Introduction
TAM receptors are a family of receptor tyrosine
kinases (RTKs) consisting of TYRO3, AXL, and MERTK that are
involved in cell survival, innate immunity, and immunosuppression
(1-6).
They are overexpressed in various types of tumor cells and immune
cells and activate several downstream signaling pathways such as
phosphatidylinositol 3-kinase, extracellular signal-regulated
kinase, and nuclear factor kappa-light-chain-enhancer of activated
B cells pathways. These downstream signaling pathways play a role
in promoting the growth and metastasis of tumor cells. Therefore,
activation of TAM receptors has shown the correlations with tumor
progression and poor prognosis in many cancers and is also
recognized as a mediator of acquired resistance to chemotherapy and
targeted therapy (1,2,7).
With regard to its immunomodulatory role, previous studies using a
syngeneic breast cancer model showed that conditional knockout of
MERTK caused an increase in tumor-infiltrating lymphocytes and
inflammatory cytokines, showing that antitumor immunity could be
promoted. Numerous studies suggest that genetic modification or
pharmacologic inhibition of TAM receptors can act as therapeutic
targets to prevent tumor progression (8-12).
Several TAM tyrosine kinase inhibitors have been
developed, and some are undergoing clinical trials for cancer
treatment. The first-in-class AXL inhibitor R428, is being actively
investigated in several cancers including acute myeloid leukemia,
non-small cell lung cancer, and melanoma (13). Another MERTK inhibitor, UNC2025,
has a 40-fold higher inhibitory activity on MERTK than AXL. Mice
treated with UNC2025 show a significant reduction in tumor size and
increased survival compared with vehicle-treated mice in leukemia
models (14,15). However, other studies have
indicated that inhibition of a single TAM receptor can result in
drug resistance by inducing intrinsic and adaptive compensatory
mechanisms by activating other subunits. R428 showed an increase in
the expression of MERTK protein both in vitro and in
vivo in proportion to AXL inhibition, thereby reducing the
efficacy of the drug. In addition, the combined targeting of MERTK
and AXL showed a synergistic antitumor effect. Based on these
results, it has been hypothesized that targeting multiple members
of the TAM family would be a more effective cancer treatment than
single-target agents (16,17).
Pan-TAM inhibitors, such as BMS-777607 and
INCB081776, are under development for the treatment of cancer, but
it will take some time to achieve Food and Drug Administration
(FDA) approval. Despite the development of high-throughput
screening technologies, both a considerable cost and a long
development time are required for a drug to be approved and
released to the market. Above all, a significant number of novel
drug candidates have failed in preclinical or clinical trials for
various reasons, such as poor pharmacokinetics, unexplained
toxicity, and lack of efficacy. Therefore, drug repurposing, which
repurposes existing drugs into new treatments for intractable
diseases, is being proposed as one of the methods to overcome the
difficulties of drug development from novel compounds. Another
advantage of drug repurposing is that existing drugs have a proven
safety profile, so there are relatively few concerns about safety
issues. Recently, there have been many attempts in the drug
repurposing area that are expected to increase the success rate of
drug development using advanced in silico strategies
(18,19). Virtual screening using machine
learning and artificial intelligence (AI) provides insights into
finding new anticancer drug candidates from a large-scale
drug–target interaction (DTI) dataset, including BindingDB
(20), PubChem (21), and ChEMBL (22). Our group has developed a new deep
DTI prediction model called Molecule Transformer (MT)-DTI (23). In 2020, using MT-DTI, we predicted
that atazanavir and remdesivir were drugs that could inhibit
SARS-CoV-2(24). We then searched
for a drug among commercially available medications that could
simultaneously target AXL, MERTK, and TYRO3, using the MT-DTI
algorithm. As a result, fostamatinib, one of the spleen tyrosine
kinase (Syk) inhibitors, was predicted to inhibit TAM family
members effectively, and we further verified this result using
several cancer cell lines. Fostamatinib has been approved as a drug
for treating chronic immune thrombocytopenia (ITP) (25), but its inhibitory effect on TAM
kinases has not been reported. Here, we demonstrate that the
inhibitory effect on the TAM family is a unique function of
fostamatinib that is not observed with other Syk inhibitors. Our
results suggest not only the possibility of expanding the use of
fostamatinib by revealing a new function but also suggest the
possibility of identifying new drug mechanisms through AI.
