation was performed. Before euthanasia, the
health and behavior of these animals, including their mental and
breathing state, and activity, and whether these animals exhibited
anxiety, restlessness and squeaking, were monitored every 15 min.
To minimize suffering and distress, the animals were rapidly
decapitated using a clean, sharp and regularly maintained
guillotine device (cat. no. ZK-ZSQ-SD; ChiCo JX Co., Ltd.) by a
skilled operator, and then, the brain tissue was removed under
aseptic conditions. Subsequently, the hippocampal tissue was
isolated, digested with trypsin and centrifuged (310 x g; 7 min;
25˚C) to obtain the neurons. The neurons were inoculated into
six-well culture plates at a density of 5x105 cells/ml
and incubated with Dulbecco's modified Eagle's medium containing
10% fetal bovine serum at 37˚C in an atmosphere of 5%
CO2 and saturated humidity for 8 h. Then, Dulbecco's
modified Eagle's medium was changed to neurobasal medium containing
glutamine (0.5 mmol/l), B27 (2%), streptomycin (100 U/ml) and
penicillin (100 U/ml) at 37˚C for 48 h. Subsequently, cytarabine
(10 µM) was added to inhibit glial cell growth. The solution was
changed every 3 days. The rat hippocampal neurons, which were
observed to be maturing and forming networks after ~15 days of
incubation at 37˚C, were ready for subsequent experiments.
Drug administration
Drug administration was performed at 25˚C and
repeated 6 times. The cultured neurons were divided into a blank
control group, a model group and a ginsenoside Rg1 treatment group.
The neurons of the model group were treated with Aβ1-42
(final concentration, 8 µM) (24)
for 24 h, whereas the neurons of the ginsenoside Rg1 group was
pretreated with ginsenoside Rg1 (final concentration, 60 µM)
(25) for 1 h and subsequently
co-treated with Aβ1-42 (final concentration, 8 µM) for
24 h; by contrast, no drugs were administered to the neurons of the
blank control group. To further confirm that ginsenoside Rg1 exerts
anti-Aβ effects through acting on ERK to regulate PPARγ
phosphorylation, the ERK inhibitor PD98059 was used to inhibit ERK,
and the effects of ginsenoside Rg1 on Aβ1-42-treated
neurons were observed. The neurons were divided into three groups
as follows: i) Model group, cultured neurons were treated with
Aβ1-42 (final concentration, 8 µM) for 24 h; ii) PD98059
group, the cultured neurons were pretreated with PD98059 (final
concentration, 20 µM) (26) for 1
h, and subsequently co-treated with Aβ1-42 (final
concentration, 8 µM) for 24 h; and iii) ginsenoside Rg1 + PD98059
group, the cultured neurons were treated with PD98059 (final
concentration, 20 µM) for 0.5 h, subsequently ginsenoside Rg1
(final concentration, 60 µM) was added for pretreatment for 1 h,
and finally, Aβ1-42 (final concentration, 8 µM) was
added for co-treatment for 24 h.
TUNEL staining
After cultured cells were placed on the
poly-L-lysine-coated slides (cell density, 70-80%), they were dried
at 25˚C. Subsequently, cells were immersed in 4% paraformaldehyde
solution and fixed for 30 min at 25˚C. Next, 0.1% Triton X-100/10
mM PBS was added dropwise to permeabilize the cells for 5 min.
After rinsing with PBS, the TUNEL reaction solution was added
dropwise to the cells, and the mixture was incubated for 60 min at
37˚C with humidification away from light. Subsequently, cells were
rinsed with PBS, followed by counterstaining with DAPI (5.7 µM) in
the dark for 5 min at 25˚C. After having been rinsed with PBS
again, the slides were sealed with a fluorescence quencher. Neurons
were counted by two pathologists under a fluorescence microscope
(cat. no. BX53; Olympus Corporation) at a magnification of x400 in
a blinded manner. Each pathologist observed four random fields of
view of each slide. First, the total numbers of neurons
(DAPI+ neurons) and apoptotic neurons (TUNEL+
neurons) in each field of view were determined to obtain the
percentage of apoptotic neurons. Then, the average percentage of
apoptotic neurons was evaluated for the four fields of view using
the results from each pathologist. Finally, the average percentage
from the two pathologists was calculated as the final result.
Immunofluorescence staining
After fixing the neurons with 4% paraformaldehyde
for 15 min at 25˚C, 0.1% Triton X-100 was added for
permeabilization for 30 min at 25˚C. 10% goat serum was
subsequently added dropwise for blocking for 15 min at 25˚C. The
rabbit anti-rat ERK antibody (1:200) was then added for a 12 h
incubation at 4˚C. After washing with PBS, Cy3-labeled goat
anti-rabbit IgG (for red fluorescence; 1:200) was added, with a
further incubation for 60 min at 25˚C away from the light.
Subsequently, the nuclei were subjected to counterstaining with
DAPI in the dark for 5 min at 25˚C. Finally, slides were sealed by
dropwise addition of fluorescence quencher. ERK subcellular
localization in the neurons was observed under a fluorescence
microscope (BX53; Olympus Corporation).
To observe the binding of ERK to PPARγ,
immunofluorescence double staining was performed for ERK and PPARγ.
The neurons were fixed with 4% paraformaldehyde for 15 min at 25˚C,
and then permeabilized with 0.1% Triton X-100 for 30 min at 25˚C.
After blocking the neurons with 10% goat serum for 15 min at 25˚C,
rabbit anti-rat polyclonal PPARγ antibody (1:200) and mouse
anti-rat ERK monoclonal antibody (1:200) were added for incubation
overnight at 4˚C. After washing with PBS, Cy3-labeled goat
anti-rabbit IgG (for red fluorescence; 1:200) and FITC-labeled goat
anti-mouse IgG (for green fluorescence; 1:200) were added for
incubation for 60 min at 25˚C in the dark. After rinsing with PBS,
the nuclei were counterstained with DAPI in the dark for 5 min at
25˚C. After rinsing a further time with PBS, the fluorescence
quencher was added dropwise to mount the slides. Finally, the sites
of expression of ERK and PPARγ in the neurons were observed under a
fluorescence microscope (cat. no. BX53; Olympus Corporation).
ELISA
The cultured cells were collected and lysed on ice
for 15 min by adding a lysis solution of RIPA and PMSF (ratio,
100:1). Subsequently, the cells were centrifuged at 12,000 x g for
10 min at 4˚C. After separating the supernatant from the pellet,
the level of intracellular Aβ was detected using a rat
Aβ1-42 ELISA kit (Wuhan USCN Business Co., Ltd.)
according to the manufacturer's instructions. To detect the level
of extracellular Aβ1-42, the culture medium of each
group was collected and centrifuged at 3,000 x g for 10 min at 4˚C
to obtain the supernatant. The concentration of Aβ1-42
in the supernatant was detected using the aforementioned rat
Aβ1-42 ELISA kit according to the manufacturer's
instructions.
Western blotting
The neuronal nuclear protein, cytoplasmic protein
and total protein fractions were extracted respectively using
nuclear and cytoplasmic protein extraction and whole cell lysis
assay kits (Wanleibio Co., Ltd.) according to the instructions of
these kits. A BCA assay was performed to determine protein
concentration (Wanleibio Science Co., Ltd.). Subsequently, the
proteins were denatured by heating at 95˚C, and 40 µg protein was
loaded each electrophoretic lane and separated by electrophoresis
on 8% SDS-PAGE gels. After having been transferred to
nitrocellulose membranes, the separated proteins were subjected to
blocking using 5% skimmed milk powder at 37˚C for 1 h.
Subsequently, the levels of nuclear and cytoplasmic ERK protein,
nuclear PPARγ, nuclear p-PPARγ-Ser112 protein and whole-cell IDE
protein were separately measured. Nitrocellulose membranes were
incubated with rabbit anti-rat ERK, p-PPARγ-Ser112, PPARγ, IDE,
Histone H3, GAPDH and β-actin polyclonal antibodies (diluted in 5%
skimmed milk powder; 1:600, 1:500, 1:500, 1:800, 1:800, 1:800 and
1:1,000, respectively), overnight at 4˚C, followed by washes with
Tris-buffered saline containing 0.05% Tween 20. Subsequently,
nitrocellulose membranes were treated with horseradish
peroxidase-labeled goat anti-rabbit secondary antibody (1:900) at
37˚C for 45 min. Protein bands were visualized using an ECL
chemiluminescent reagent. Gel-Pro-Analyzer software (version 4.0;
Media Cybernetics, Inc.) was used to analyze the optical density
values of the target bands, with Histone H3 as the loading control
for nuclear ERK, p-PPARγ-Ser112 and PPARγ proteins, with GAPDH on
cytoplasmic ERK protein and with β-actin on whole IDE protein.
Statistical analysis
Data are presented as the mean ± SEM obtained from 6
independent experiments. Statistical analyses were performed using
SPSS (version 19.0; IBM Corp.). One-way ANOVA was used for
comparisons among groups, followed by the least significant
difference post hoc test for pairwise comparisons between groups.
P<0.05 was considered to indicate a statistically
significant difference.
