Introduction
Acute myocardial infarction (AMI) is a prevalent
cardiovascular disease with a high morbidity and mortality rate. It
is characterized by the blockage of coronary arteries, leading to
restricted blood supply to the heart and resulting in tissue
hypoxia. Following an AMI, various strategies are employed to
reduce the area of myocardial infarction and improve clinical
outcomes. These strategies include early primary percutaneous
coronary intervention or timely and successful myocardial
reperfusion using thrombolytic therapy. However, the process of
restoring myocardial blood flow and reoxygenation can have
detrimental effects on the therapeutic outcome, leading to
myocardial ischemia-reperfusion injury (MIRI) (1). As a result, hypoxia/reoxygenation
(H/R) plays a crucial role in the pathological process associated
with MIRI (2). The most commonly
used method to simulate MIRI in vitro involves establishing
a cellular H/R model (3).
As research into the underlying pathophysiology of
MIRI progresses, the pathological mechanisms are gradually becoming
clearer. These mechanisms primarily involve the excessive
production of reactive oxygen species (ROS), autophagy, apoptosis,
necroptosis and the generation of inflammatory factors.
Collectively, these processes contribute to myocardial cell death
and subsequent myocardial dysfunction (4-7).
During MIRI, cardiomyocytes are stimulated to produce large amounts
of ROS, resulting in oxidative damage and significant mitochondrial
dysfunction. Concurrently, the mitochondrial apoptotic pathway is
activated, leading to irreversible apoptosis (8). Despite ongoing efforts, MIRI remains
a significant clinical challenge with no definitive therapeutic
solution. Therefore, there is an urgent need to further understand
the pathogenesis and explore novel treatment strategies to improve
clinical outcomes for patients with AMI.
Glycyrrhiza uralensis Fisch, a traditional
Chinese medicinal plant, holds significant commercial and medicinal
value as both a medicine and a food source (9-11).
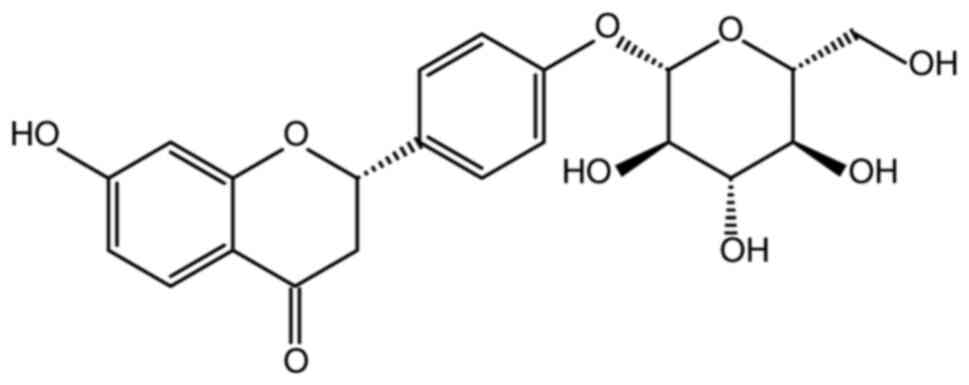
Within this plant, a flavonoid known as liquiritin (LIQ) has been
identified and its chemical structure has been determined (Fig. 1). LIQ demonstrates various
pharmacological activities, including cardioprotective effects
(12). A previous study reported
the potential of LIQ as a therapeutic strategy for repairing
injured myocardium by reducing the expression of inflammatory
mediators, inhibiting cardiac oxidative stress and preventing
apoptosis (13). Furthermore, a
study indicated that LIQ protects the myocardium from H/R injury by
mitigating mitochondrial Ca2+ overload and preserving
mitochondrial mass (14). A
research group has previously validated the protective effect of
LIQ in a rat model of myocardial infarction (15). The current study aimed to
investigate the specific mechanisms through which LIQ safeguards
cardiomyocytes from H/R injury using in vitro assays,
contributing to the advancement of targeted drugs for treating
cardiovascular diseases.
Network pharmacology is an interdisciplinary
approach that combines bioinformatics and pharmacology to
systematically uncover the connections between Chinese medicines,
compounds and various diseases. Computational integration and
analysis of data enable the exploration of drug action mechanisms
(16). Based on the aforementioned
theoretical background, the present study innovatively integrated
network pharmacology and cellular experiments to elucidate the
mechanism of action of LIQ in preventing and treating cardiomyocyte
H/R injury. Specifically, network pharmacological methods were
employed to preliminarily predict the key targets and biological
pathways involved in LIQ therapy for MIRI. Subsequently, cellular
experiments are conducted to demonstrate specific underlying
mechanisms through various perspectives including oxidative stress,
apoptosis, inflammation, mitochondrial damage and molecular
pathways. The present study provided a scientific foundation for
understanding the molecular-level protective effect of LIQ on
myocardium.
Materials and methods
Database and analysis software
Traditional Chinese Medicine Systems Pharmacology
Database v2.3 (TCMSP; https://old.tcmsp-e.com/index.php); UniProt database
v2020_01 (https://www.uniprot.org/); Swiss
Target Prediction v2019 (http://www.swisstargetprediction.ch/); PharmMapper
v2017 (http://www.lilab-ecust.cn/pharmmapper/index.html);
GeneCards database v5.5 (https://www.genecards.org/); E Venn Diagram website
(http://www.ehbio.com/test/venn/); STRING
database v11.0 (https://string-db.org/); DAVID v6.8 (https://david.ncifcrf.gov/tools.jsp);
Microbiology website (http://www.bioinformatics.com.cn/) and Cytoscape
software (version 3.7.2) (17)
were employed.
Network pharmacology methods.
Acquisition of potential targets for LIQ and MIRI
The compound characteristics and physicochemical
parameters of LIQ were obtained using CAS No. 551-15-5 from the
TCMSP. Additionally, the corresponding targets were obtained. The
SDF files of LIQ were acquired from the PubChem database and
subsequently imported into the Swiss Target Prediction and
PharmMapper websites to predict the targets associated with LIQ.
The results from these three databases were then consolidated. The
UniProt database was used to gather the gene names of the
respective LIQ targets, with a species restriction set to ‘Homo
sapiens’. Finally, a search was conducted in the GeneCards database
using ‘Myocardial ischemia-reperfusion injury’ as a keyword to
identify potential targets.
Drug-disease common target screening
and interoperability network construction
Venn diagrams were created by intersecting the
targets associated with MIRI and those associated with LIQ, using
an online Venn diagram tool. This facilitated the identification of
common targets shared between the drug and the disease. Following
this, a protein-protein interaction (PPI) network was constructed
for these common targets using the STRING 11.0 database. The
resulting TSV files were downloaded and then imported into the
Cytoscape software for visualization.
Core target screening
The ‘NetworkAnalyzer’ plug-in within Cytoscape
software was used to analyze the Degree values of each target in
the PPI network. This analysis enabled the identification of core
targets by prioritizing them based on their Degree values. It is
important to highlight that targets with higher Degree values hold
greater significance within the network.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) enrichment
The common targets were imported into the DAVID
database to gain deeper insights into the functions of the common
targets resulting from the intersection of LIQ and MIRI. Following
this, GO and KEGG enrichment analyses were performed using the
DAVID database by entering a list of target gene names and
specifying the species as human. The GO analysis encompassed
biological process (BP), cellular component (CC) and molecular
function (MF) categories. The obtained results were subsequently
analyzed and visualized using the Microbiology website.
Chemicals
LIQ was obtained from Chengdu Alfa Biotechnology
Co., Ltd. CoCl2 was purchased from Toronto Research
Chemicals. Cell Counting Kit-8 (CCK-8; cat. no. G02111) was
acquired from Dongren Chemical Technology Co., Ltd. High-glucose
Dulbecco's modified Eagle medium (DMEM; cat. no. 12430047) and
fetal bovine serum (FBS; cat. no. 10093188) were purchased from
Gibco (Thermo Fisher Scientific, Inc.). TNF-α (cat. no. 88-7340-88)
and IL-6 (cat. no. 88-50625-88) ELISA kits were obtained from
Thermo Fisher Scientific, Inc. The remaining ELISA kits, including
lactated dehydrogenase (LDH; cat. no. A02022), creatine kinase
isoenzyme-MB (CK-MB; cat. no. H197-1-1), superoxide dismutase (SOD;
cat. no. A00132), catalase (CAT; cat. no. A007-11), glutathione
peroxidase (GSH-Px; cat. no. A005-1-2) and malondialdehyde (MDA;
cat. no. A00311), were from Nanjing Jiancheng Bioengineering
Institute. Unless stated otherwise, all other reagents used in this
experiment were purchased from Sigma-Aldrich (Merck KGaA).
Cell culture
H9c2 cells (Bluefbio Biotechnology Development,
Inc.; cat. no. BFN60804388) were maintained in high-glucose DMEM
supplemented with 10% FBS and 100 U/ml penicillin/streptomycin. The
cells were incubated at 37˚C, 5% CO2 and 95% air in an
incubator. The culture medium was refreshed every 2-3 days. When
the cells reached ~70-80% confluence, they were detached using 0.25
g/l trypsin, passaged and diluted to the appropriate cell
suspension concentration. Subsequently, the cells were cultured in
96-, 24- and 6-well plates until they adhered.
Proliferation inhibition assay of H9c2
cells by LIQ
H9c2 cells cultured in 96-well plates were exposed
to various concentrations of LIQ (1, 3, 10, 30, 100 and 300
µmol/l). After a 24-h incubation period, 10 µl of CCK-8 solution
was added and absorbance was measured at 450 nm using an enzyme
labeling instrument (Thermo Scientific Varioskan LUX; Thermo Fisher
Scientific, Inc.). The protective concentrations of LIQ were
determined based on CCK-8 calculations, confirming 3 and 10 µmol/l
as the effective concentrations.
Determination of hypoxic dose and
reoxygenation time
Experiments were conducted with H9c2 cells seeded in
96-well plates. The cells were exposed to different concentrations
(200, 400, 600, 800 and 1,000 µmol/l) of CoCl2 solutions
prepared in complete medium for 24 h. The optimal hypoxic
concentration was determined based on the results obtained from the
CCK-8 assay. Following the hypoxic phase, the cells were incubated
in complete medium for varying time intervals (1, 2, 3, 4, 5 and 6
h) to determine the appropriate reoxygenation duration. The optimal
reoxygenation time was established using the results from the CCK-8
kit. In summary, the H/R injury model was established by subjecting
the cells to 400 µmol/l CoCl2-induced hypoxia for 24 h,
followed by reoxygenation for 3 h. The experimental groups were
categorized as follows: i) Control (CON); ii) H/R; iii) H/R + 3
µmol/l (L-)LIQ; iv) H/R + 10 µmol/l (H-)LIQ; v) LIQ (10
µmol/l).
Measurement of LDH and CK-MB
Following the aforementioned treatment, the cell
culture medium supernatant was collected. Subsequently, the levels
of LDH and CK-MB were measured in each group following the
instructions provided with the respective kits.
Measurement of SOD, CAT, GSH-Px and
MDA activities
At the conclusion of the experiment, the cells were
scraped and resuspended in 500 µl of PBS solution. A cell
homogenate was then prepared and the levels of SOD, CAT, GSH-Px and
MDA were measured using the respective kits, following the provided
instructions.
Measurement of ROS generation
2,7-Dichlorodihydro-fluorescein diacetate (DCFHDA,
Cayman Chemical Company; cat. no. 85155) itself does not emit green
fluorescence. However, intracellular ROS oxidizes the
non-fluorescent DCFH to green-fluorescent DCF. The intensity of the
resulting green fluorescence indirectly reflects the level of
intracellular ROS. H9c2 cells were seeded in 24-well culture plates
and allowed to grow until they reached ~80% confluence.
Subsequently, the cells were treated as required. After the
treatment, the cells were rinsed twice with PBS and incubated with
10 µmol/l DCFH-DA dye at 37˚C for 20 min. Images were captured
using a fluorescence microscope (Nikon Eclipse C1; Nikon
Corporation) and the green fluorescence intensity was
semi-quantitatively analyzed using ImagePro Plus 6.0 software
(Media Cybernetics, Inc.). The green fluorescence intensity was
then analyzed semi-quantitatively and statistically for each sample
within each group.