Materials and methods
Prediction of bioactivity and binding
affinity for TAM kinases
The Deargen MT-DTI was used for the chemical
prediction to target triple TAM kinases simultaneously (23). Based on a manually curated public
database, the model was trained on bioactivity data
(IC50/EC50) and affinity data (Kd)
from the ChEMBL database (22),
Drug Target Common database (26),
and BindingDB database (27).
MT-DTI uses a GCN (28) algorithm
to extract molecular features and a ProtBert (29) algorithm to extract protein
features. With these features, our model was trained to predict
each parameter (IC50, EC50, and Kd)
simultaneously as multitask learning (30). It allowed prediction of the
IC50, EC50, and Kd values for one
drug-protein pair.
Connectivity map analysis
The Connectivity Map (CMap) CLUE web application
(https://clue.io) was used to assess the connectivity
of gene expression signatures between chemical and genetic
perturbations (31). The
Touchstone app of the CLUE database provided connectivity scores
ranging from -100 to +100, where positive scores corresponded to
the positive correlation of gene signatures between the two
perturbagens. Signatures with connectivity scores >40 (similar)
were further investigated in this study.
Cell culture and reagents
NCI-H1299 (H1299; human non-small cell lung cancer
cells), MDA-MB-231 (MB231; human breast adenocarcinoma cells), SCC4
(human tongue squamous cell carcinoma cells), and CAL27 (human
tongue squamous cell carcinoma cells) cells were obtained from the
Korean Cell Line Bank (Seoul, Korea) and American Type Culture
Collection (Manassas, VA, USA). H1299 and MB231 cells were
maintained in RPMI 1640 (HyClone, Logan, UT, USA), and SCC4 cells
were maintained in DMEM: F12 medium (Gibco, Waltham, MA, USA)
containing 10% fetal bovine serum (Gibco) and 1% antibiotics at
37˚C in the presence of 5% CO2. Fostamatinib (#S2625),
BMS-777607 (#S1561), TAK-659 (#S8442), and MG-132 (#M7449) were
purchased from Selleckchem (Houston, TX, USA).
Cell viability assay
Cell viability was evaluated using a CellTiter
96® AQueous One Solution Cell Proliferation Assay
(Promega, San Luis Obispo, CA, USA). Cells were seeded at
3x103 cells/well in 96-well plates and then exposed to
fostamatinib, BMS-777607, or TAK-659 for 48 h. Assays were
performed by adding CellTiter 96® AQueous One Solution
Reagent directly to the wells, incubating them for 2 h, and then
measuring absorbance at 490 nm with a 96-well plate reader.
Western blotting and densitometric
analysis
Cells were lysed in RIPA buffer (Elpis Biotech,
Daejeon, Korea; #EBA-1149). Cell lysates were separated using
SDS-PAGE, transferred onto PVDF membranes, and then incubated with
primary antibodies followed by HRP-conjugated second antibodies
(Genetex, Irvine, CA, USA). Chemiluminescent signals were
visualized using the NEW Clarity ECL substrate (GE Healthcare,
Chicago, IL, USA). Antibodies for phospho-AXL (#5724), AXL (#8661),
and TYRO3 (#5585) were purchased from Cell Signaling Technology
(Beverly, MA, USA). Antibodies for phospho-MERTK (#ab14921), MERTK
(#ab52968), phospho-Syk (#ab62338), Syk (#ab3993), and α-tubulin
(#ab4074) were purchased from Abcam (Cambridge, MA, USA). Western
blot quantification was performed using ImageJ software (version
1.51, National Institutes of Health, Bethesda, MD, USA). The
intensity of each protein band was quantified by measuring the
integrated density value using ImageJ software. Background
intensity was subtracted from each band intensity. The relative
protein expression was calculated by normalizing the target protein
intensity to that of α-tubulin from the same sample. Data from
three independent experiments were analyzed and presented as mean ±
standard deviation (SD).
Statistical analysis
All experiments were performed independently at
least three times. Results are presented as the mean ± SD.