Results
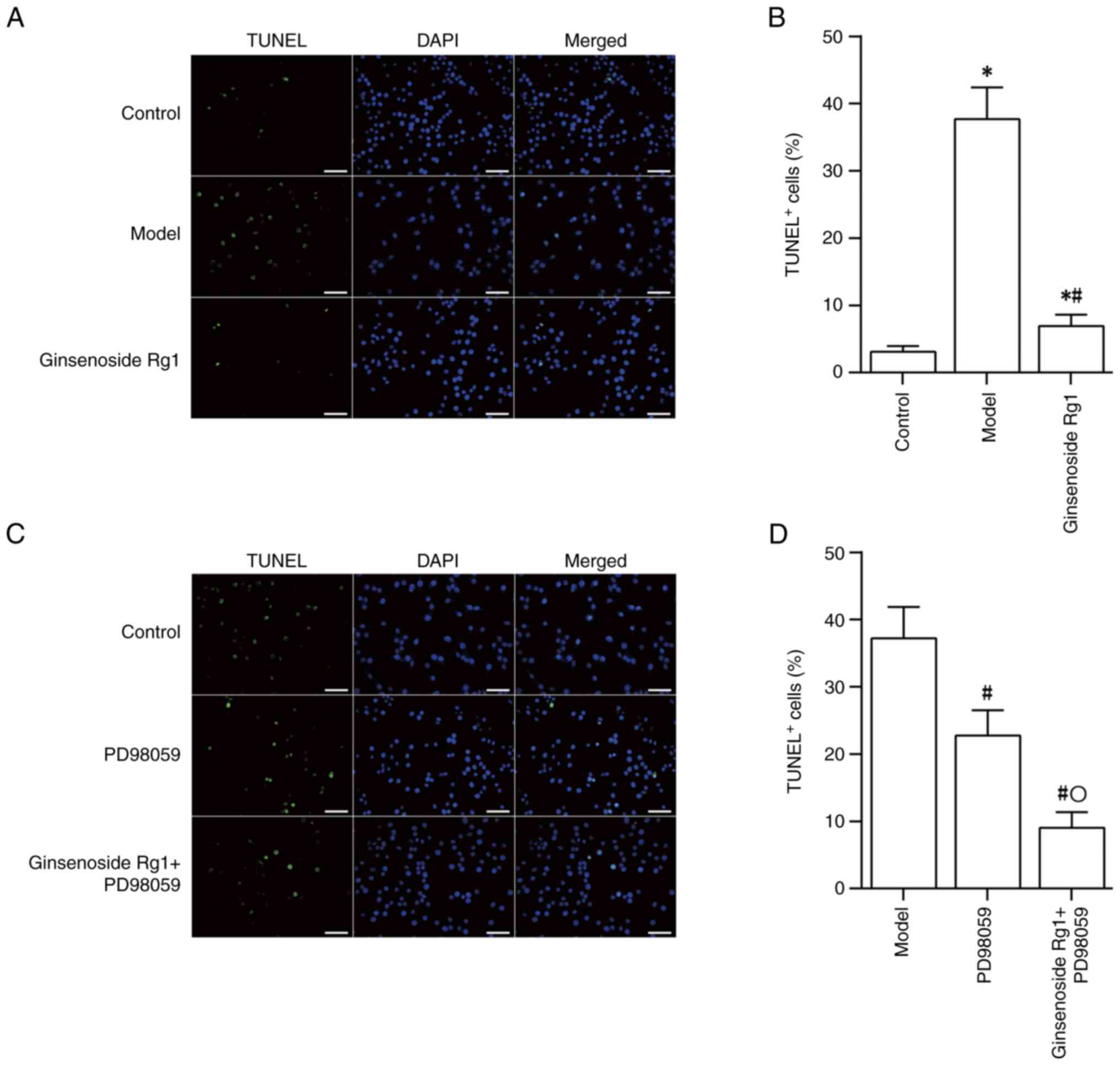
Neuroprotective effects of ginsenoside
Rg1 on an AD neuronal model
After treating primary cultured hippocampal neurons
with Aβ1-42, number of TUNEL+ neurons, as
detected by TUNEL staining, was increased significantly (control
group vs. model group; P<0.05), which suggested that
Aβ1-42 could induce neuronal apoptosis. Pretreatment
with ginsenoside Rg1 prior to treatment with Aβ1-42 was
found to reduce the TUNEL+ rate in neurons (ginsenoside
Rg1 group vs. model group; P<0.05), indicating that ginsenoside
Rg1 could effectively reduce neuronal apoptosis in the AD model
(Fig. 1A and B). The use of the ERK inhibitor PD98059
also led to a reduction in the rate of neuronal apoptosis induced
by Aβ1-42 (model group vs. PD98059 group; P<0.05).
Treatment with PD98059 followed by ginsenoside Rg1 resulted in a
further decrease in neuronal apoptosis compared with that resulting
from treatment with PD98059 alone (PD98059 group vs. ginsenoside
Rg1 + PD98059; P<0.05; Fig. 1C
and D).
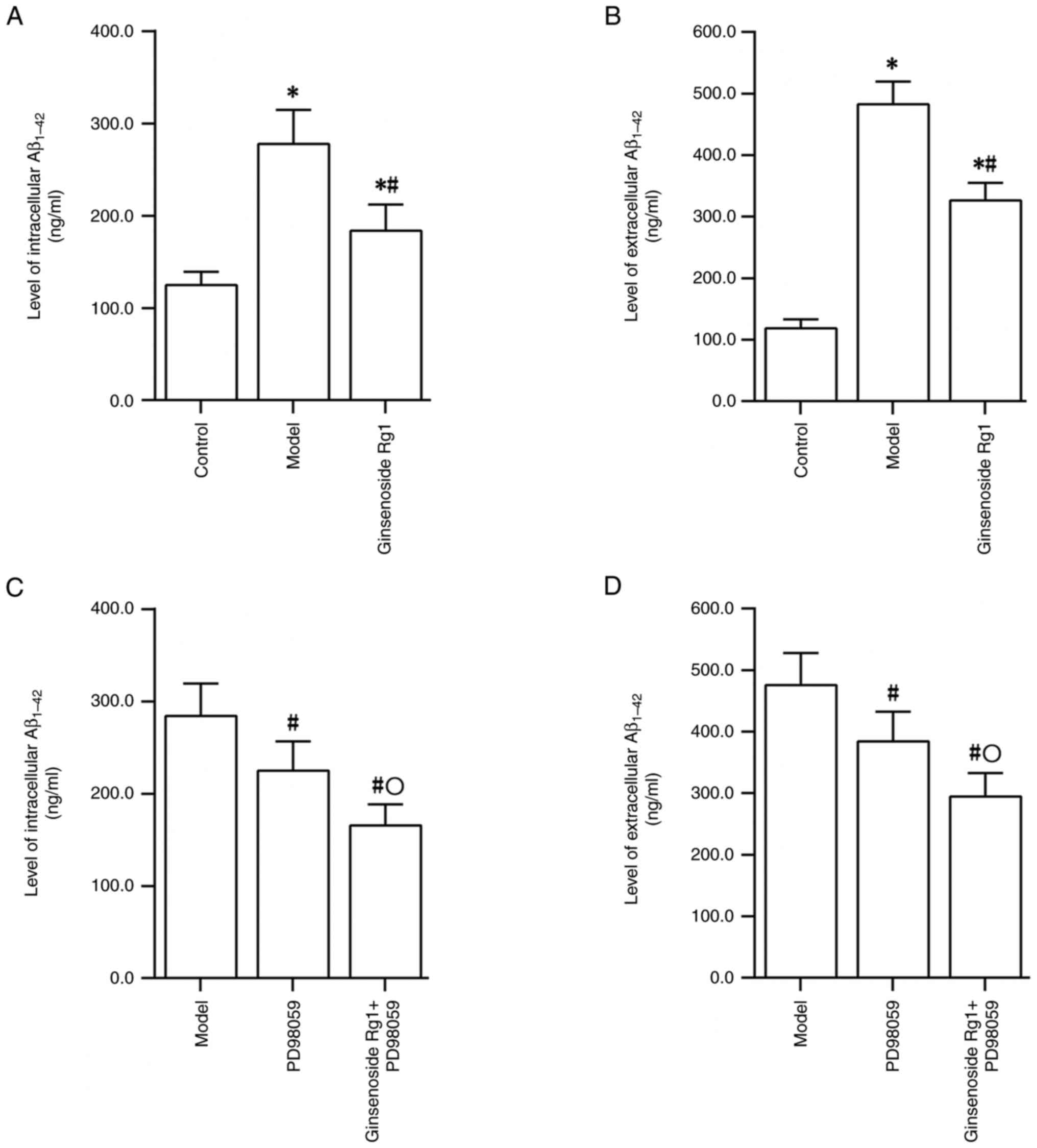
Ginsenoside Rg1 reduces the level of
Aβ in an AD neuronal model
ELISA results demonstrated that treatment of primary
cultured rat hippocampal neurons with Aβ1-42 could lead
to significantly increased intra- and extracellular levels of
Aβ1-42 (control group vs. model group; P<0.05).
However, treatment with ginsenoside Rg1 reduced the intra- and
extracellular Aβ1-42 levels in the AD neuronal model
(model group vs. ginsenoside Rg1 group; P<0.05; Fig. 2A and B). Treatment with PD98059 also reduced
the intra- and extracellular levels of Aβ1-42 in the AD
neuronal model (model group vs. PD98059 group; P<0.05; Fig. 2C and D). Following ERK inhibition by PD98059 in
the AD neuronal model, treatment with ginsenoside Rg1 resulted in a
further decrease in the intra- and extracellular Aβ1-42
levels compared with the levels detected following treatment with
PD98059 alone (PD98059 group vs. ginsenoside Rg1 + PD98059 group;
P<0.05; Fig. 2C and D).