Measurement of mitochondrial membrane
potential (MtMP)
Rhodamine 123 (Shanghai Yuanye Biotechnology Co.,
Ltd; cat. no. S19123) is a fluorescent dye that selectively
accumulates in mitochondria based on their transmembrane potential.
When the cells reached ~80% confluence, various conditioned media
were applied as per the experimental requirements. Following
treatment, the cells were washed twice with PBS and then incubated
at 37˚C for 20 min in a serum-free medium containing 100 µg/l
Rh123. The green fluorescence observed around the nuclei indicated
the uptake of Rh123 by the mitochondria. The green fluorescence
intensity, representing Rh123 uptake by mitochondria, was
semi-quantitatively analyzed using ImagePro Plus 6.0 software
(Media Cybernetics, Inc.). Statistical analysis was conducted on
the green fluorescence intensity for each sample within each
group.
Measurement of intracellular
inflammatory cytokines
H9c2 cells were seeded in 6-well plates and treated
according to the experimental group requirements once they reached
the desired confluency. The treated cells were collected as the
test specimens. ELISA procedures were carried out following the
instructions provided with the respective kits. After the reaction
was completed, the absorbance values of each well were measured
using a multifunctional microplate reader. The levels of TNF-α and
IL-6 were determined following the provided kit instructions.
Measurement of apoptosis
H9c2 cells were cultured in 24-well plates and
treated as required once they reached the desired density.
Hoechst-33258 dye (Beijing Solarbio Science & Technology Co.,
Ltd; cat. no. IH0060) buffer at a concentration of 5 mg/l was
applied to the cells, followed by a 15-min incubation in a cell
incubator. The nuclei of apoptotic cells displayed either condensed
solidified forms or granular fluorescence. Quantification of
apoptotic cells was performed using ImagePro Plus 6.0 software
(Media Cybernetics, Inc.).
Western blotting
The core targets and key signaling pathways
predicted through network pharmacology were validated using the
western blotting. H9c2 cells were seeded in 6-well plates and
treated according to the experimental requirements. The cells were
washed twice with pre-cooled PBS and then lysed using cell lysate
(Beijing Solarbio Science & Technology Co., Ltd.; cat. no.
R0020). The lysates were centrifuged at 12,000 x g for 10 min at
4˚C. The supernatant was quantified using the BCA method. Total
protein (10 µg) was separated using 10% SDS-PAGE and transferred
onto PVDF membranes. The PVDF membranes were subsequently incubated
in 5% skimmed milk powder for 1.5 h to block nonspecific binding at
37˚C. Next, the membranes were incubated overnight at 4˚C with the
corresponding primary antibodies, as follows: TNF-α receptor type 1
(TNFR1, Affinity Biosciences; diluted at 1:1,000; cat. no. AF0282),
NF-κB (Affinity Biosciences; diluted at 1:1,000; cat. no. BF8005),
phosphorylated (p-)NF-κB (Affinity Biosciences; diluted at 1:1,000;
cat. no. AF2006), MMP9 (Affinity Biosciences; diluted at 1:1,000;
cat. no. AF5228) and β-actin (Affinity Biosciences; diluted at
1:10,000; cat. no. AF7018). The following day, the membranes were
washed three times with TBST (0.1% Tween 20) for an average of 10
min per wash, followed by incubation with the corresponding
secondary antibody at room temperature for 1 h. After an additional
three washes, grayscale values were measured using Visioncapt
software (Fusion FX5 Spectra; Wilbur Bioimaging).
Statistical analysis
GraphPad Prism 8.0.2 software (Dotmatics) was used
for statistical analyses. Data were presented as mean ± standard
error of mean and one-way analysis of variance followed by Tukey's
post hoc test was employed to assess statistically significant
differences between groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Network pharmacology analysis predicts
results. Physicochemical properties of LIQ
The data pertaining to the physicochemical and
pharmacological molecular properties of LIQ were sourced from the
TCMSP database. Drug-like properties (DL) serve as a valuable
indicator for assessing the potential of an ingredient in drug
development, while oral availability (OB) is employed to gauge the
ease of absorption of the ingredient into the bloodstream. The
obtained OB value was 65.69% and the DL properties were measured at
0.74. These results demonstrated that LIQ exhibited an OB value of
≥30% and a DL value of ≥0.18, indicating its strong potential for
drug development, as it is well-absorbed and possesses favorable
drug-like properties.
LIQ and MIRI's potential target
acquisition
A comprehensive analysis identified 315 targets for
LIQ through predictions derived from the TCMSP database, Swiss
Target Prediction and PharmMapper databases. In parallel, 1,052
targets for MIRI were predicted based on data from the Genecards
database. The intersection of these two datasets revealed a total
of 99 shared targets, as illustrated in Fig. 2A. Detailed information pertaining
to specific target sites can be found in Fig. 2B.
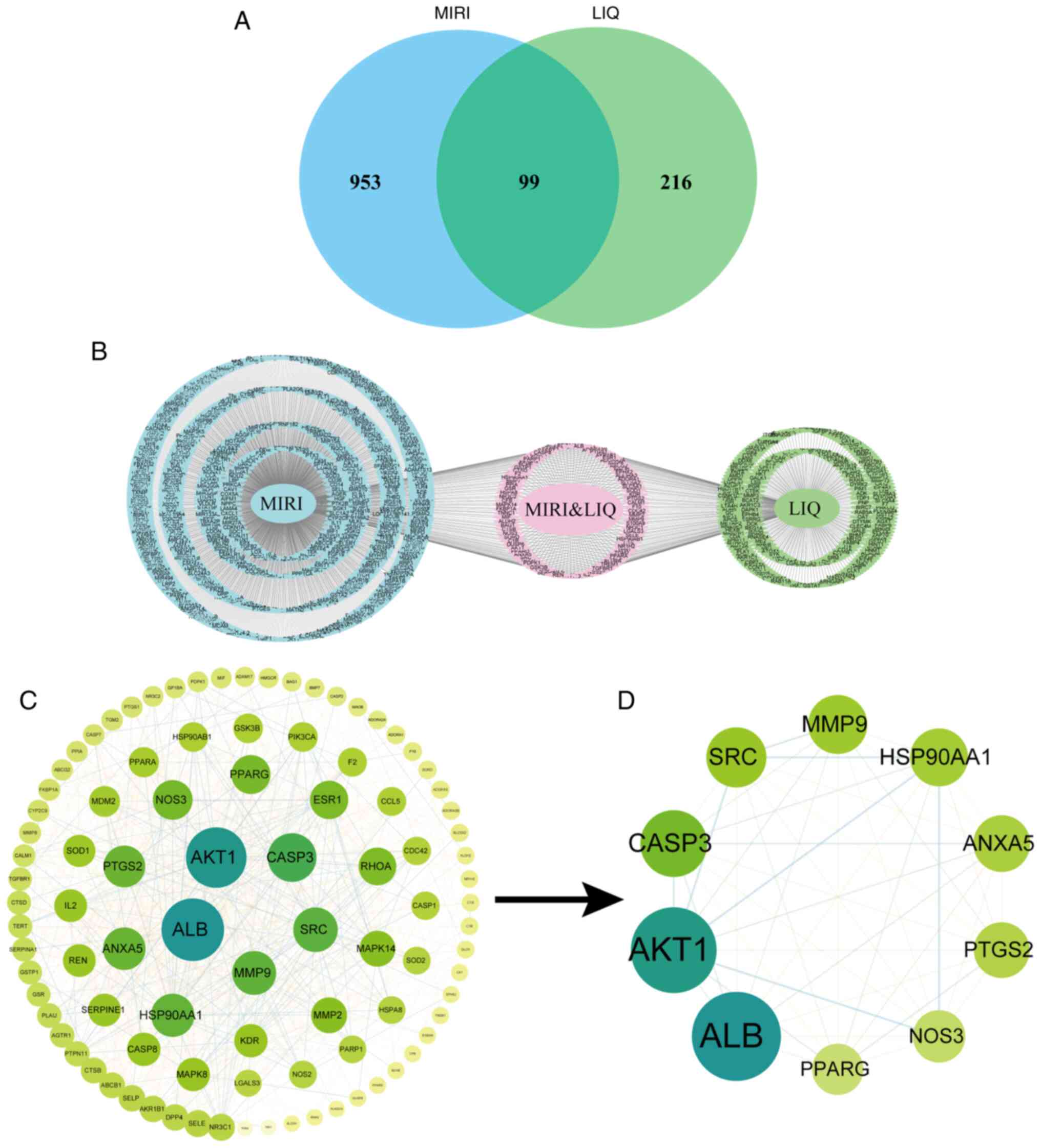 | Figure 2Core target analysis diagram on
overlapping focal points of LIQ for MIRI. (B) Composite target
display and (A) Venn diagram of LIQ and MIRI-related targets. (C)
Core Target Interaction Network Diagram. (D) Top 10 targets of the
core targets. LIQ, liquiritin; MIRI, myocardial
ischemia-reperfusion injury; AKT1, Protein Kinase B α; CASP3,
cystatin protease 3; ALB, albumin; SRC, proto-oncogene
tyrosine-protein kinase Src; MMP9, Matrix metalloproteinase-9;
HSP90AA1, heat shock protein 90α family class A member 1; ANXA5,
membrane-linked protein A5; PTGS2, prostaglandin-endoperoxide
synthase 2; NOS3, nitric oxide synthase 3; PPARG, peroxisome
proliferator-activated receptor γ. |
Construction of PPI networks
The PPI network was established by entering the
shared targets into the STRING database with subsequently analysis
to identify its core network, as depicted in Fig. 2C. Within this core network, the top
10 targets were identified (Fig.
2D), which included AKT1, cystatin protease 3 (CASP3), albumin
(ALB), proto-oncogene tyrosine-protein kinase Src (SRC), MMP9, heat
shock protein 90α family class A member 1 (HSP90AA1),
membrane-linked protein A5 (ANXA5), prostaglandin-endoperoxide
synthase 2 (PTGS2), nitric oxide synthase 3 (NOS3) and peroxisome
proliferator-activated receptor γ (PPARG). MMP9 was selected as the
primary focus of this research among these targets.