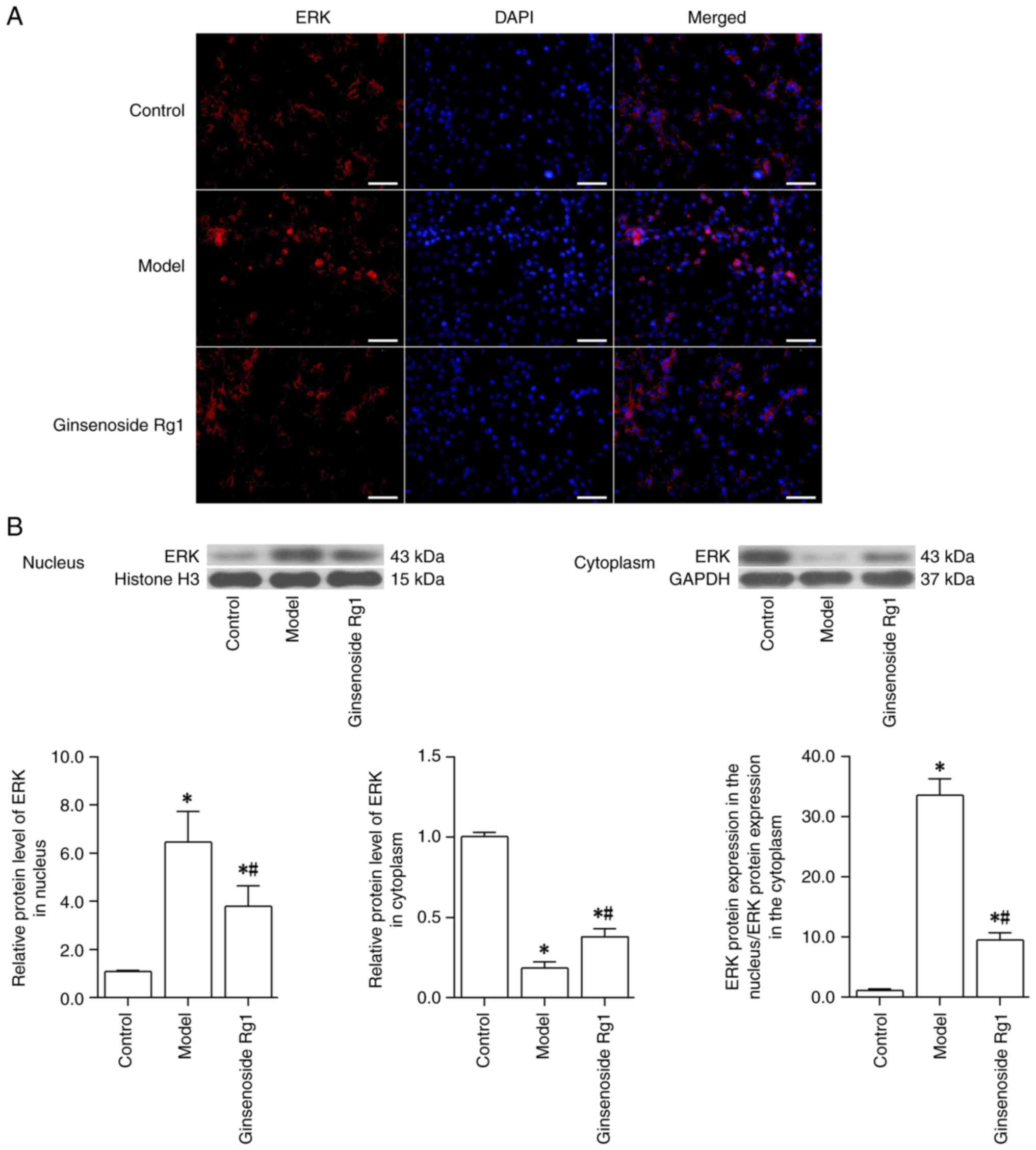
Ginsenoside Rg1 prevents ERK
translocation from the cytoplasm to the nucleus in an AD neuronal
model
To examine the effect of Aβ and ginsenoside Rg1 on
ERK translocation, ERK expression and its subcellular localization
in neurons were first observed using immunofluorescence staining.
The results revealed that, when compared with the intensity of red
fluorescent staining in normal neurons, the ERK red fluorescent
staining in the cytoplasm of neurons treated with Aβ1-42
was diminished, whereas that in the nucleus was enhanced. Compared
with that in the Aβ1-42-treated neurons (model group),
pretreatment with ginsenoside Rg1 prior to Aβ1-42
treatment resulted in enhanced ERK red fluorescence in the
cytoplasm and reduced red fluorescence in the nuclei of the neurons
(Fig. 3A). To further examine the
translocation of ERK, the expression levels of the ERK protein in
the cytoplasm and the nucleus were also detected using western
blotting, and the ratio of the ERK protein expression in the
nucleus relative to that in the cytoplasm was calculated. These
results indicated that compared with those in the control group,
ERK protein expression in the cytoplasm of neurons was decreased,
whereas that in the nucleus was increased in the model group (all
P<0.05); therefore, the ratio of ERK protein expression in the
nucleus to that in the cytoplasm was increased following
Aβ1-42 treatment (control group vs. model group;
P<0.05). However, pretreatment with ginsenoside Rg1 effectively
inhibited the Aβ1-42-induced changes in ERK expression
in the cytoplasm and in the nucleus; ERK protein expression in the
cytoplasm was increased, whereas that in the nucleus was decreased,
indicating that the ratio of ERK protein expression in the nucleus
to that in the cytoplasm was decreased (model group vs. ginsenoside
Rg1 group; all P<0.05; Fig.
3B). Taken together, these results suggested that
Aβ1-42 could promote the translocation of ERK from the
cytoplasm to the nucleus, whereas ginsenoside Rg1 was able to
inhibit this translocation of ERK.
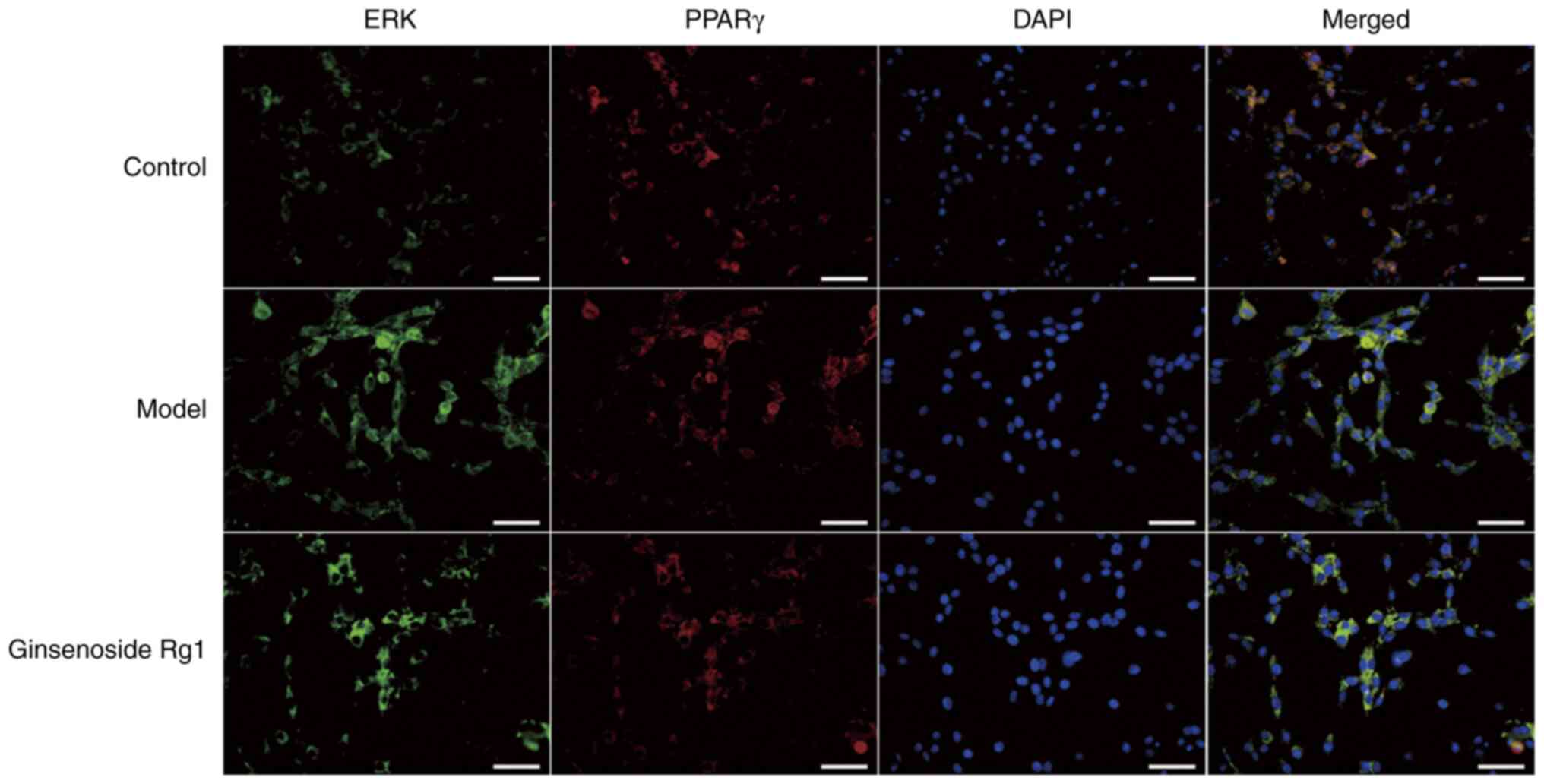
Ginsenoside Rg1 inhibits the
co-expression of ERK and PPARγ in an AD neuronal model
To further confirm the binding of ERK to its target
molecule, PPARγ, following translocation from the cytoplasm to the
nucleus, immunofluorescence double staining of ERK and PPARγ was
performed to detect their co-expression levels. The co-expression
of ERK and PPARγ in the nuclei of Aβ1-42-treated
hippocampal neurons was increased compared with that in the nuclei
of normal neurons (model group vs. control), whereas the
co-expression of ERK and PPARγ was lower in the nuclei of neurons
that were pretreated with ginsenoside Rg1 prior to
Aβ1-42 treatment compared with that in the nuclei of
neurons treated with Aβ1-42 alone (ginsenoside Rg1 group
vs. model group). These findings suggested that ginsenoside Rg1
could inhibit the co-expression of ERK and PPARγ in the AD neuronal
model (Fig. 4).
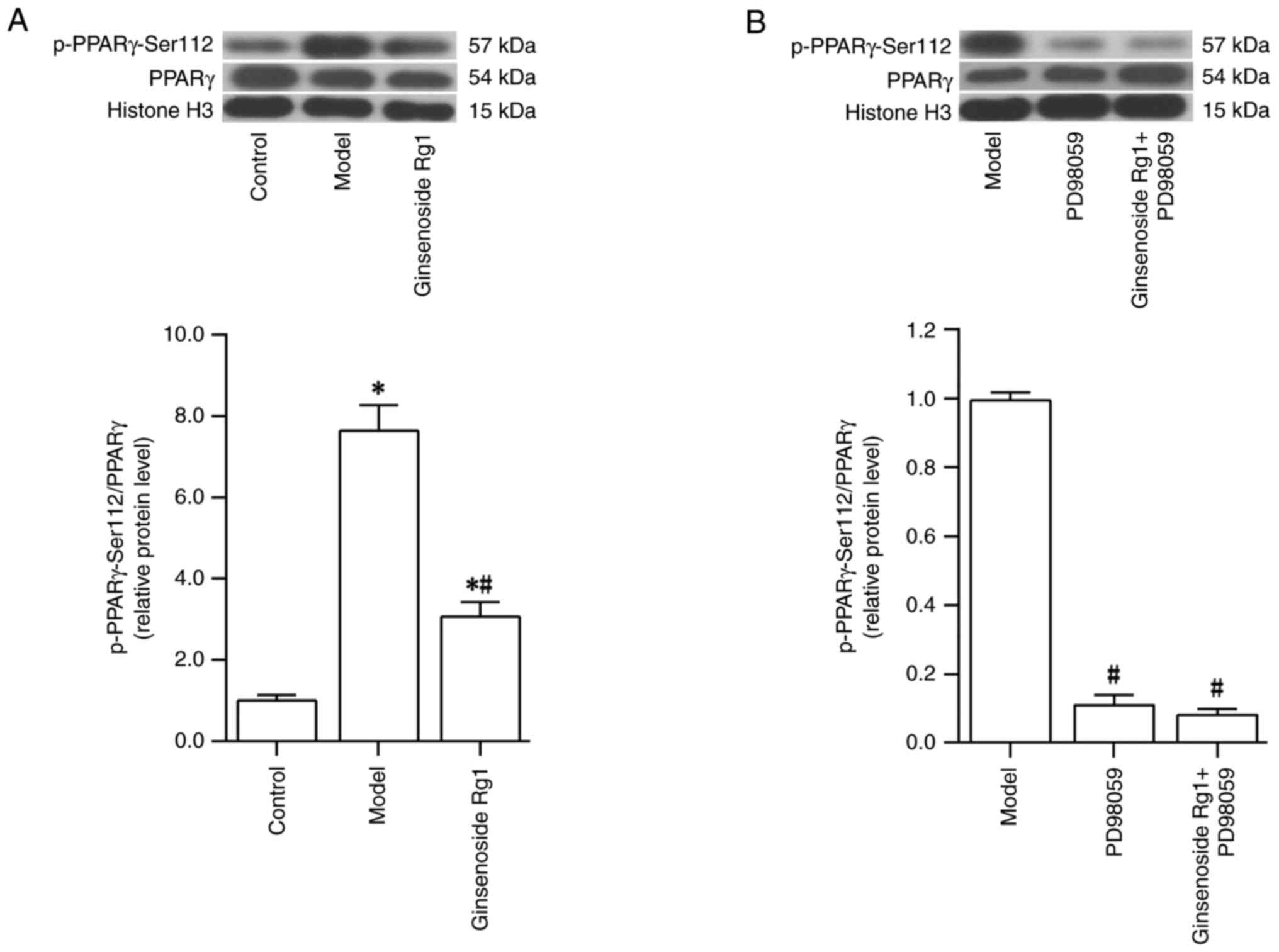
Ginsenoside Rg1 inhibits the
phosphorylation of PPARγ at Ser112 in an AD neuronal model
The phosphorylation level of PPARγ at Ser112
(p-PPARγ-Ser112/PPARγ) in the nucleus of the AD neuronal model
following Aβ1-42 and ginsenoside Rg1 treatment was
investigated. The results revealed that the level of p-PPARγ-Ser112
was increased after treatment of primary cultured hippocampal
neurons with Aβ1-42 (control group vs. model group;
P<0.05; Fig. 5A). However,
pretreatment with ginsenoside Rg1 prior to treatment with
Aβ1-42 reduced the level of p-PPARγ at Ser112 (model
group vs. ginsenoside Rg1 group; P<0.05; Fig. 5A). Following treatment with the ERK
inhibitor PD98059 in the AD neuronal model, it was found that the
level of p-PPARγ at Ser112 was reduced (model group vs. PD98059
group; P<0.05; Fig. 5B).
Following ERK inhibition by PD98059 and treatment with ginsenoside
Rg1 in the AD neuronal model, the level of p-PPARγ at Ser112 was
not further decreased compared with that of the group treated with
PD98059 alone (PD98059 group vs. ginsenoside Rg1 + PD98059;
P>0.05; Fig. 5B). Taken
together, these findings suggested that PD98059 and ginsenoside Rg1
were able to effectively inhibit the phosphorylation of PPARγ at
Ser112 in the nuclei of the AD neuronal model.
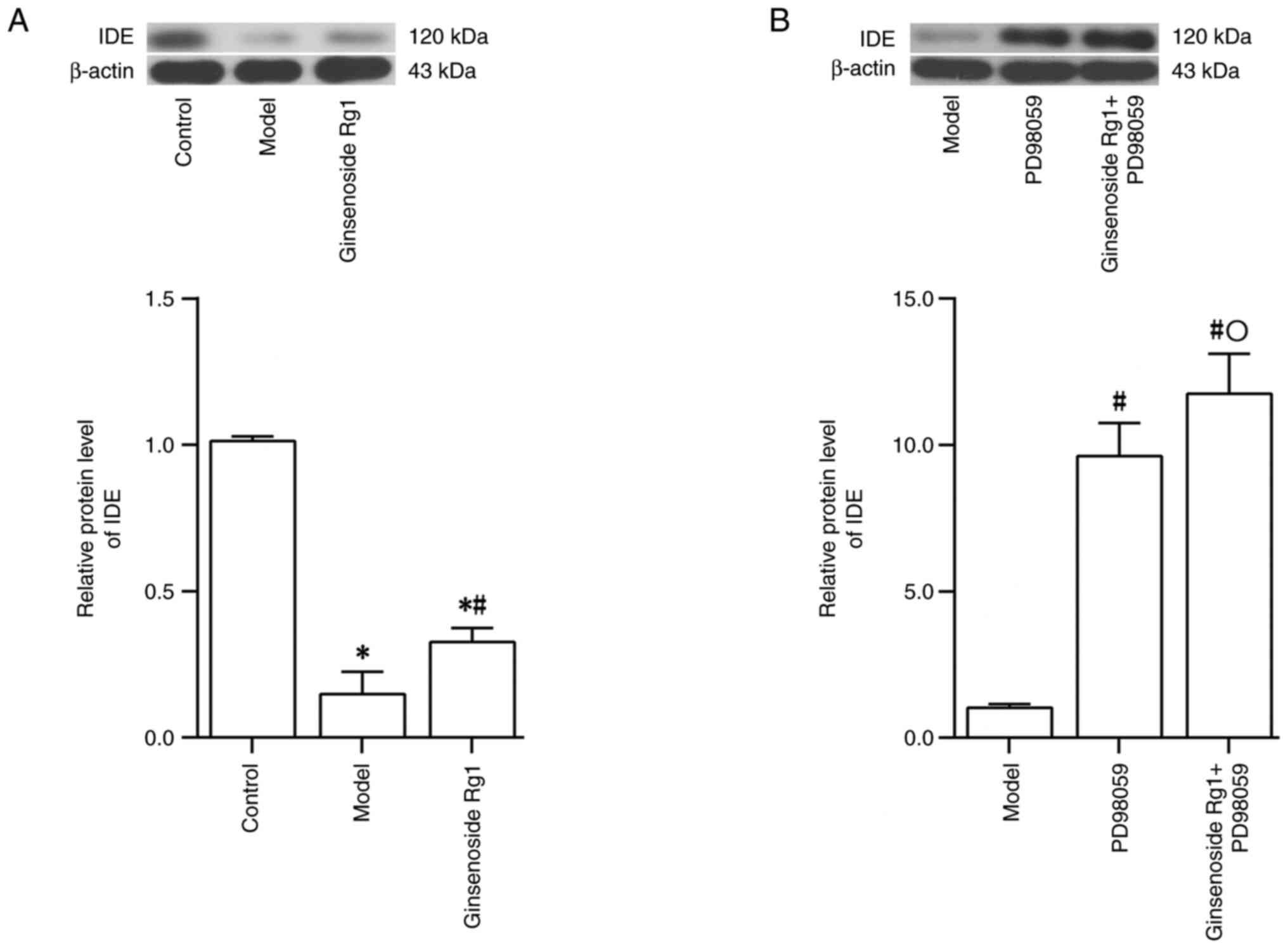
Ginsenoside Rg1 increases IDE protein
expression in an AD neuronal model
In a subsequent series of experiments, the effect of
ginsenoside Rg1 on IDE protein expression in the AD neuronal model
was examined using western blotting. The results demonstrated that
IDE protein expression in the AD neuronal model, prepared by
treating hippocampal neurons with Aβ1-42, was decreased
(control group vs. model group; P<0.05; Fig. 6A). However, IDE protein expression
in the AD neuronal model was increased following treatment with
ginsenoside Rg1 (model group vs. ginsenoside Rg1 group; P<0.05;
Fig. 6A). IDE protein expression
in the AD neuronal model was increased following treatment with
PD98059 (model group vs. PD98059 group; P<0.05; Fig. 6B). However, IDE protein expression
was increased further following pretreatment with PD98059 combined
with treatment with ginsenoside Rg1 in the AD neuronal model
(PD98059 group vs. ginsenoside Rg1 + PD98059 group; P<0.05;
Fig. 6B).