GO functional analysis and KEGG
pathway analysis
GO analysis, conducted using the DAVID database,
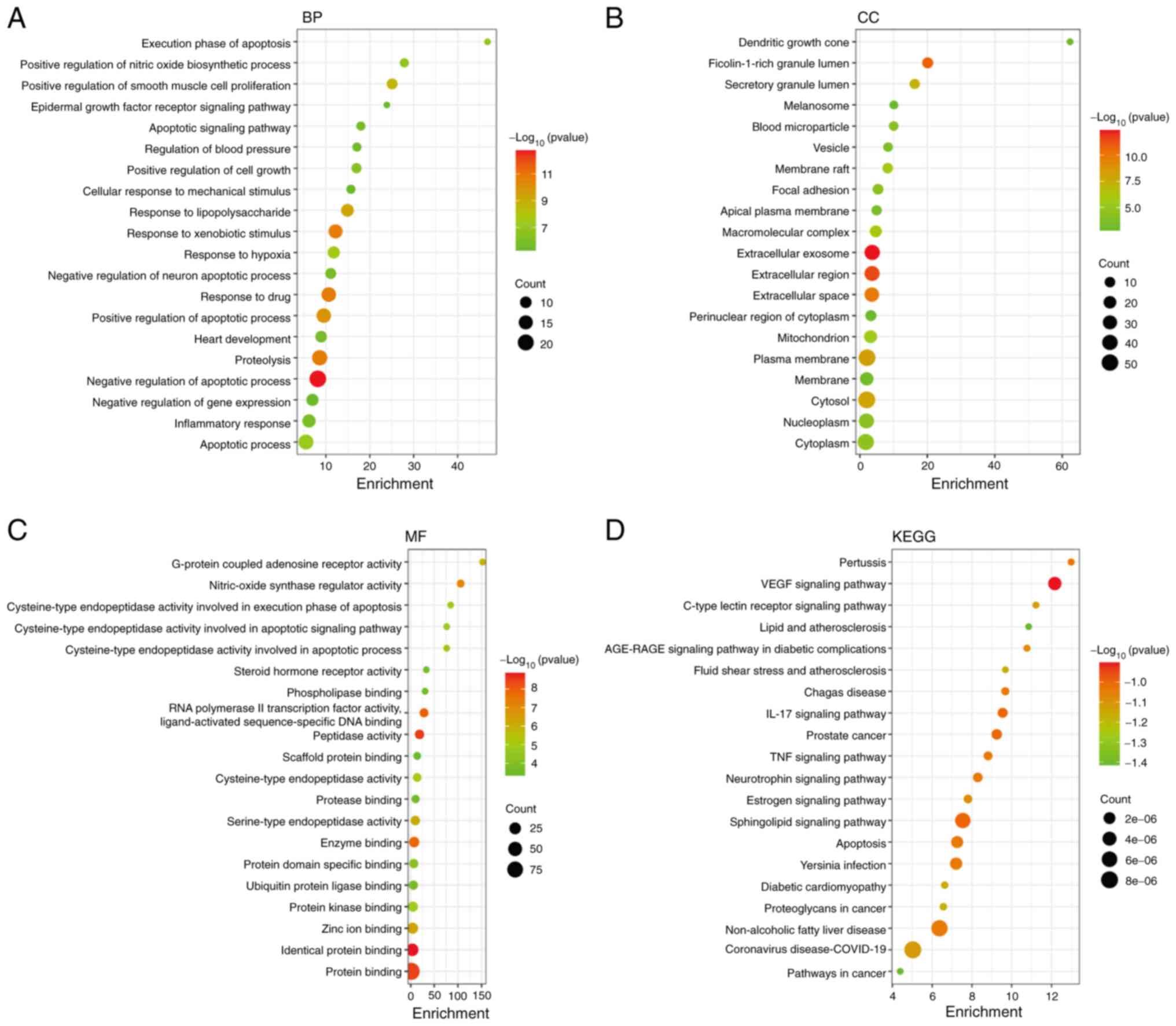
yielded a total of 541 GO entries. Examination of the 394 BP
categories unveiled the involvement of processes such as hypoxia,
apoptosis, inflammation and oxidative stress. Within the CC
entries, a total of 48 terms were enriched, primarily connected to
extracellular exosomes, extracellular regions, plasma membranes,
cytosols and mitochondria. The MF entries were also enriched,
featuring 99 terms primarily associated with functions such as
identical protein binding, peptidase activity, protein binding,
zinc ion binding and cysteine-type endopeptidase activity involved
in the execution phase of apoptosis, as depicted in Fig. 3A-C.
The KEGG pathway enrichment analysis identified a
total of 125 pathways (P<0.01). These pathways were
predominantly related to lipid and atherosclerosis, pathways in
cancer, the TNF signaling pathway, the IL-17 signaling pathway and
apoptosis. To enhance clarity, the top 20 pathways were selected
for visualization, as depicted in Fig.
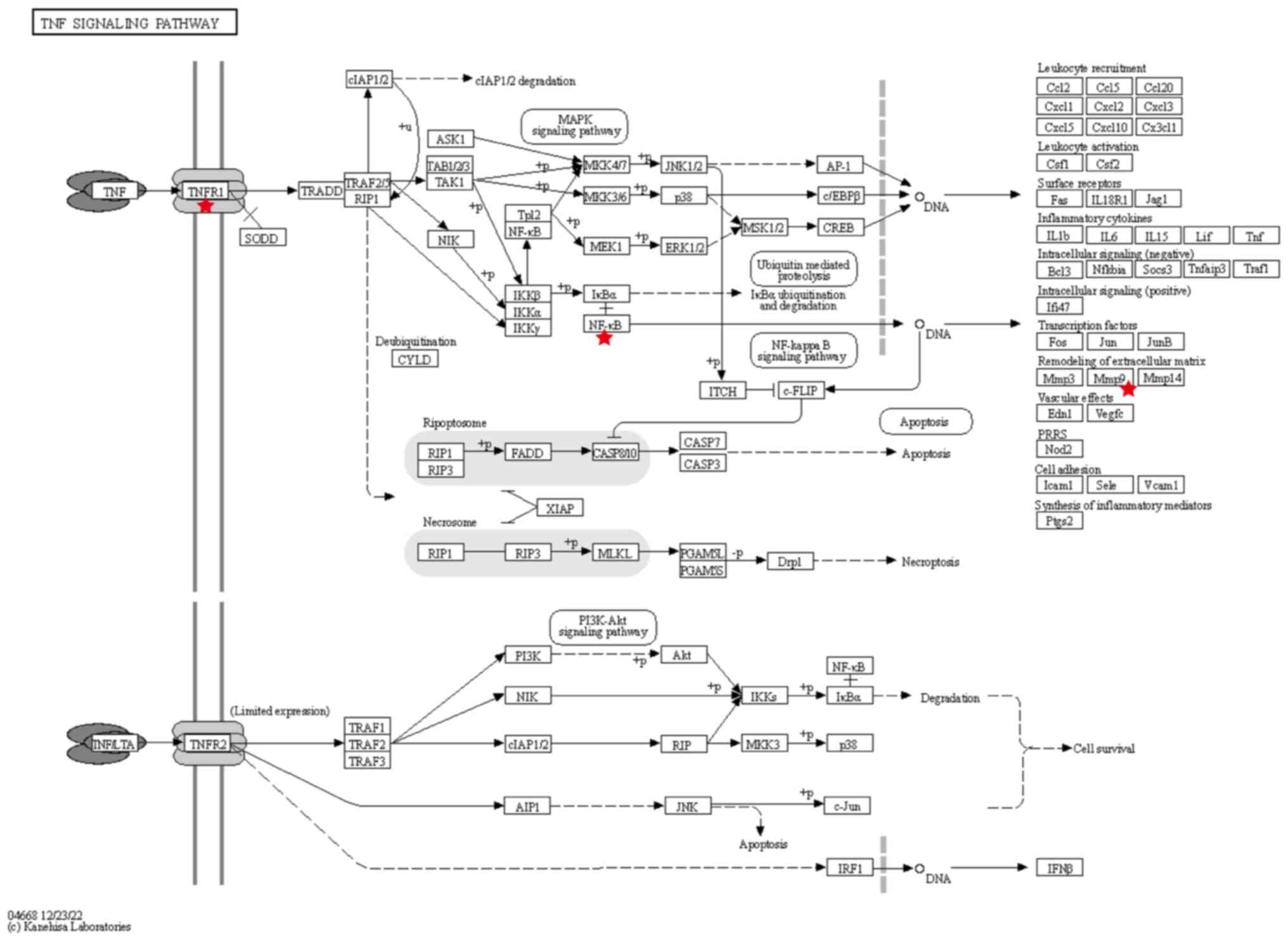
3D. Notably, among these pathways, 11 targets of LIQ were found
to be involved in the regulation of the TNF signaling pathway, with
the core target MMP9 among them. Details of the predicted targets
associated with the TNF signaling pathway can be found in Fig. 4.
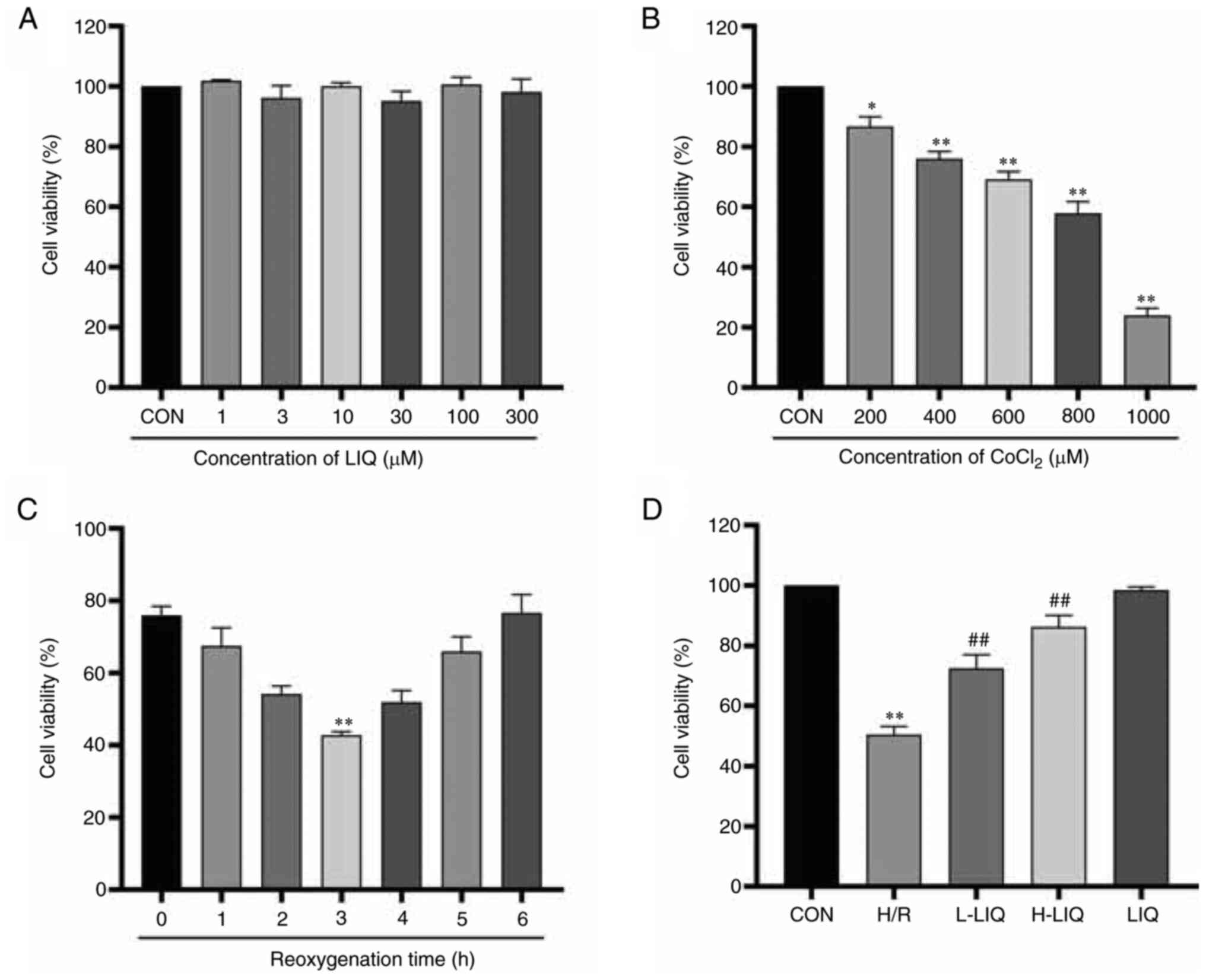
Effect of LIQ on cell viability
As shown in Fig.
5A, no significant changes in cell survival were observed when
H9c2 cells were exposed to various concentrations of LIQ. To
determine the optimal pre-protective concentrations for LIQ, 3
µmol/l and 10 µmol/l were selected. As depicted in Fig. 5B, cell viability decreased as the
CoCl2 concentration increased when H9c2 cells were
subjected to varying CoCl2 concentrations. To maintain
cell survival at ~50% following hypoxia/reoxygenation, 400 µmol/l
of CoCl2 was selected to induce hypoxia (P<0.01),
followed by a 3-h reoxygenation period (Fig. 5C; P<0.01). As illustrated in
Fig. 5D, the viability of cells in
the H/R group was significantly lower than that in the CON group
(P<0.01). However, LIQ pre-treatment effectively mitigated the
inhibitory effect of CoCl2 on H9c2 cell viability
(P<0.01).
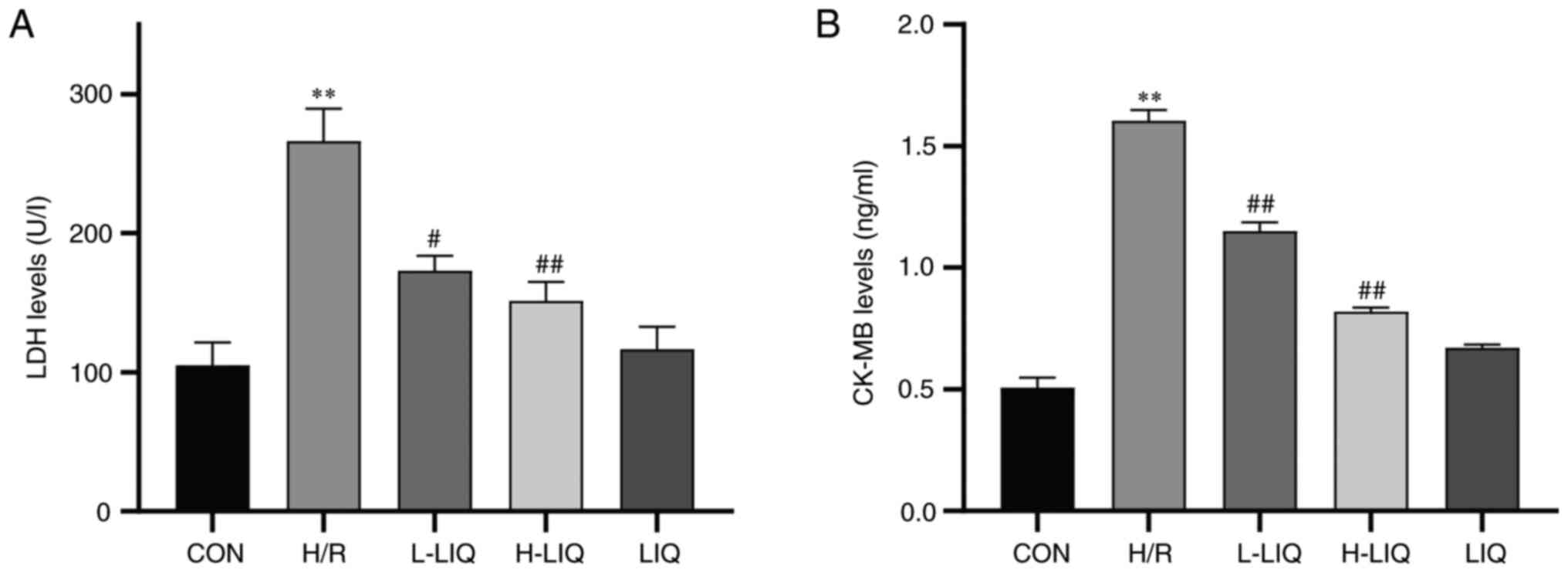
Effect of LIQ on the release of LDH
and CK-MB
As depicted in Fig.
6A and B, the levels of LDH
and CK-MB exhibited a significant increase in the H/R group
compared with the CON group (P<0.01). However, treatment with
LIQ resulted in a notable reduction in the levels of LDH and CK-MB
in cardiomyocytes when compared with the H/R group (P<0.05 or
0.01). Additionally, it was notable that the reductions in LDH and
CK-MB levels were more pronounced at higher LIQ doses, suggesting
that LIQ pre-treatment effectively mitigated the damage to
cardiomyocytes caused by H/R.
Effect of LIQ on the levels of SOD,
CAT, GSH-Px and MDA
As illustrated in Fig.
7, the levels of MDA, SOD, CAT and GSH-Px and can serve as
indicators of oxidative stress damage. In comparison to the CON
group, the H/R group displayed notably lower levels of SOD, CAT and
GSH-Px (P<0.01), while the levels of MDA were significantly
higher (P<0.01). However, the pre-protective effect of LIQ
alleviated the damage induced by H/R, as evidenced by significantly
higher levels of SOD, CAT and GSH-Px in the LIQ group in comparison
to the H/R group (P<0.05 or 0.01), along with significantly
lower levels of MDA (P<0.01).
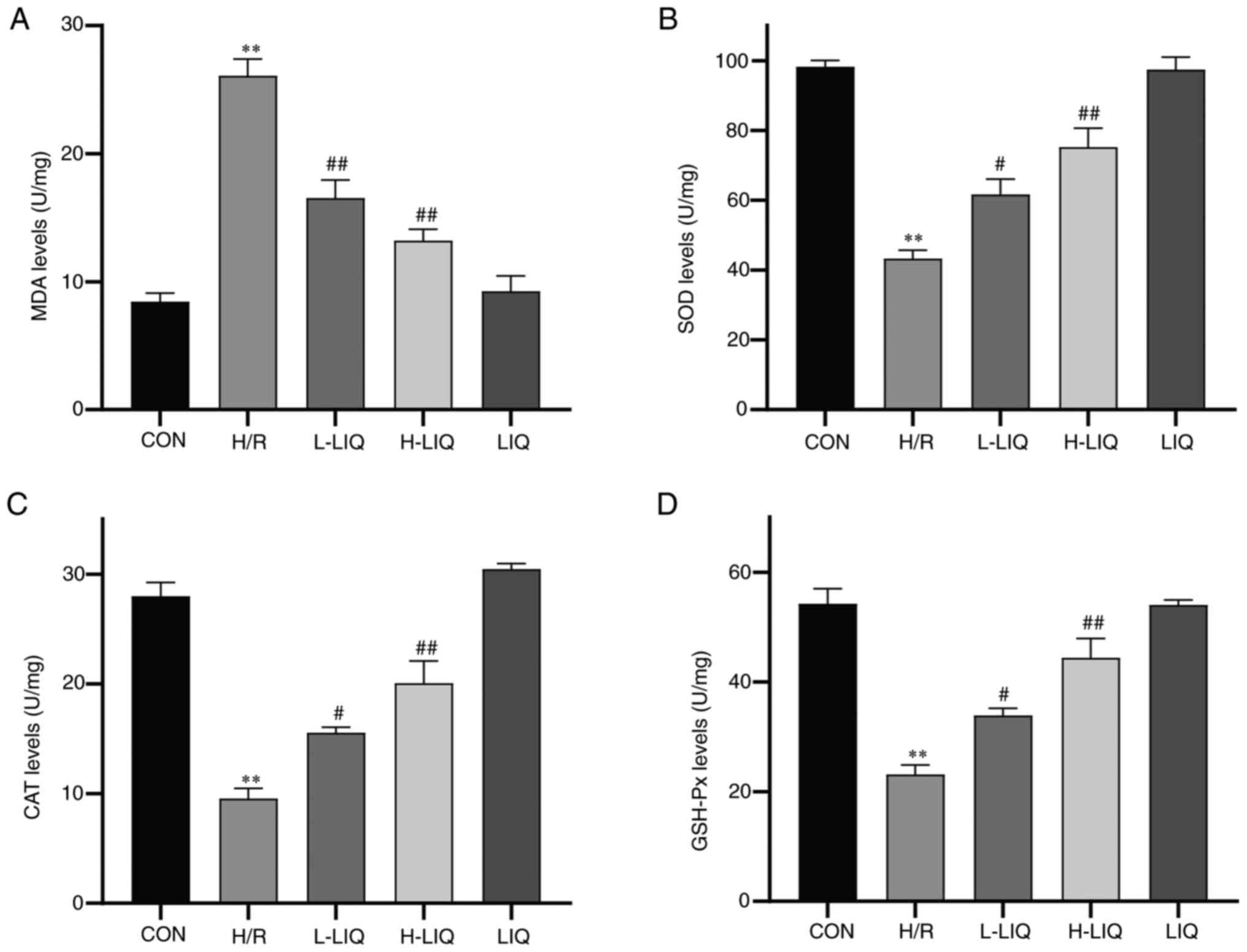 | Figure 7Effects of LIQ on H/R-induced levels
of oxidative stress. (A) MDA, (B) SOD. (C) CAT and (D) GSH-Px
levels. Data are represented as mean ± standard error of mean.
**P<0.01 vs. CON; #P<0.05,
##P<0.01 vs. H/R group, n=3. LIQ, liquiritin; H/R,
hypoxia/reoxygenation; MDA, malondialdehyde; SOD, superoxide
dismutase; CAT, catalase; GSH-Px, glutathione peroxidase; CON,
control; H/R, hypoxia/reoxygenation; L-LIQ, 3 µmol/l; H-LIQ, 10
µmol/l. |
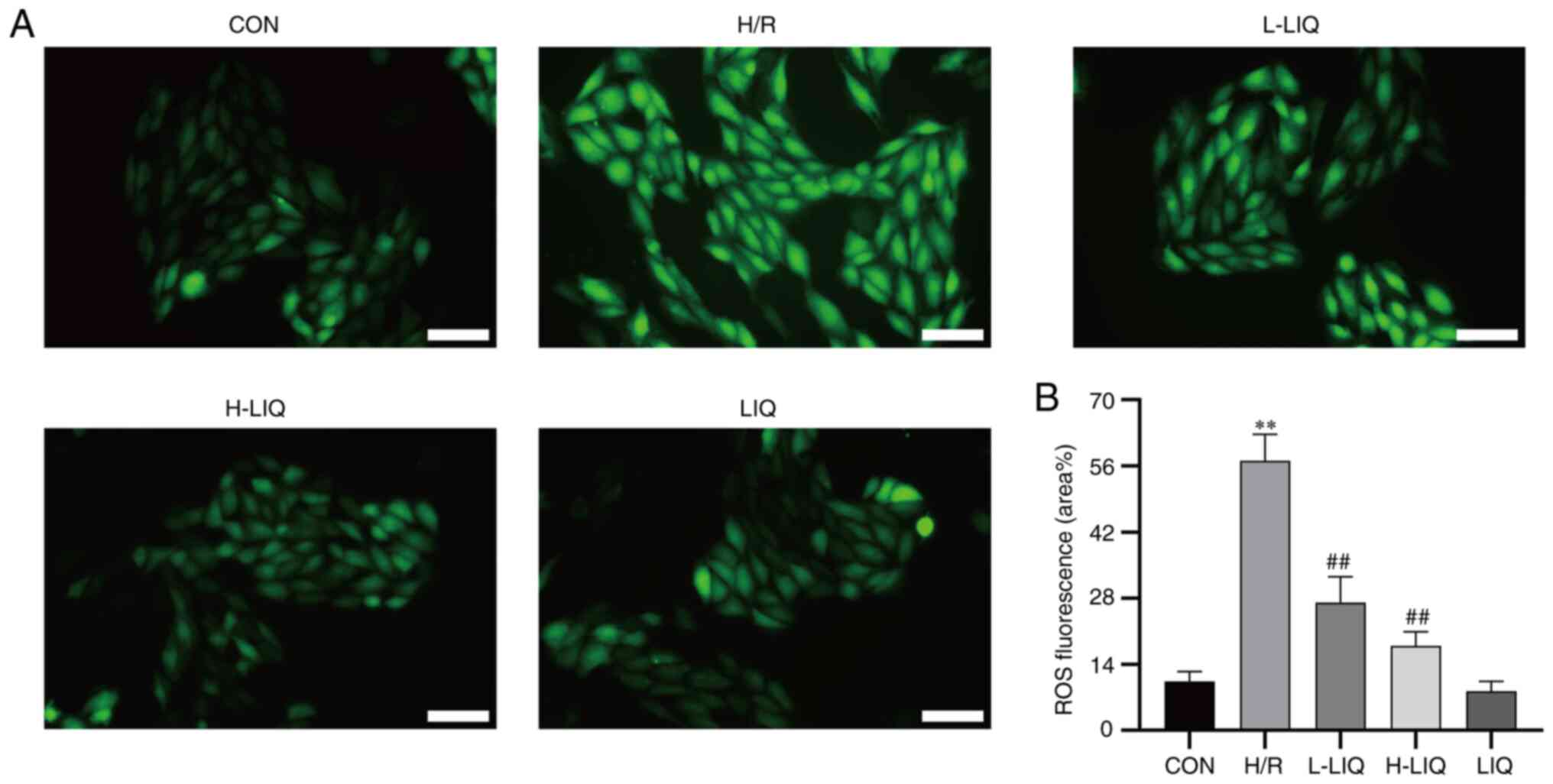
Effect of LIQ on ROS generation
The intracellular ROS level and the intensity of
green fluorescence demonstrated a direct proportional relationship.