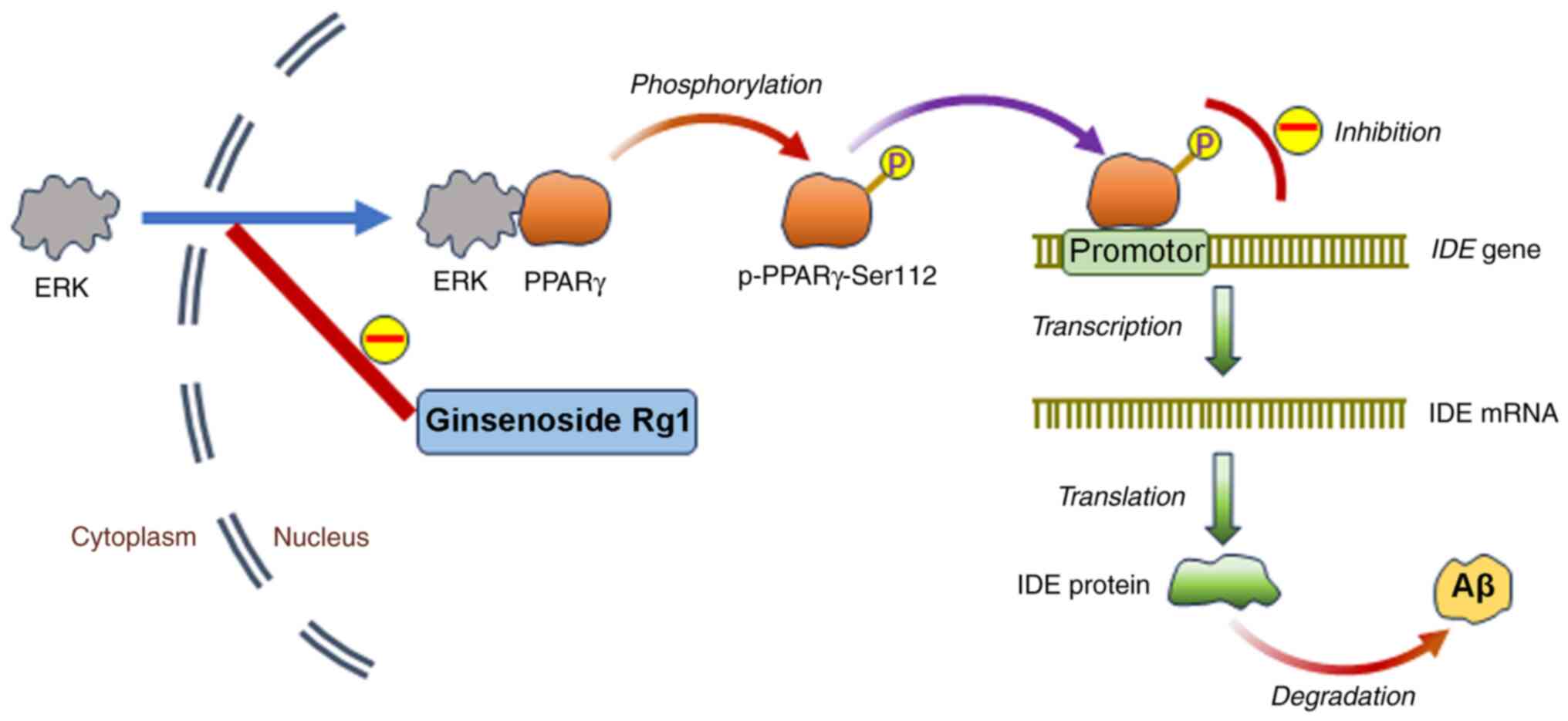
Discussion
The findings presented in the current study further
demonstrated the scavenging effect of ginsenoside Rg1 on Aβ, and
the results suggested that the underlying mechanism might be
associated with the inhibition of the ERK-mediated phosphorylation
of PPARγ. Ginsenoside Rg1 prevents ERK translocation from the
cytoplasm to the nucleus in an Alzheimer's disease neuronal model,
resulting in the inhibition of PPARγ phosphorylation at Ser112 and
the promotion of the transcriptional activity of PPARγ. As a
result, the target gene of PPARγ IDE expression increases to
promote Aβ degradation (Fig. 7).
ERK regulates numerous downstream transcription factors, such as
PPARγ (27), NF-κB (28), E-twenty-six-like transcription
factor 1(29), c-Myc (30) and c-Fos (31). Some molecules, such as BACE1
(32,33) and IDE (10,11),
can be regulated transcriptionally by PPARγ, and are involved in
the pathogenesis of AD. Therefore, PPARγ was chosen as the subject
of the present study. PPARγ is a ligand-activated nuclear
transcription factor that binds to retinoic X receptors to form
heterodimers, subsequently binding to its ligand, and thereby
regulating the expression of downstream target genes that are
involved in the regulation of numerous physiological responses,
including those associated with lipid metabolism, cell fate,
glucose homeostasis, insulin sensitivity, immune responses and
inflammation (34-36).
Previous studies have also demonstrated that PPARγ is not only
associated with obesity, diabetes, inflammation and tumors, but is
also closely associated with AD (37,38).
Furthermore, a previous study (39) demonstrated that PPARγ activation
improved spatial memory in animal models of AD, and it was
accomplished by inhibiting β-secretase expression and reducing Aβ
production through modulating the responses of the microglia to Aβ
deposition, thereby increasing the phagocytosis of Aβ and reducing
cytokine release (40,41). Furthermore, it also accomplished
this by improving mitochondrial function in AD models (42), reducing oxidative stress (43) and improving impaired synaptic
plasticity (44).
The functions of PPARγ are regulated by
post-translational modifications, including ubiquitination,
phosphorylation, acetylation, SUMOylation and O-GlcNAcylation
(45). As aging progresses, PPARγ
phosphorylation inactivation occurs in various tissues, such as the
kidney, cerebral cortex and adipose tissue (46). Bartl et al (47) showed that PPARγ phosphorylation is
also present in the brains of patients with AD, and the number of
p-PPARγ+ cells was found to be notably increased in the
cortex and hippocampus of deceased patients with AD compared with
those in age-matched control patients without dementia. In
addition, a previously published in vitro study confirmed
that the presence of Aβ can result in the phosphorylation of PPARγ
at Ser273(19). However, in the
present study, Aβ treatment of the primary cultured rat hippocampal
neurons resulted in an increase in the level of PPARγ
phosphorylation at Ser112. At present, no definitive answer may be
provided as to whether PPARγ phosphorylation leads to the onset of
AD, or whether PPARγ phosphorylation occurs after the onset of
AD.
Previous studies have demonstrated that certain
stimuli such as epidermal growth factor, transforming growth factor
β, insulin and prostaglandin F2α can trigger PPARγ phosphorylation
at different sites through the activation of MAPK, thereby leading
to the increase or decrease of transcriptional activity of PPARγ
(45,48,49).
MAPK comprises a family of serine/threonine protein kinases, which
are able to transmit numerous extracellular signals to the nucleus
to regulate various cellular processes, including cell
proliferation, gene expression, apoptosis, differentiation and
stress responses (20,21). The MAPK signaling pathway includes
three main kinases, including MAPK, MAPK kinase (MAPKK) and MAPKK
kinase (MAPKKK). Stimuli, such as growth factor receptors, receptor
tyrosine kinase and G-protein-coupled receptors, are able to
activate Ras, which in turn activates MAPKKK. Activated MAPKKK
subsequently phosphorylates and activates MAPKK which
phosphorylates and activates MAPK (50). Following activation, MAPK is
translocated from the cytoplasm into the nucleus where it binds to
its target molecules via docking-mediated interactions, followed by
serine/threonine-residue phosphorylation of multiple substrate
proteins and the modification of their activity (22). The main members of the MAPK family
are ERKs, p38 and JNK. It has been demonstrated that ERK and p38
mediate the Ser112 phosphorylation of the A/B structural domain of
PPARγ, thereby inhibiting its transcriptional activity (23,27,51).