As depicted in Fig. 8, the
intracellular fluorescence intensity and ROS level in H9c2 cells
within the H/R group were markedly higher than in the CON group
(P<0.01). By contrast, the fluorescence intensity and ROS level
in the LIQ group were significantly lower than those in the H/R
group (P<0.01). Additionally, it was observed that the
antagonistic effect on H/R injury became more pronounced with
higher doses of LIQ.
Effects of LIQ on MtMP
The extent of MtMP damage showed an inverse
correlation with the aggregation of Rh123. As depicted in Fig. 9, the aggregation of Rh123 in H9c2
cells of the H/R group was significantly lower compared with the
CON group, indicating substantial MtMP damage (P<0.01). In
contrast, the aggregation of Rh123 in H9c2 cells was significantly
increased in the LIQ-treated group compared with the H/R group
(P<0.05 or 0.01). This suggests that LIQ can effectively reduce
MtMP damage in H/R injury with a concentration-dependent effect.
The higher the concentration, the more pronounced the effect.
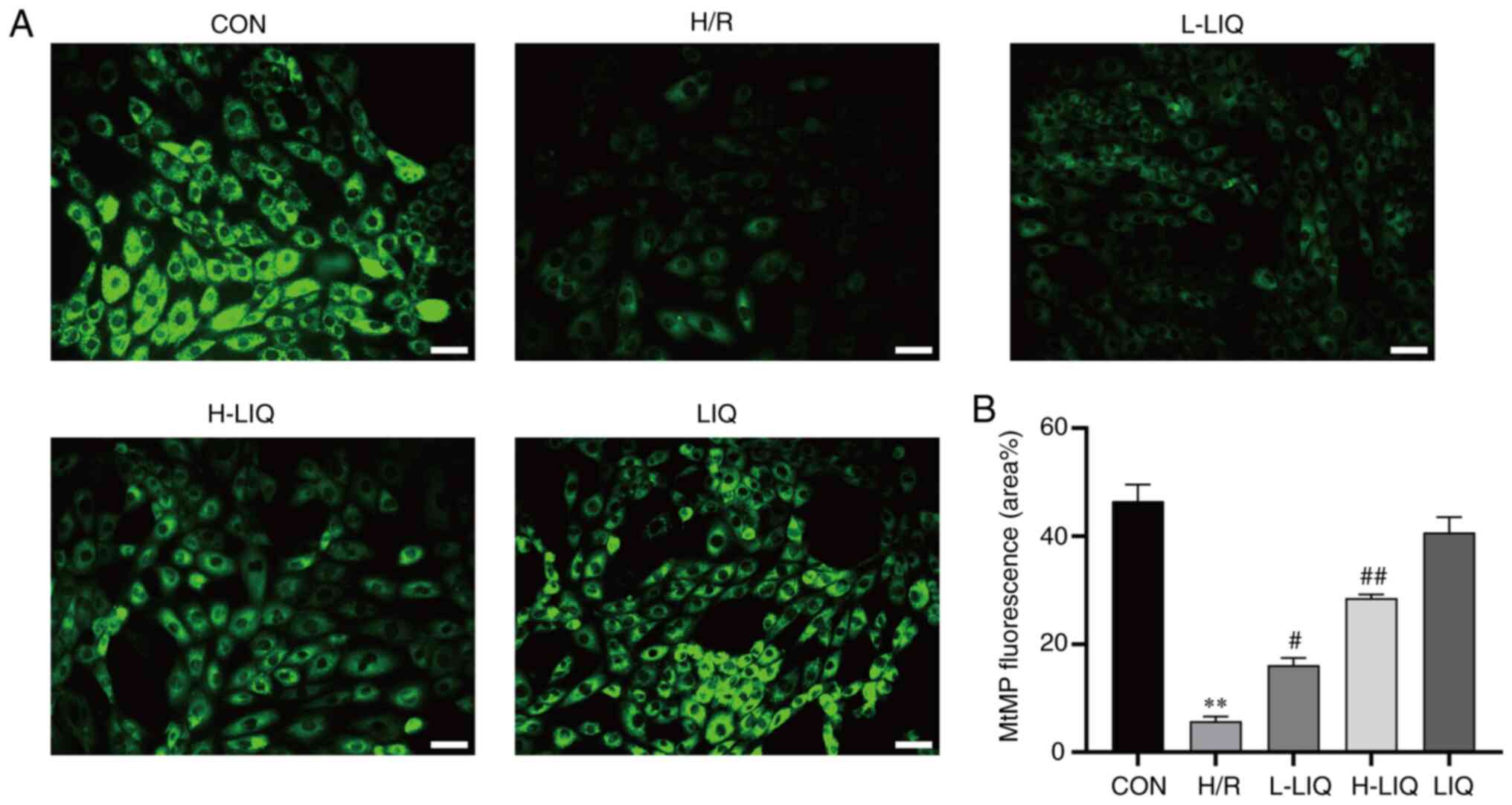 | Figure 9Effects of LIQ on H/R-induced
mitochondrial damage in H9c2 cells. (A) Morphological changes of
mitochondria were observed by Rhodamine 123 (scale bar, 100 µm;
magnification, x200). (B) Statistical analysis of mitochondrial
fluorescence was presented. Data are represented as mean ± standard
error of mean. **P<0.01 vs. CON;
#P<0.05, ##P<0.01 vs. H/R group, n=3.
LIQ, liquiritin; H/R, hypoxia/reoxygenation; CON, control; H/R,
hypoxia/reoxygenation; L-LIQ, 3 µmol/l; H-LIQ, 10 µmol/l; MtMP,
mitochondrial membrane potential. |
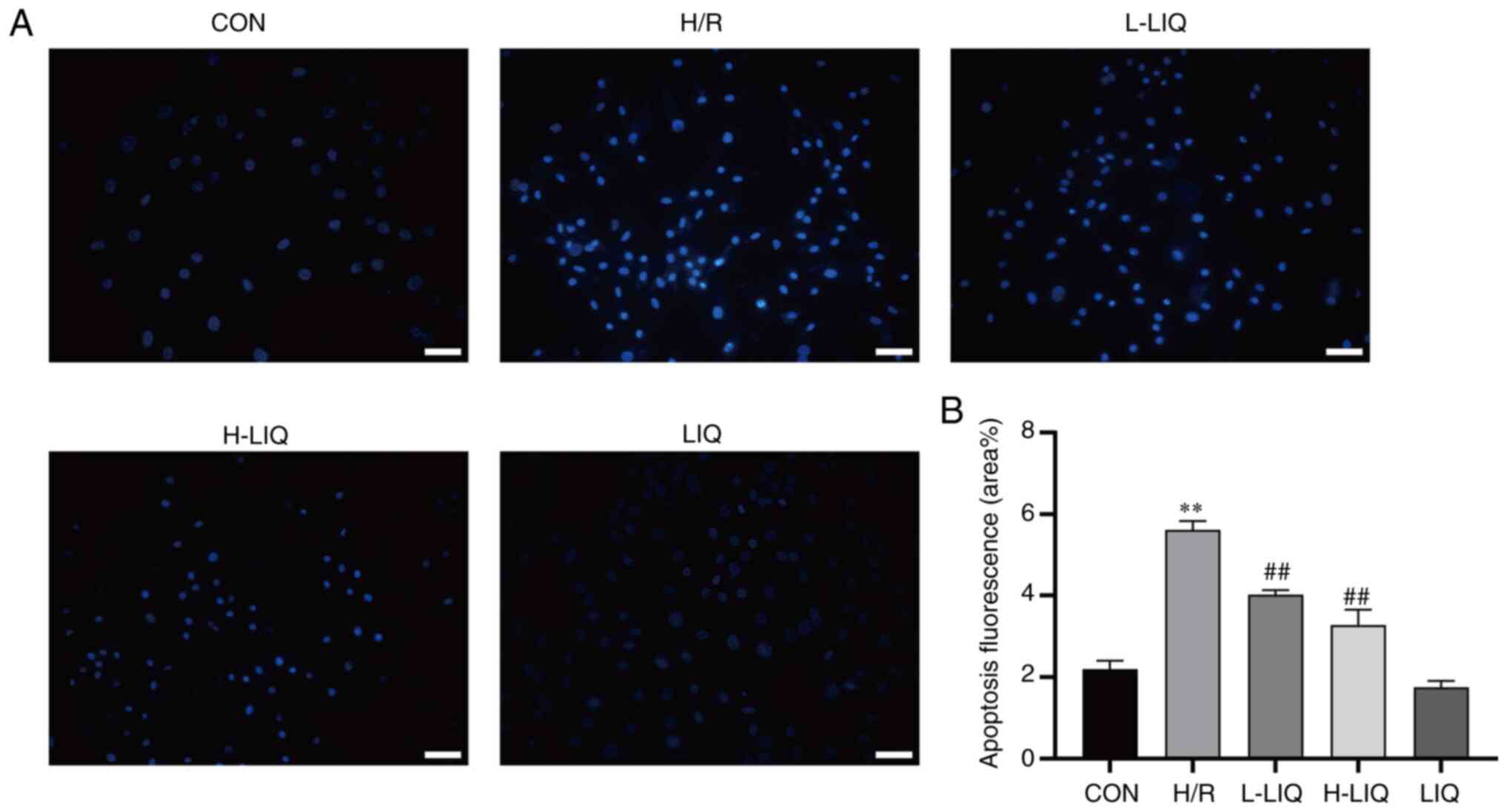
Effects of LIQ on apoptosis
During the process of apoptosis, morphological
abnormalities often manifest, including cell shrinkage, chromatin
condensation, cell membrane rupture and nuclear fragmentation
(18). To verify the presence of
apoptosis, the morphology of abnormal cells was examined using
Hoechst 33258 staining and fluorescence microscopy. In normal
cells, the nuclei displayed uniform blue fluorescence, while
apoptotic cells exhibited densely stained blue nuclei or fragmented
densely stained blue nuclei.
As illustrated in Fig.
10, when compared with the CON group, cells in the H/R group
exhibited pronounced apoptotic morphology when observed under
fluorescence microscopy following ultraviolet light excitation
(P<0.01). However, in the LIQ-treated group, the apoptotic
morphology was significantly reduced compared with the H/R group
(P<0.01), indicating that LIQ attenuated H/R-induced apoptosis
and enhanced the survival rate of H9c2 cells.
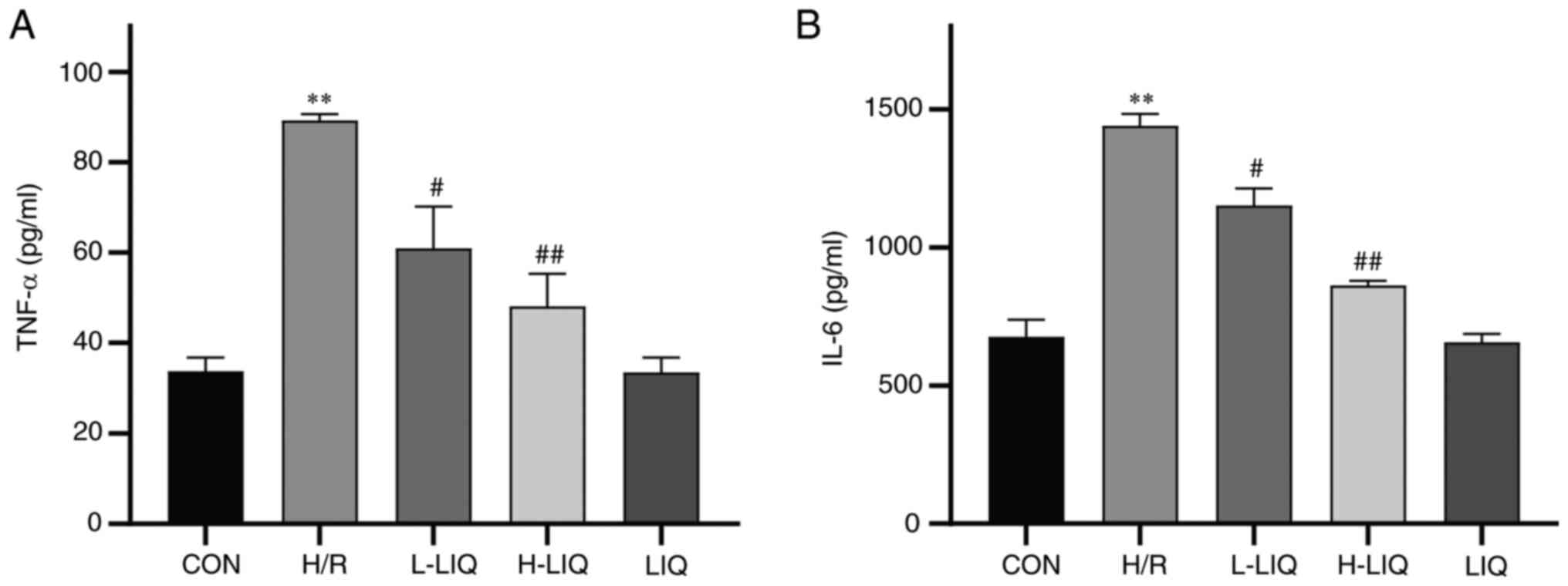
Effects of LIQ on the levels of TNF-α
and IL-6
The results depicted in Fig. 11 indicated a significant increase
in the secretion levels of TNF-α and IL-6 in the H/R group when
compared with the CON group (P<0.01). However, LIQ treatment
notably reduced the secretion levels of TNF-α and IL-6 (P<0.05
or 0.01). These findings suggested that LIQ can effectively
mitigate the inflammatory damage induced by H/R, with a more
pronounced antagonistic effect observed at higher doses.
Effect of LIQ on the protein
expression levels of TNFR1, p-NF-κB/NF-κB and MMP9
The results of western blotting, showing TNFR1,
p-NF-κB/NF-κB and MMP9 protein expression in H9c2 cells, are
presented in Fig. 12. The
expression levels of TNFR1, p-NF-κB/NF-κB and MMP9 proteins were
markedly higher in the H/R group compared with the CON group
(P<0.01). However, in the LIQ-treated group, the expression
levels of these proteins were significantly lower than in the H/R
group, demonstrating significant differences (P<0.05 or 0.01).
These findings indicated that LIQ effectively mitigated the
abnormally elevated expression of TNFR1, p-NF-κB/NF-κB and MMP9
proteins.
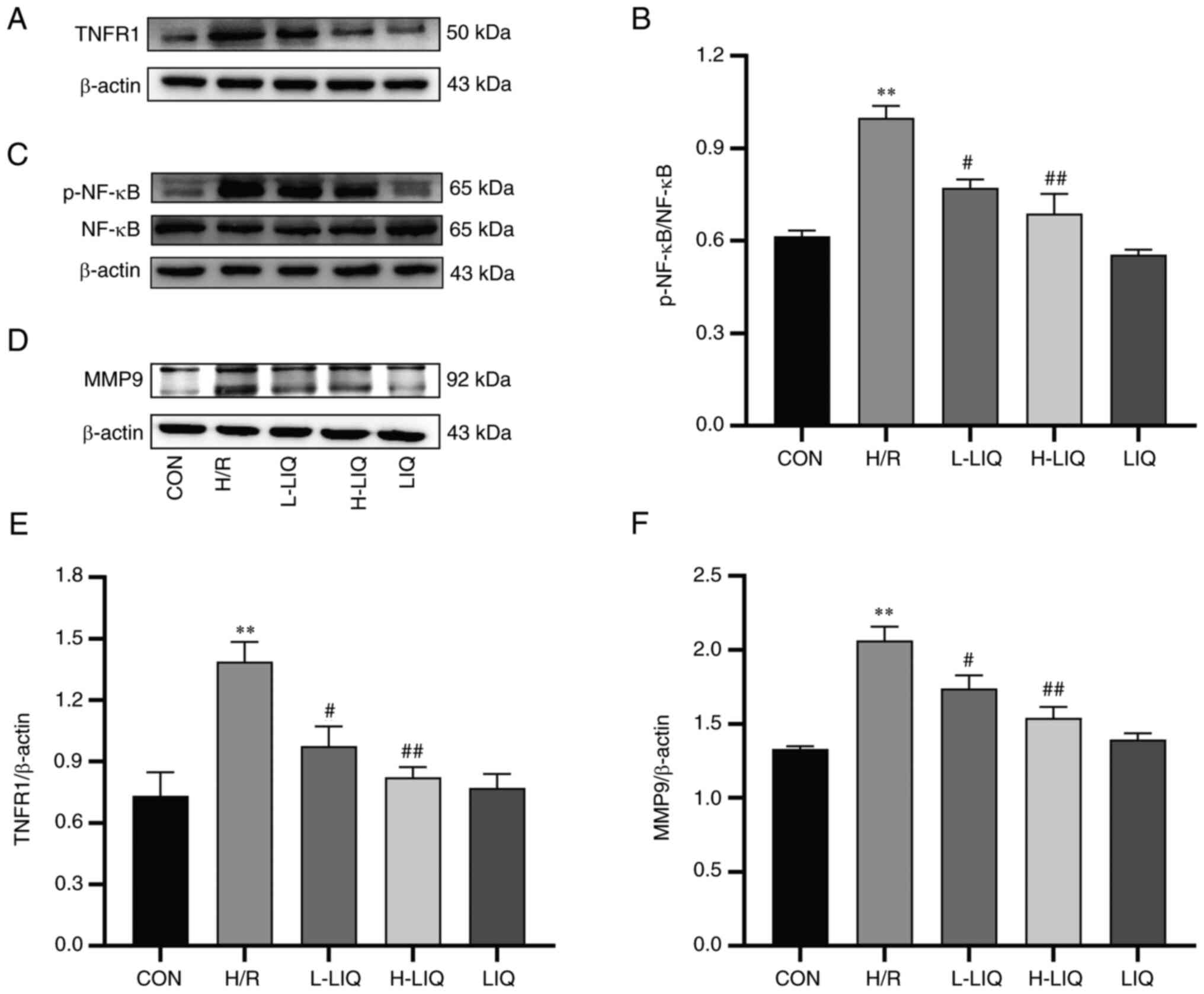 | Figure 12Effects of LIQ on the (A and E)
TNFR1, (C and B) p-NF-κB/NF-κB and (D and F) MMP9 protein
expressions. Data are represented as mean ± standard error of mean.
**P<0.01 vs. CON; #P<0.05,
##P<0.01 vs. H/R group, n=3. LIQ, liquiritin; TNFR1,
TNF-α receptor type 1; p-, phosphorylated; CON, control; H/R,
hypoxia/reoxygenation; L-LIQ, 3 µmol/l; H-LIQ, 10 µmol/l. |
Discussion
The present study proposes the use of network
pharmacology to predict potential targets and mechanisms of LIQ in
the treatment of MIRI. An in vitro H/R model was employed to
simulate MIRI and cellular experiments were conducted to validate
the protective mechanisms of LIQ on H/R-injured cardiomyocytes. The
investigation focused on various perspectives, including oxidative
stress, inflammation, apoptosis, mitochondria and molecular
mechanisms (Fig. 13). These
observations suggested that LIQ exhibited multiple biological
properties, implying its potential as an effective therapeutic
approach or adjunct in the treatment of MIRI.
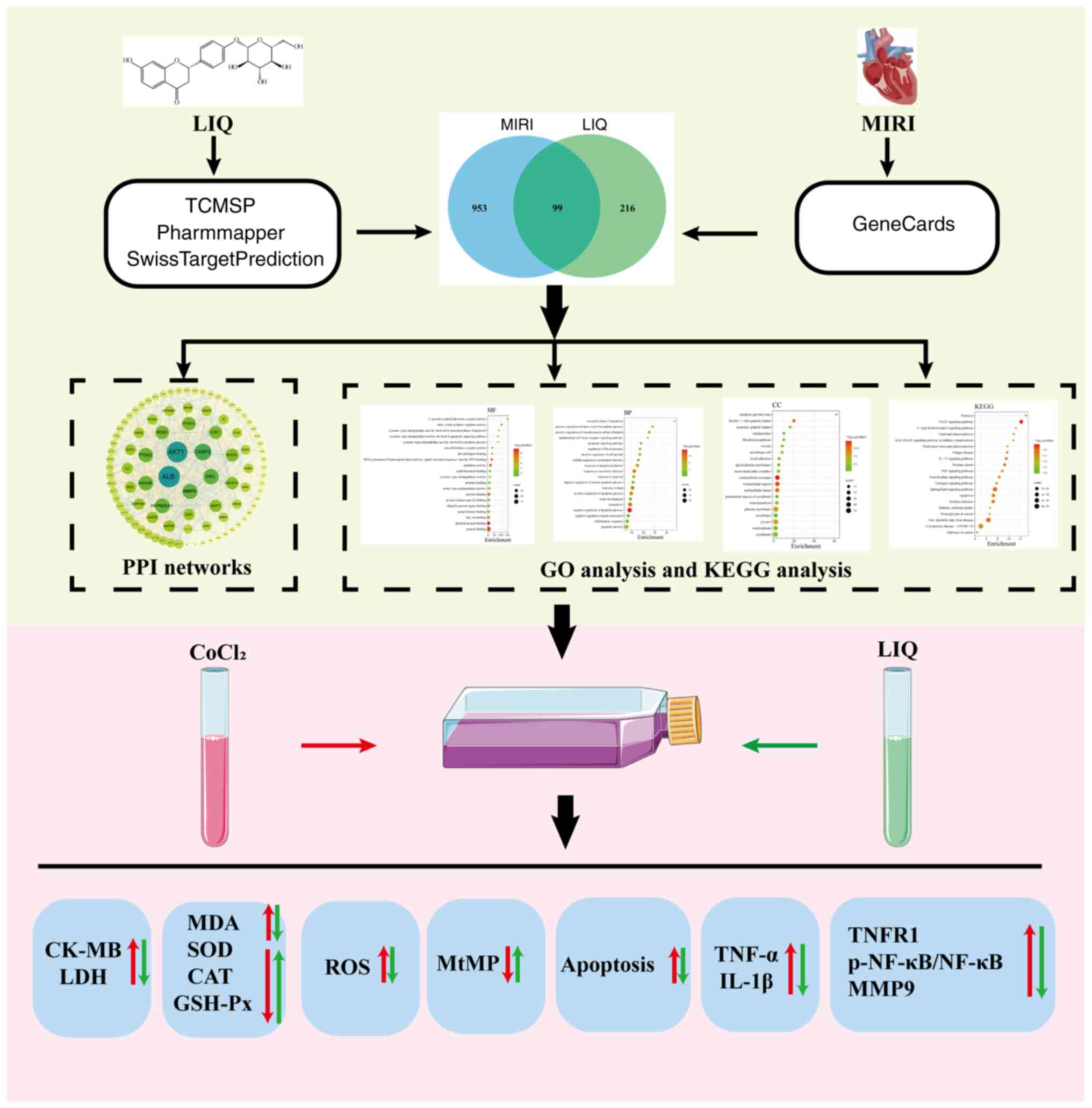 | Figure 13Workflow diagram for studying the
mechanism of LIQ resistance to H/R-induced damage. LIQ, liquiritin;
H/R, hypoxia/reoxygenation; L-LIQ, 3 µmol/l; H-LIQ, 10 µmol/l.
MIRI, myocardial ischemia-reperfusion injury; TCMSP, Traditional
Chinese Medicine Systems Pharmacology Database; PPI,
protein-protein interaction; GO, Gene Ontology; KEGG, Kyoto
Encyclopedia of Genes and Genomes; CK-MB, creatine kinase
isoenzyme-MB; LDH, lactated dehydrogenase; MDA, malondialdehyde;
SOD, superoxide dismutase; CAT, catalase; GSH-Px, glutathione
peroxidase; ROS, reactive oxygen species; TNFR1, TNF-α receptor
type 1; p-, phosphorylated; MtMP, mitochondrial membrane
potential. |
The physicochemical parameters of LIQ, which are
crucial for subsequent drug analysis, were sourced from the TCMSP
database. These parameters highlighted the potential of LIQ as a
candidate for effective drug development. The Venn diagram revealed
that LIQ may target inflammation through 99 identified targets. By
mining core targets, the present study was able to pinpoint more
critical targets among the common ones, including AKT1, CASP3, ALB,
SRC and MMP9, from the 95 targets identified through Cytoscape
analysis. Additionally, GO and KEGG analyses suggested that the
anti-MIRI effects of LIQ were associated with multiple mechanisms,
such as oxidative stress, apoptosis, mitochondria and inflammation.