In addition, JNK can mediate the phosphorylation of PPARγ at Ser82,
also inhibiting its transcriptional activity (52). IDE is a zinc metalloprotease and is
regulated transcriptionally by PPARγ (10). Previous studies have demonstrated
that, in addition to its ability to degrade insulin, IDE is also
able to effectively degrade Aβ, including intra- and extracellular
Aβ (53,54). In the present study, the following
observations were made after treating primary cultured rat
hippocampal neurons with Aβ: ERK was translocated from the
cytoplasm to the nucleus, the phosphorylation level of PPARγ at
Ser112 was increased, IDE expression was decreased and neuronal
apoptosis was increased. These findings suggested that Aβ could
stimulate ERK translocation to the nucleus, thereby mediating the
phosphorylation of PPARγ, and consequently affecting the
PPARγ-mediated regulation of IDE expression. In the present study,
treatment with ginsenoside Rg1 effectively inhibited the
translocation of ERK from the cytoplasm to the nucleus and
inhibited the co-expression of ERK and PPARγ. It was also found
that the phosphorylation of PPARγ at Ser112 was reduced, IDE
expression was increased, and both intra- and extracellular levels
of Aβ were reduced, suggesting that ginsenoside Rg1 might exert an
anti-Aβ effect by inhibiting the ERK-mediated phosphorylation of
PPARγ. However, the present study did have certain limitations.
First, the mechanisms through which Aβ affects the translocation of
ERK from the cytoplasm to the nucleus, how ginsenoside Rg1 inhibits
ERK or whether the MAPK signaling pathway is involved in this
process have not been revealed. In addition to its inhibitory
effect on PPARγ via the phosphorylation of Ser112, ERK can also
induce nuclear export of PPARγ via direct interactions with PPARγ,
thereby modulating the nucleo-cytoplasmic compartmentalization of
PPARγ and attenuating the transactivation function of PPARγ
(55). It is also unclear whether
Aβ promotes the aforementioned ERK-mediated nuclear export of
PPARγ, or whether ginsenoside Rg1 may inhibit the ERK-mediated
nuclear export of PPARγ. Relevant experiments will be performed in
the future to address these questions.
PPARγ belongs to a family of ligand-regulated
nuclear receptors with other members including PPARα, PPARβ and
PPARδ (56). Previous studies have
demonstrated that PPARα, PPARβ, PPARδ are also associated with AD
(56-60).
PPARα is able to regulate the expression of genes encoding enzymes
that are engaged in amyloid precursor protein metabolism and
downregulate BACE1 expression to reduce Aβ generation (56). By contrast, PPARβ and PPARδ can
alleviate AD through their insulin-sensitizing, anti-inflammatory
and myelin sheath-stabilizing effects, thereby decreasing Aβ
deposition (57). It has been
demonstrated that PPARδ activation may exert a neuroprotective
effect in AD models by inhibiting the inflammation and the
amelioration of Aβ1-42-induced hippocampal neurotoxicity
(58,59). Additionally, PPARδ has been
demonstrated to suppress the generation of neurotoxic Aβ by
attenuating BACE1 expression via the cytokine signaling 1-mediated
inhibition of signal transducer and activator of transcription 1
signaling (60). In the present
study, only the effects of ginsenoside Rg1 on PPARγ were observed.
Therefore, it is not yet clear whether ginsenoside Rg1 also exerts
any effects on PPARα, PPARβ, PPARδ.
The present study revealed that ERK pathway
activation could induce cellular apoptosis, and that the inhibitor
PD98059 inhibited cell apoptosis, findings that were consistent
with those of numerous previous studies (61-63).
The ERK pathway not only mediates cell proliferation but can also
induce apoptosis (64). Activation
of the ERK signaling pathway is able to promote the proliferation
of tumor cells and enhance the processes of tumor cell migration
and invasion (65-68),
all of which leads to an acceleration of tumor progression. PD98059
could exert a positive protective effect, as it can inhibit tumor
cell proliferation, migration and invasion (66,69-71).
In addition, activation of the ERK pathway has been demonstrated to
mediate cell apoptosis, and this effect can be blocked by PD98059
(61-63).
In cardiomyocytes, ERK activation is involved in
doxorubicin-induced cardiomyocyte apoptosis, which has been
demonstrated to be blocked by the knockdown of ERK (61). In rats with pre-eclampsia,
activation of the ERK signaling pathway has been demonstrated to
induce trophoblast apoptosis (62). Furthermore, in IL-1β-stimulated
chondrocytes, ERK has been demonstrated to be involved in the
dynamin-related protein 1-mediated induction of apoptosis, and
apoptosis can be inhibited by PD98059(63). However, it appears that several
studies have obtained contrary findings. For example, in colorectal
cancer cells, treatment with PD98059 combined with paclitaxel led
to an increase in apoptosis (72).
Another study showed that activation of ERK improve the cell
viability of H2O2-treated bone marrow-derived
mesenchymal stem cells, and the effect could be blocked by PD98059
and ERK small interfering RNA (73). Therefore, further studies are
required to fully clarify whether the effects of ERK activation and
PD98059 on apoptosis are inhibitory or stimulatory, and these
studies will be performed in the future.
In the present study, use of the ERK inhibitor
PD98059, followed by treatment with ginsenoside Rg1, was found to
result in significant improvements in Aβ levels, neuronal apoptosis
and IDE expression in an AD neuronal model compared with the
effects of PD98059 treatment alone. This suggested that, in
addition to the ERK/PPARγ pathway, ginsenoside Rg1 might also
increase the levels and activity of IDE and promote Aβ degradation
through other pathways. Besides PPARγ, IDE levels have been shown
to be regulated by other molecules and signaling factors/processes,
including circulating insulin, glucose, L-lactate and free fatty
acids (74), nucleoside
triphosphates (75), sex steroids
(76) and insulin-mediated Akt
activation (77). However, further
studies are required to confirm whether ginsenoside Rg1 may affect
IDE expression through these pathways or act on IDE directly. In
addition to IDE, certain other proteases are known to degrade Aβ,
including neprilysin, MMP-9 and MMP-2(78). In other studies, it has been shown
that ginsenoside Rg1 is able to i) decrease accumulation of
neurofibrillary tangles in the retina by regulating the activities
of neprilysin and protein kinase A in the retinal cells of an AD
mouse model (79); ii) inhibit
tumor cell invasion and migration by inhibiting NF-κB-dependent
MMP-9 expression (80); and iii)
inhibit myocardial remodeling in an animal model of chronic
thromboembolic pulmonary hypertension through upregulating MMP-2
and MMP-9 expression in myocardial tissue (81). However, to the best of our
knowledge, whether ginsenoside Rg1 removes Aβ by acting on these
molecules is unknown, and further studies are required to
investigate this possibility.
There are two further limitations of the present
study. First, only immunofluorescence double staining was used to
detect the combination of ERK and PPARγ in the AD neuronal model
used in the present study. It would have been useful to investigate
the effect of Aβ and ginsenoside Rg1 on the combination of ERK and
PPARγ using co-immunoprecipitation. In addition, only in
vitro experiments were performed in the present study and,
although this does not substantially affect conclusions, the
results of the present study would be more impactful, and the
conclusion would be strengthened if animal experiments had been
conducted.
In conclusion, the present study demonstrated that
ginsenoside Rg1 may exert neuroprotective effects on AD by
inhibiting the ERK/PPARγ phosphorylation pathway, which thereby
upregulated the expression levels of the PPARγ-targeted gene
IDE and promoted Aβ degradation. These findings have
presented evidence in favor of a novel mechanism of action of
ginsenoside Rg1 against AD, also providing a theoretical basis for
a further application of ginsenoside Rg1 in the treatment of
AD.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Natural Science
Basic Research Program of Shaanxi (grant no. 2021JM-288) and
Science and Technology Plan Project of Xi'an City (grant no.
22YXYJ0106).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HY, QQ and XL designed the study. QQ and HY wrote
the manuscript. QQ, XM and ML performed the experiments. HY, QQ and
XM collected and analyzed the data. HY and QQ confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All experimental procedures in the present study
were approved by The Ethics Committee of Xi'an Jiaotong University
Health Science Center (approval no. 2020-942; Xi'an, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Murphy MP and LeVine H III: Alzheimer's
disease and the amyloid-beta peptide. J Alzheimers Dis. 19:311–323.
2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Seino Y, Kawarabayashi T, Wakasaya Y,
Watanabe M, Takamura A, Yamamoto-Watanabe Y, Kurata T, Abe K, Ikeda
M, Westaway D, et al: Amyloid β accelerates phosphorylation of tau
and neurofibrillary tangle formation in an amyloid precursor
protein and tau double-transgenic mouse model. J Neurosci Res.
88:3547–3554. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chen Y, Fu AKY and Ip NY: Synaptic
dysfunction in Alzheimer's disease: Mechanisms and therapeutic
strategies. Pharmacol Ther. 195:186–198. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jawhar S, Trawicka A, Jenneckens C, Bayer
TA and Wirths O: Motor deficits, neuron loss, and reduced anxiety
coinciding with axonal degeneration and intraneuronal Aβ
aggregation in the 5XFAD mouse model of Alzheimer's disease.
Neurobiol Aging. 33:196.e29–e40. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cai Z, Hussain MD and Yan LJ: Microglia,
neuroinflammation, and beta-amyloid protein in Alzheimer's disease.
Int J Neurosci. 124:307–321. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Esteras N and Abramov AY: Mitochondrial
calcium deregulation in the mechanism of beta-amyloid and tau
pathology. Cells. 9(2135)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Caruso G, Spampinato SF, Cardaci V, Caraci
F, Sortino MA and Merlo S: β-amyloid and oxidative stress:
Perspectives in drug development. Curr Pharm Des. 25:4771–4781.
2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Huang Z, Yan Q, Wang Y, Zou Q, Li J, Liu Z
and Cai Z: Role of mitochondrial dysfunction in the pathology of
amyloid-β. J Alzheimers Dis. 78:505–514. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tran MH, Yamada K and Nabeshima T: Amyloid
beta-peptide induces cholinergic dysfunction and cognitive
deficits: A minireview. Peptides. 23:1271–1283. 2002.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Du J, Zhang L, Liu SB, Zhang C, Huang XQ,
Li J, Zhao NM and Wang Z: PPARgamma transcriptionally regulates the
expression of insulin-degrading enzyme in primary neurons. Biochem
Biophys Res Commun. 383:485–490. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sahoo BR, Panda PK, Liang W, Tang WJ,
Ahuja R and Ramamoorthy A: Degradation of Alzheimer's amyloid-β by
a catalytically inactive insulin-degrading enzyme. J Mol Biol.
433(166993)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kurochkin IV, Guarnera E and Berezovsky
IN: Insulin-degrading enzyme in the fight against Alzheimer's
disease. Trends Pharmacol Sci. 39:49–58. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hu BY, Liu XJ, Qiang R, Jiang ZL, Xu LH,
Wang GH, Li X and Peng B: Treatment with ginseng total saponins
improves the neurorestoration of rat after traumatic brain injury.
J Ethnopharmacol. 155:1243–1255. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bai L, Gao J, Wei F, Zhao J, Wang D and
Wei J: Therapeutic potential of ginsenosides as an adjuvant
treatment for diabetes. Front Pharmacol. 9(423)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fang F, Chen X, Huang T, Lue LF, Luddy JS
and Yan SS: Multi-faced neuroprotective effects of ginsenoside Rg1
in an Alzheimer mouse model. Biochim Biophys Acta. 1822:286–292.
2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yang Y, Wang L, Zhang C, Guo Y, Li J, Wu
C, Jiao J and Zheng H: Ginsenoside Rg1 improves Alzheimer's disease
by regulating oxidative stress, apoptosis, and neuroinflammation
through Wnt/GSK-3β/β-catenin signaling pathway. Chem Biol Drug Des.
99:884–896. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kwan KKL, Yun H, Dong TTX and Tsim KWK:
Ginsenosides attenuate bioenergetics and morphology of mitochondria
in cultured PC12 cells under the insult of amyloid beta-peptide. J
Ginseng Res. 45:473–481. 2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang L, Lu J, Zeng Y, Guo Y, Wu C, Zhao H,
Zheng H and Jiao J: Improving Alzheimer's disease by altering gut
microbiota in tree shrews with ginsenoside Rg1. FEMS Microbiol
Lett. 367(fnaa011)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Quan Q, Li X, Feng J, Hou J, Li M and
Zhang B: Ginsenoside Rg1 reduces β-amyloid levels by inhibiting
CDΚ5-induced PPARγ phosphorylation in a neuron model of Alzheimer's
disease. Mol Med Rep. 22:3277–3288. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Urushibara N, Mitsuhashi S, Sasaki T,
Kasai H, Yoshimizu M, Fujita H and Oda A: JNK and p38 MAPK are
independently involved in tributyltin-mediated cell death in
rainbow trout (Oncorhynchus mykiss) RTG-2 cells. Comp Biochem
Physiol C Toxicol Pharmacol. 149:468–475. 2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y and
Hu LL: ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med.
19:1997–2007. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Turjanski AG, Vaqué JP and Gutkind JS: MAP
kinases and the control of nuclear events. Oncogene. 26:3240–3253.
2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ge C, Cawthorn WP, Li Y, Zhao G,
Macdougald OA and Franceschi RT: Reciprocal control of osteogenic
and adipogenic differentiation by ERK/MAP kinase phosphorylation of
Runx2 and PPARγ transcription factors. J Cell Physiol. 231:587–596.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yang EJ, Ahn S, Ryu J, Choi MS, Choi S,
Chong YH, Hyun JW, Chang MJ and Kim HS: Phloroglucinol attenuates
the cognitive deficits of the 5XFAD mouse model of Alzheimer's
disease. PLoS One. 10(e0135686)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li Y, Guan Y, Wang Y, Yu CL, Zhai FG and
Guan LX: Neuroprotective effect of the ginsenoside Rg1 on cerebral
ischemic injury in vivo and in vitro is mediated by PPARγ regulated
antioxidative and anti-inflammatory pathways. Evid Based Complement
Alternat Med. 2017(7842082)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kiwanuka E, Junker JP and Eriksson E:
Transforming growth factor β1 regulates the expression of CCN2 in
human keratinocytes via Smad-ERK signalling. Int Wound J.
14:1006–1018. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
El Ouarrat D, Isaac R, Lee YS, Oh DY,
Wollam J, Lackey D, Riopel M, Bandyopadhyay G, Seo JB,
Sampath-Kumar R and Olefsky JM: TAZ is a negative regulator of
PPARγ activity in adipocytes and TAZ deletion improves insulin
sensitivity and glucose tolerance. Cell Metab. 31:162–173.e5.
2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jiang B, Xu S, Hou X, Pimentel DR, Brecher
P and Cohen RA: Temporal control of NF-kappaB activation by ERK
differentially regulates interleukin-1beta-induced gene expression.
J Biol Chem. 279:1323–1329. 2004.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Mut M, Lule S, Demir O, Kurnaz IA and
Vural I: Both mitogen-activated protein kinase
(MAPK)/extracellular-signal-regulated kinases (ERK) 1/2 and
phosphatidylinositide-3-OH kinase (PI3K)/Akt pathways regulate
activation of E-twenty-six (ETS)-like transcription factor 1
(Elk-1) in U138 glioblastoma cells. Int J Biochem Cell Biol.
44:302–310. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zuo Z, Liu J, Sun Z, Cheng YW, Ewing M,
Bugge TH, Finkel T, Leppla SH and Liu S: ERK and c-Myc signaling in
host-derived tumor endothelial cells is essential for solid tumor
growth. Proc Natl Acad Sci USA. 120(e2211927120)2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Monje P, Hernández-Losa J, Lyons RJ,
Castellone MD and Gutkind JS: Regulation of the transcriptional
activity of c-Fos by ERK. A novel role for the prolyl isomerase
PIN1. J Biol Chem. 280:35081–35084. 2005.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lin N, Chen LM, Pan XD, Zhu YG, Zhang J,
Shi YQ and Chen XC: Tripchlorolide attenuates β-amyloid generation
via suppressing PPARγ-regulated BACE1 activity in N2a/APP695 cells.
Mol Neurobiol. 53:6397–6406. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sadleir KR, Eimer WA, Cole SL and Vassar
R: Aβ reduction in BACE1 heterozygous null 5XFAD mice is associated
with transgenic APP level. Mol Neurodegener. 10(1)2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wagner N and Wagner KD: The role of PPARs
in disease. Cells. 9(2367)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Szanto A, Balint BL, Nagy ZS, Barta E,
Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, et al: STAT6
transcription factor is a facilitator of the nuclear receptor
PPARγ-regulated gene expression in macrophages and dendritic cells.
Immunity. 33:699–712. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Vallée A, Lecarpentier Y, Guillevin R and
Vallée JN: Effects of cannabidiol interactions with Wnt/β-catenin
pathway and PPARγ on oxidative stress and neuroinflammation in
Alzheimer's disease. Acta Biochim Biophys Sin (Shanghai).
49:853–866. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Janani C and Ranjitha Kumari BD: PPAR
gamma gene-a review. Diabetes Metab Syndr. 9:46–50. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Prashantha Kumar BR, Kumar AP, Jose JA,
Prabitha P, Yuvaraj S, Chipurupalli S, Jeyarani V, Manisha C,
Banerjee S, Jeyabalan JB, et al: Minutes of PPAR-γ agonism and
neuroprotection. Neurochem Int. 140(104814)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Rodriguez-Rivera J, Denner L and Dineley
KT: Rosiglitazone reversal of Tg2576 cognitive deficits is
independent of peripheral gluco-regulatory status. Behav Brain Res.
216:255–261. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Heneka MT, Sastre M, Dumitrescu-Ozimek L,
Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van
Leuven F and Landreth GE: Acute treatment with the PPARgamma
agonist pioglitazone and ibuprofen reduces glial inflammation and
Abeta1-42 levels in APPV717I transgenic mice. Brain. 128:1442–1453.
2005.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yamanaka M, Ishikawa T, Griep A, Axt D,
Kummer MP and Heneka MT: PPARγ/RXRα-induced and CD36-mediated
microglial amyloid-β phagocytosis results in cognitive improvement
in amyloid precursor protein/presenilin 1 mice. J Neurosci.
32:17321–17331. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zolezzi JM, Silva-Alvarez C, Ordenes D,
Godoy JA, Carvajal FJ, Santos MJ and Inestrosa NC: Peroxisome
proliferator-activated receptor (PPAR) γ and PPARα agonists
modulate mitochondrial fusion-fission dynamics: Relevance to
reactive oxygen species (ROS)-related neurodegenerative disorders?
PLoS One. 8(e64019)2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Nicolakakis N, Aboulkassim T, Ongali B,
Lecrux C, Fernandes P, Rosa-Neto P, Tong XK and Hamel E: Complete
rescue of cerebrovascular function in aged Alzheimer's disease
transgenic mice by antioxidants and pioglitazone, a peroxisome
proliferator-activated receptor gamma agonist. J Neurosci.
28:9287–9296. 2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Xu S, Liu G, Bao X, Wu J, Li S, Zheng B,
Anwyl R and Wang Q: Rosiglitazone prevents amyloid-β
oligomer-induced impairment of synapse formation and plasticity via
increasing dendrite and spine mitochondrial number. J Alzheimers
Dis. 39:239–251. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Brunmeir R and Xu F: Functional regulation
of PPARs through post-translational modifications. Int J Mol Sci.