These network pharmacology findings underscore the integrative
nature of Chinese medicine.
H9c2, a rodent ventricular cardiomyocyte cell line
with biochemical and physiological properties similar to human
cardiomyocytes, has become a preferred choice for in vitro
studies investigating the electrophysiological and biochemical
characteristics of myocardial tissues (19). The most commonly utilized method
for inducing hypoxia in laboratory settings is chemical
hypoxia-induced by CoCl2. This method offers advantages
in terms of cost-effectiveness and convenience compared with
physical hypoxia methods. The mechanism involves the replacement of
Fe2+ in prolyl hydroxylase (PHD) with Co2+
from CoCl2, resulting in the inactivation of PHD.
Consequently, the degradation of hypoxia-inducible factor (HIF) is
blocked. HIF plays a critical role in regulating oxygen homeostasis
in mammals. Notably, even under normoxic conditions, HIF
degradation is halted, remaining stable for several hours. This
experimental stability provides operators with more time for sample
manipulation and analysis (20).
The present study employed the CCK-8 assay to assess cell viability
and determine the optimal hypoxic concentration and reoxygenation
time. Based on the results, it established the H/R model by
subjecting cells to 400 µmol/l CoCl2-induced hypoxia,
followed by reoxygenation for 3 h.
The levels of LDH and CK-MB released served as
indicators of cell damage and the integrity of the cell membrane.
These enzymes are released when the cell membrane is disrupted and
therefore their levels reflect the extent of damage and cellular
necrosis (21). The results
further substantiate this observation, as they revealed a
significant increase in LDH and CK-MB release following H/R injury
when compared with the CON group. However, pre-treatment with LIQ
mitigated this injury.
Oxidative stress is a pivotal pathological mechanism
within the complex network of systems involved in H/R pathology
(22). The normal balance between
intracellular ROS production and the activity of oxidant-scavenging
enzyme systems (such as CAT, SOD and GSH) is disrupted, leading to
oxidative stress (23). Hypoxia
induces a reduction in endogenous ROS scavengers and an increase in
ROS production due to residual oxygen, resulting in ROS
accumulation in the myocardium. Upon reperfusion, the sudden influx
of excessive oxygen further exacerbates ROS levels, causing
oxidative damage to cellular structures, including lipid
peroxidation (24). Among the
sources of ROS in cardiac myocytes, mitochondria are the primary
contributors. Insufficient oxygen supply disrupts mitochondrial
coupling, leading to abnormal electron transfer in the
mitochondrial aerobic metabolic respiratory chain. The subsequent
elevation in ROS levels damages cells and can trigger apoptosis
(25). MDA, the most crucial
product of lipid peroxidation, reflects the extent of lipid
peroxidation and the severity of cellular exposure to free
radicals, making it a commonly used indicator of oxidative stress.
SOD, CAT and GSH-Px, integral components of the oxidant scavenger
enzyme system, constitute the primary cellular defense against
oxidative damage and their activity reflects the ability of the
body to eliminate excess ROS (24). The findings of the present study
align with the aforementioned processes. As depicted in Figs. 7 and 8, H/R-treated H9c2 cells exhibited a
significant increase in MDA and ROS levels, along with decreased
SOD, CAT and GSH-Px activity. Conversely, pre-treatment with LIQ
yielded the opposite results.
Cardiomyocytes house numerous mitochondria,
comprising ~30% of their volume. These mitochondria play a pivotal
role in providing adenosine triphosphate to the cell through
glucose degradation and other pathways (26), rendering them essential for
maintaining cardiomyocyte function (27). Mitochondria are intricately
involved in the complex pathological mechanisms of H/R injury.
Significant factors include excessive ROS production, dysfunction
in the electron transport chain, dysregulation of calcium
homeostasis, abnormal opening of the mitochondrial permeability
transition pore (mPTP), depolarization of MtMP and inappropriate
activation of apoptosis (28).
During myocardial hypoxia, there is a gradual impairment of
mitochondrial electron transport system function and compromised
mitochondrial energy metabolism (29). Although energy metabolism can be
restored during reperfusion, early ROS accumulation can promote the
opening of the mPTP, leading to MtMP collapse and ultimately
triggering apoptosis (30). This
underscores the critical role of mitochondrial damage in the
progression of MIRI to heart failure (26). The present study used Rhodamine
123, a cationic fluorescent dye designed specifically to monitor
mitochondrial function in living cells (31). The findings indicated that LIQ
pre-treatment improved mitochondrial function, thus protecting H9c2
cells from H/R-induced damage.
Apoptosis is recognized as a significant
characteristic in the pathogenesis of MIRI (32) and a primary mode of cell death
(33) within the complex
pathophysiological processes involved. A previous study has
indicated that myocardial cells undergo apoptosis during hypoxia
and reperfusion exacerbates its severity (34). The current study demonstrated that
LIQ effectively reduced apoptosis levels, thus emphasizing its
anti-apoptotic effect. During H/R injury, a reciprocal causal
relationship exists between inflammation and apoptosis (35).
Numerous studies have highlighted the crucial
regulatory role of the inflammatory response in H/R injury. Hypoxic
necrotic cardiomyocytes trigger the transcriptional activity of
inflammatory factors, including TNF-α and IL-6, which further
exacerbates the inflammatory response (36,37).
The findings of the present study align with prior research,
demonstrating that H/R injury induced the release of TNF-α and IL-6
in cardiomyocytes, while LIQ pre-treatment significantly inhibited
the abnormal expression of TNF-α and IL-6. Moreover, the increase
in TNF-α activated xanthine oxidase, leading to ROS production and
upregulation of inflammatory factor expression, thereby
intensifying the inflammatory response. These processes not only
worsen myocardial MIRI but also contributed to structural
remodeling of the ventricles, ultimately resulting in the
deterioration of cardiac function (38).
The pharmacology section of the network analysis
produced a PPI network, revealing 95 common targets for LIQ and
MIRI. Among these targets, AKT1, CASP3, ALB, SRC and MMP9 were
identified as core predicted targets. KEGG analysis indicated a
close association between LIQ and the TNF signaling pathway, with
MMP9 identified as a core target within this pathway. Previous
research has highlighted TNF as an effective pro-inflammatory
cytokine that promotes and exacerbates inflammation through the
induction of apoptosis or necroptosis (39,40).
TNFR1 binds to TNF, activating the conventional NF-κB pathway
(39). NF-κB regulation in
cardiovascular diseases involves various pathological mechanisms,
including immunity and inflammation (41).
Furthermore, MMP9, a member of the matrix
metalloproteinase family, is implicated in the pathogenesis of
various cardiovascular conditions. Research has shown elevated
myocardial levels of MMP9 in both ischemic and non-ischemic
cardiomyopathy (42-44).
Alterations in NF-κB expression can also affect the regulation of
the MMP9 gene (45). As a result,
cellular experiments aimed at validating molecular mechanisms
primarily revolve around the TNFR1/NF-κB/MMP9 signaling
pathway.
TNFR1 is expressed in nearly all cell types,
including cardiomyocytes and plays a crucial role in both
pro-inflammatory and programmed cell death pathways (46). Therefore, TNFR1 was chosen as the
focal point of the present study. In line with this, the results
revealed an elevated expression of TNFR1 protein in
CoCl2-induced H/R injury in H9c2 cells.
Within the TNFR1 signaling pathway, the activation
of NF-κB is a pivotal event that induces the expression of
inflammatory genes and initiates an anti-apoptotic transcriptional
program to counteract the pro-apoptotic pathway (47). Under normal conditions, the
intracellular region of TNFR1 is inhibited by binding to the
silencer of the death domain (SODD). When TNFR1 binds to TNF-α,
SODD dissociates from TNFR1, enabling proteins such as TNF
Receptor-Associated Death Domain, Receptor Interacting Protein 1,
TNF Receptor-Associated Factor 2 and other proteins to assemble and
form complex I. In its inactive state, the p50/65 dimer of NF-κB is
localized in the cytoplasm and concealed by IκB, which binds to the
p50/65 dimer, forming a trimer. Activation of complex I leads to
the activation of the IκB kinase complex (Ikk), which
phosphorylates the serine residue at the N-terminal end of IκB.
Subsequently, the phosphorylated IκB protein undergoes
ubiquitination and degradation, thereby releasing the active p50/65
dimer. Consequently, NF-κB is liberated and translocates from the
cytoplasm to the nucleus (48-50),
where it acts as a transcription factor, binding to specific DNA
sequences and inducing the transcription and expression of relevant
genes (51,52). A Previous study has identified a
regulatory, structural domain of NF-κB in the 670 bp promoter
region of MMP-9(53). TNF-α
induces MMP9 expression in mammalian cells through the NF-κB
signaling pathway and activates the MMP9 promoter at varying TNF-α
concentrations (54). MMP9 has
been implicated in myocardial injury following AMI. During cardiac
remodeling following AMI, MMP9 participates in extracellular matrix
degradation, increases capillary permeability and activates
inflammatory mediators. Moreover, in animal models of
cardiovascular diseases, elevated myocardial MMP9 expression has
been observed and the deletion or inhibition of MMP9 has shown
beneficial effects (55). The
present study also demonstrated that H/R significantly increased
NF-κB phosphorylation and MMP9 expression and LIQ pre-treatment
effectively alleviated their upregulation.
While the present study showed the beneficial
effects of LIQ in mitigating H/R injury, it is important to
acknowledge certain limitations. First, although H9c2 cells share
morphological features and signaling pathway components with
primary cardiomyocytes (56,57),
they do not fully replicate the complex regulatory processes that
occur in the heart in vivo. Hence, further validation
through in vivo experiments is warranted. Additionally, the
number of targets validated in the present study was limited.
Further investigation is necessary to confirm the regulatory
effects of LIQ on other crucial targets involved in H/R injury.
Another limitation of the present study is the absence of
established positive controls, which were not included due to the
previously established cardiac protective effect of LIQ using such
controls (15). This aspect will
be further investigated in our future research.
The present study employed a combination of network
pharmacology and cellular experiments to illustrate that LIQ has
the potential to mitigate MIRI and provide protection to the heart.
It accomplished this by activating the TNFR1/NF-κB/MMP9 signaling
pathway, inhibiting oxidative stress, enhancing MtMP regulation,
managing inflammation and suppressing apoptosis. These findings
enhance our comprehension of the pharmacological mechanism of LIQ
and introduce a novel therapeutic approach for the clinical
treatment of AMI.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Research
Foundation of Natural Science Foundation of Hebei Province Youth
Project (grant no. H2021423027), Hebei Province high-level talent
funding project (grant no. C20231060) and The Hebei Natural Science
Foundation (grant no. H2022423306).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL, LB, SG, XH and HW were involved in the
conception and planning of the current study. HL, LB, XS and XC
performed the experiments. HL, XS and LB interpreted the data. HL,
XC, YX and JS were involved in the data analysis. XC, YX, JS and YL
provided guidance for software and figures. HL wrote the original
draft. LB, XS, XC, MZ, YL, XH, SG and HW reviewed and edited the
manuscript. XH, SG and HW supervised the project. SG identified
resources. HL and SG confirm the authenticity of all the raw data.
All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pollard TJ: The acute myocardial
infarction. Prim Care. 27:631–649. 2000.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Eltzschig HK, Bonney SK and Eckle T:
Attenuating myocardial ischemia by targeting A2B adenosine
receptors. Trends Mol Med. 19:345–354. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yang H, Wang C, Zhang L, Lv J and Ni H:
Rutin alleviates hypoxia/reoxygenation-induced injury in myocardial
cells by up-regulating SIRT1 expression. Chem Biol Interact.