19(1738)2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ye P, Zhang XJ, Wang ZJ and Zhang C:
Effect of aging on the expression of peroxisome
proliferator-activated receptor gamma and the possible relation to
insulin resistance. Gerontology. 52:69–75. 2006.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Bartl J, Monoranu CM, Wagner AK, Kolter J,
Riederer P and Grünblatt E: Alzheimer's disease and type 2
diabetes: Two diseases, one common link? World J Biol Psychiatry.
14:233–240. 2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Hu E, Kim JB, Sarraf P and Spiegelman BM:
Inhibition of adipogenesis through MAP kinase-mediated
phosphorylation of PPARgamma. Science. 274:2100–2103.
1996.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Camp HS and Tafuri SR: Regulation of
peroxisome proliferator-activated receptor gamma activity by
mitogen-activated protein kinase. J Biol Chem. 272:10811–10816.
1997.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Irnaten M, Duff A, Clark A and O'Brien C:
Intra-cellular calcium signaling pathways (PKC, RAS/RAF/MAPK, PI3K)
in lamina cribrosa cells in glaucoma. J Clin Med.
10(62)2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Stechschulte LA, Hinds TD Jr, Khuder SS,
Shou W, Najjar SM and Sanchez ER: FKBP51 controls cellular
adipogenesis through p38 kinase-mediated phosphorylation of GRα and
PPARγ. Mol Endocrinol. 28:1265–1275. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Camp HS, Tafuri SR and Leff T: c-Jun
N-terminal kinase phosphorylates peroxisome proliferator-activated
receptor-gamma1 and negatively regulates its transcriptional
activity. Endocrinology. 140:392–397. 1999.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Vingtdeux V, Chandakkar P, Zhao H, Blanc
L, Ruiz S and Marambaud P: CALHM1 ion channel elicits amyloid-β
clearance by insulin-degrading enzyme in cell lines and in vivo in
the mouse brain. J Cell Sci. 128:2330–2338. 2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Quan Q, Qian Y, Li X and Li M: CDK5
participates in amyloid-β production by regulating PPARγ
phosphorylation in primary rat hippocampal neurons. J Alzheimers
Dis. 71:443–460. 2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Burgermeister E and Seger R: MAPK kinases
as nucleo-cytoplasmic shuttles for PPARgamma. Cell Cycle.
6:1539–1548. 2007.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Wójtowicz S, Strosznajder AK, Jeżyna M and
Strosznajder JB: The novel role of PPAR alpha in the brain:
Promising target in therapy of Alzheimer's disease and other
neurodegenerative disorders. Neurochem Res. 45:972–988.
2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Strosznajder AK, Wójtowicz S, Jeżyna MJ,
Sun GY and Strosznajder JB: Recent insights on the role of PPAR-β/δ
in neuroinflammation and neurodegeneration, and tts potential
target for therapy. Neuromolecular Med. 23:86–98. 2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Malm T, Mariani M, Donovan LJ, Neilson L
and Landreth GE: Activation of the nuclear receptor PPARδ is
neuroprotective in a transgenic mouse model of Alzheimer's disease
through inhibition of inflammation. J Neuroinflammation.
12(7)2015.PubMed/NCBI View Article : Google Scholar
|
|
59
|
An YQ, Zhang CT, Du Y, Zhang M, Tang SS,
Hu M, Long Y, Sun HB and Hong H: PPARδ agonist GW0742 ameliorates
Aβ1-42-induced hippocampal neurotoxicity in mice. Metab Brain Dis.
31:663–671. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Lee WJ, Ham SA, Lee GH, Choi MJ, Yoo H,
Paek KS, Lim DS, Hong K, Hwang JS and Seo HG: Activation of
peroxisome proliferator-activated receptor delta suppresses BACE1
expression by up-regulating SOCS1 in a JAK2/STAT1-dependent manner.
J Neurochem. 151:370–385. 2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Zhang DX, Ma DY, Yao ZQ, Fu CY, Shi YX,
Wang QL and Tang QQ: ERK1/2/p53 and NF-κB dependent-PUMA activation
involves in doxorubicin-induced cardiomyocyte apoptosis. Eur Rev
Med Pharmacol Sci. 20:2435–2442. 2016.PubMed/NCBI
|
|
62
|
Song XP, Zhang YM, Sui SA, Li XY and Huang
Y: Activation of the ERK1/2 signaling pathway enhances
proliferation and apoptosis of trophoblast in preeclampsia rats.
Eur Rev Med Pharmacol Sci. 25:598–604. 2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Ansari MY, Novak K and Haqqi TM:
ERK1/2-mediated activation of DRP1 regulates mitochondrial dynamics
and apoptosis in chondrocytes. Osteoarthritis Cartilage.
30:315–328. 2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Mebratu Y and Tesfaigzi Y: How ERK1/2
activation controls cell proliferation and cell death: Is
subcellular localization the answer? Cell Cycle. 8:1168–1175.
2009.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Yan Z, Ohuchida K, Fei S, Zheng B, Guan W,
Feng H, Kibe S, Ando Y, Koikawa K, Abe T, et al: Inhibition of
ERK1/2 in cancer-associated pancreatic stellate cells suppresses
cancer-stromal interaction and metastasis. J Exp Clin Cancer Res.
38(221)2019.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Wang H, Du S, Cai J, Wang J and Shen X:
Apolipoprotein E2 promotes the migration and invasion of pancreatic
cancer cells via activation of the ERK1/2 signaling pathway. Cancer
Manag Res. 12:13161–13171. 2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Chen S, Li Z, Wang Y and Fan S: BTN3A3
inhibits the proliferation, migration and invasion of ovarian
cancer cells by regulating ERK1/2 phosphorylation. Front Oncol.
12(952425)2022.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Xu H, Zhao H and Yu J: HOXB5 promotes
retinoblastoma cell migration and invasion via ERK1/2
pathway-mediated MMPs production. Am J Transl Res. 10:1703–1712.
2018.PubMed/NCBI
|
|
69
|
Wang G, Yin L, Peng Y, Gao Y, Gao H, Zhang
J, Lv N, Miao Y and Lu Z: Insulin promotes invasion and migration
of KRASG12D mutant HPNE cells by upregulating MMP-2
gelatinolytic activity via ERK- and PI3K-dependent signalling. Cell
Prolif. 52(e12575)2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Xu Y, Gao F, Zhang J, Cai P and Xu D:
Fibroblast growth factor receptor 2 promotes the proliferation,
migration, and invasion of ectopic stromal cells via activation of
extracellular-signal-regulated kinase signaling pathway in
endometriosis. Bioengineered. 13:8360–8371. 2022.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Cheng XD, Gu JF, Yuan JR, Feng L and Jia
XB: Suppression of A549 cell proliferation and metastasis by
calycosin via inhibition of the PKC-α/ERK1/2 pathway: An in
vitro investigation. Mol Med Rep. 12:7992–8002. 2015.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Li Y and Yang Q: Effect of PD98059 on
chemotherapy in patients with colorectal cancer through ERK1/2
pathway. J BUON. 24:1837–1844. 2019.PubMed/NCBI
|
|
73
|
Fang J, Zhao X, Li S, Xing X, Wang H,
Lazarovici P and Zheng W: Protective mechanism of artemisinin on
rat bone marrow-derived mesenchymal stem cells against apoptosis
induced by hydrogen peroxide via activation of
c-Raf-Erk1/2-p90rsk-CREB pathway. Stem Cell Res Ther.
10(312)2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
González-Casimiro CM, Cámara-Torres P,
Merino B, Diez-Hermano S, Postigo-Casado T, Leissring MA,
Cózar-Castellano I and Perdomo G: Effects of fasting and feeding on
transcriptional and posttranscriptional regulation of
insulin-degrading enzyme in mice. Cells. 10(2466)2021.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Camberos MC, Pérez AA, Udrisar DP,
Wanderley MI and Cresto JC: ATP inhibits insulin-degrading enzyme
activity. Exp Biol Med (Maywood). 226:334–341. 2001.PubMed/NCBI View Article : Google Scholar
|
|
76
|
George S, Petit GH, Gouras GK, Brundin P
and Olsson R: Nonsteroidal selective androgen receptor modulators
and selective estrogen receptor β agonists moderate cognitive
deficits and amyloid-β levels in a mouse model of Alzheimer's
disease. ACS Chem Neurosci. 4:1537–1548. 2013.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Zhao L, Teter B, Morihara T, Lim GP,
Ambegaokar SS, Ubeda OJ, Frautschy SA and Cole GM:
Insulin-degrading enzyme as a downstream target of insulin receptor
signaling cascade: Implications for Alzheimer's disease
intervention. J Neurosci. 24:11120–11126. 2004.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Humpel C: Organotypic vibrosections from
whole brain adult Alzheimer mice (overexpressing
amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as
a model to study clearance of beta-amyloid plaques. Front Aging
Neurosci. 7(47)2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
He Y, Zhao H and Su G: Ginsenoside Rg1
decreases neurofibrillary tangles accumulation in retina by
regulating activities of neprilysin and PKA in retinal cells of AD
mice model. J Mol Neurosci. 52:101–106. 2014.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Li L, Wang Y, Qi B, Yuan D, Dong S, Guo D,
Zhang C and Yu M: Suppression of PMA-induced tumor cell invasion
and migration by ginsenoside Rg1 via the inhibition of
NF-κB-dependent MMP-9 expression. Oncol Rep. 32:1779–1786.
2014.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Li CY, Deng W, Liao XQ, Deng J, Zhang YK
and Wang DX: The effects and mechanism of ginsenoside Rg1 on
myocardial remodeling in an animal model of chronic thromboembolic
pulmonary hypertension. Eur J Med Res. 18(16)2013.PubMed/NCBI View Article : Google Scholar
|