297:44–49. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv
F, Liu Y, Zheng W, Shang H, Zhang J, et al: CaMKII is a RIP3
substrate mediating ischemia- and oxidative stress-induced
myocardial necroptosis. Nat Med. 22:175–182. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Du L, Shen T, Liu B, Zhang Y, Zhao C, Jia
N, Wang Q and He Q: Shock Wave Therapy Promotes Cardiomyocyte
Autophagy and Survival during Hypoxia. Cell Physiol Biochem.
42:673–684. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gao X, Zhang H, Zhuang W, Yuan G, Sun T,
Jiang X, Zhou Z, Yuan H, Zhang Z and Dong H: PEDF and PEDF-derived
peptide 44mer protect cardiomyocytes against hypoxia-induced
apoptosis and necroptosis via anti-oxidative effect. Sci Rep.
4(5637)2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yao H, Xie Q, He Q, Zeng L, Long J, Gong
Y, Li X, Li X, Liu W, Xu Z, et al: Pretreatment with panaxatriol
saponin attenuates mitochondrial apoptosis and oxidative stress to
facilitate treatment of myocardial ischemia-reperfusion injury via
the regulation of Keap1/Nrf2 activity. Oxid Med Cell Longev.
2022(9626703)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pastorino G, Cornara L, Soares S,
Rodrigues F and Oliveira M: Liquorice (Glycyrrhiza glabra): A
phytochemical and pharmacological review. Phytother Res.
32:2323–2339. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Fan S, Gu K, Wu Y, Luo H, Wang Y, Zhang T,
Wang X, Zhang Y and Li Y: Liquiritinapioside-A
mineralocorticoid-like substance from liquorice. Food Chem.
289:419–425. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hou Y and Jiang JG: Origin and concept of
medicine food homology and its application in modern functional
foods. Food Funct. 4:1727–1741. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang Y, Zhang L, Zhang Y, Xu JJ, Sun LL
and Li SZ: The protective role of liquiritin in high
fructose-induced myocardial fibrosis via inhibiting NF-κB and MAPK
signaling pathway. Biomed Pharmacother. 84:1337–1349.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Mou SQ, Zhou ZY, Feng H, Zhang N, Lin Z,
Aiyasiding X, Li WJ, Ding W, Liao HH, Bian ZY and Tang QZ:
Liquiritin attenuates lipopolysaccharides-induced cardiomyocyte
injury via an AMP-Activated protein kinase-dependent signaling
pathway. Front Pharmacol. 12(648688)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Thu VT, Yen NTH and Ly NTH: Liquiritin
from radix glycyrrhizae protects cardiac mitochondria from
Hypoxia/Reoxygenation damage. J Anal Methods Chem.
2021(1857464)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Han X, Yang Y, Zhang M, Li L, Xue Y, Jia
Q, Wang X and Guan S: Liquiritin Protects against cardiac fibrosis
after myocardial infarction by inhibiting CCL5 expression and the
NF-kappaB signaling pathway. Drug Des Devel Ther. 16:4111–4125.
2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Boezio B, Audouze K, Ducrot P and
Taboureau O: Network-based approaches in pharmacology. Mol Inform:
36, 2017 doi: 10.1002/minf.201700048.
|
|
17
|
Otasek D, Morris JH, Bouças J, Pico AR and
Demchak B: Cytoscape Automation: Empowering workflow-based network
analysis. Genome biology. 20(185)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hu X, Jiao R, Li H, Wang X, Lyu H, Gao X,
Xu F, Li Z, Hua H and Li D: Antiproliferative hydrogen sulfide
releasing evodiamine derivatives and their apoptosis inducing
properties. Eur J Med Chem. 151:376–388. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Fang Z, Luo W and Luo Y: Protective effect
of α-mangostin against CoCl2-induced apoptosis by
suppressing oxidative stress in H9c2 rat cardiomyoblasts. Mol Med
Rep. 17:6697–6704. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Munoz-Sanchez J and Chanez-Cardenas ME:
The use of cobalt chloride as a chemical hypoxia model. J Appl
Toxicol. 39:556–570. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Amani M, Jeddi S, Ahmadiasl N, Usefzade N
and Zaman J: Effect of HEMADO on level of CK-MB and LDH enzymes
after ischemia/reperfusion injury in isolated rat heart.
Bioimpacts. 3:101–104. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Raedschelders K, Ansley DM and Chen DD:
The cellular and molecular origin of reactive oxygen species
generation during myocardial ischemia and reperfusion. Pharmacol
Ther. 133:230–255. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Farias JG, Molina VM, Carrasco RA, Zepeda
AB, Figueroa E, Letelier P and Castillo RL: Antioxidant therapeutic
strategies for cardiovascular conditions associated with oxidative
stress. Nutrients. 9(996)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Rodrigo R: Prevention of postoperative
atrial fibrillation: Novel and safe strategy based on the
modulation of the antioxidant system. Front Physiol.
3(93)2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Xiang M, Lu Y, Xin L, Gao J, Shang C,
Jiang Z, Lin H, Fang X, Qu Y, Wang Y, et al: Role of oxidative
stress in reperfusion following myocardial ischemia and its
treatments. Oxid Med Cell Longev. 2021(6614009)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Marin W, Marin D, Ao X and Liu Y:
Mitochondria as a therapeutic target for cardiac
ischemia-reperfusion injury (Review). Int J Mol Med. 47:485–499.
2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Carreira RS, Lee P and Gottlieb RA:
Mitochondrial therapeutics for cardioprotection. Curr Pharm Des.
17:2017–2035. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Su X, Zhou M, Li Y, An N, Yang F, Zhang G,
Xu L, Chen H, Wu H and Xing Y: Mitochondrial damage in myocardial
Ischemia/Reperfusion injury and application of natural plant
products. Oxid Med Cell Longev. 2022(8726564)2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lesnefsky EJ, Chen Q, Tandler B and Hoppel
CL: Mitochondrial dysfunction and myocardial Ischemia-Reperfusion:
Implications for novel therapies. Annu Rev Pharmacol Toxicol.
57:535–565. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Anzell AR, Maizy R, Przyklenk K and
Sanderson TH: Mitochondrial quality control and disease: Insights
into Ischemia-Reperfusion injury. Mol Neurobiol. 55:2547–2564.
2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chazotte B: Labeling mitochondria with
rhodamine 123. Cold Spring Harb Protoc. 2011:892–894.
2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ and
Chen Y: Protective role of melatonin in cardiac
ischemia-reperfusion injury: From pathogenesis to targeted therapy.
J Pineal Res: 64, 2018 doi: 10.1111/jpi.12471.
|
|
33
|
Yang J, Guo X, Yang J, Ding JW, Li S, Yang
R, Fan ZX and Yang CJ: RP105 Protects against apoptosis in
Ischemia/Reperfusion-Induced myocardial damage in rats by
suppressing TLR4-Mediated signaling pathways. Cell Physiol Biochem.
36:2137–2148. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li X, Hu X, Wang J, Xu W, Yi C, Ma R and
Jiang H: Short-Term hesperidin pretreatment attenuates rat
myocardial Ischemia/Reperfusion injury by inhibiting high mobility
group box 1 protein expression via the PI3K/Akt Pathway. Cell
Physiol Biochem. 39:1850–1862. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang BF, Jiang H, Chen J, Guo X, Li Y, Hu
Q and Yang S: Nobiletin ameliorates myocardial ischemia and
reperfusion injury by attenuating endoplasmic reticulum
stress-associated apoptosis through regulation of the PI3K/AKT
signal pathway. Int Immunopharmacol. 73:98–107. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Yang Y, Lv J, Jiang S, Ma Z, Wang D, Hu W,
Deng C, Fan C, Di S, Sun Y and Yi W: The emerging role of Toll-like
receptor 4 in myocardial inflammation. Cell Death Dis.
7(e2234)2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Naidu BV, Farivar AS, Woolley SM, Grainger
D, Verrier ED and Mulligan MS: Novel broad-spectrum chemokine
inhibitor protects against lung ischemia-reperfusion injury. J
Heart Lung Transplant. 23:128–134. 2004.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Chen L, Liu P, Feng X and Ma C:
Salidroside suppressing LPS-induced myocardial injury by inhibiting
ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J Cell Mol
Med. 21:3178–3189. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ting AT and Bertrand MJM: More to Life
than NF-κB in TNFR1 Signaling. Trends Immunol. 37:535–545.
2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Van der Heiden K, Cuhlmann S, Luong le A,
Zakkar M and Evans PC: Role of nuclear factor kappaB in
cardiovascular health and disease. Clin Sci (Lond). 118:593–605.
2010.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Newby AC: Dual role of matrix
metalloproteinases (matrixins) in intimal thickening and
atherosclerotic plaque rupture. Physiol Rev. 85:1–31.
2005.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Spinale FG, Coker ML, Heung LJ, Bond BR,
Gunasinghe HR, Etoh T, Goldberg AT, Zellner JL and Crumbley AJ: A
matrix metalloproteinase induction/activation system exists in the
human left ventricular myocardium and is upregulated in heart
failure. Circulation. 102:1944–1949. 2000.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Spinale FG: Matrix metalloproteinase gene
polymorphisms in heart failure: New pieces to the myocardial matrix
puzzle. Eur Heart J. 25:631–633. 2004.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Luo X, Wu J and Wu G: PPARγ activation
suppresses the expression of MMP9 by downregulating NF-κB post
intracerebral hemorrhage. Neurosci Lett. 752(135770)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kleinbongard P, Heusch G and Schulz R:
TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and
heart failure. Pharmacol Ther. 127:295–314. 2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hayden MS and Ghosh S: Regulation of NF-κB
by TNF family cytokines. Semin Immunol. 26:253–266. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Hayden MS and Ghosh S: NF-κB, the first
quarter-century: Remarkable progress and outstanding questions.
Genes Dev. 26:203–234. 2012.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Blackwell K, Zhang L, Thomas GS, Sun S,
Nakano H and Habelhah H: TRAF2 phosphorylation modulates tumor
necrosis factor alpha-induced gene expression and cell resistance
to apoptosis. Mol Cell Biol. 29:303–314. 2009.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Devin A, Cook A, Lin Y, Rodriguez Y,
Kelliher M and Liu Z: The distinct roles of TRAF2 and RIP in IKK
activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP
mediates IKK activation. Immunity. 12:419–429. 2000.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wei Q, Tu Y, Zuo L, Zhao J, Chang Z, Zou Y
and Qiu J: MiR-345-3p attenuates apoptosis and inflammation caused
by oxidized low-density lipoprotein by targeting TRAF6 via
TAK1/p38/NF-kB signaling in endothelial cells. Life Sci.
241(117142)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Howell JA and Bidwell GL III: Targeting
the NF-κB pathway for therapy of ischemic stroke. Ther Deliv.
11:113–123. 2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Gum R, Lengyel E, Juarez J, Chen JH, Sato
H, Seiki M and Boyd D: Stimulation of 92-kDa gelatinase B promoter
activity by ras is mitogen-activated protein kinase kinase
1-independent and requires multiple transcription factor binding
sites including closely spaced PEA3/ets and AP-1 sequences. J Biol
Chem. 271:10672–10680. 1996.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Wu HT, Sie SS, Kuan TC and Lin CS:
Identifying the regulative role of NF-κB binding sites within
promoter region of human matrix metalloproteinase 9 (mmp-9) by
TNF-α induction. Appl Biochem Biotechnol. 169:438–449.
2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/Reperfusion Injury following Acute
Myocardial Infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017(7018393)2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Hescheler J, Meyer R, Plant S, Krautwurst
D, Rosenthal W and Schultz G: Morphological, biochemical, and
electrophysiological characterization of a clonal cell (H9c2) line
from rat heart. Circ Res. 69:1476–1486. 1991.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Zhu A, Wei X, Zhang Y, You T, Yao S, Yuan
S, Xu H, Li F and Mao W: Propofol provides cardiac protection by
suppressing the proteasome degradation of caveolin-3 in
ischemic/reperfused rat hearts. J Cardiovasc Pharmacol. 69:170–177.
2017.PubMed/NCBI View Article : Google Scholar
|