Statistical significance was calculated by unpaired Student's t
test using GraphPad Prism (La Jolla, CA, USA).
Results
MT-DTI predicts commercially available
TAM kinase triple inhibitor candidates
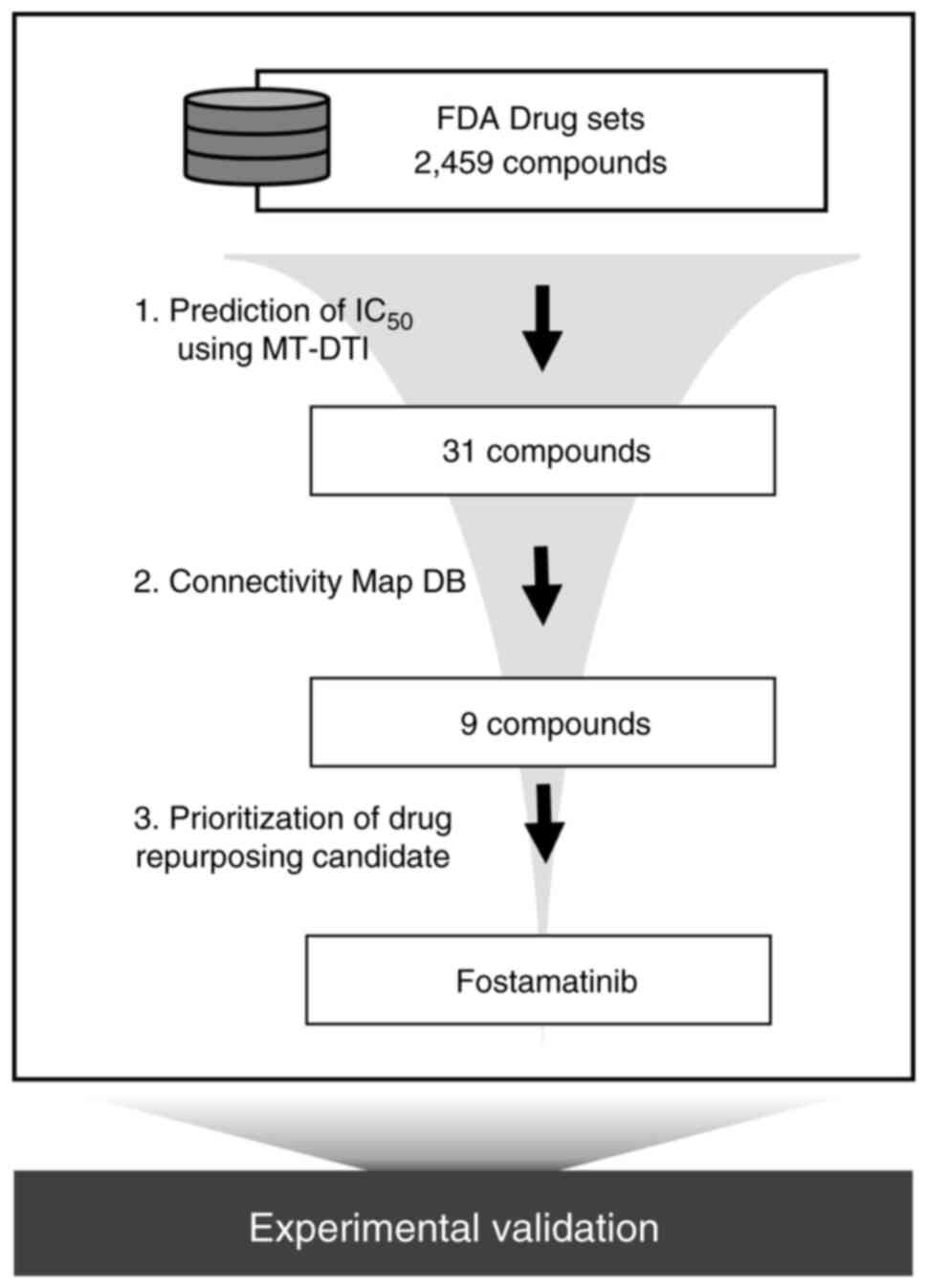
To find an FDA-approved drug that can simultaneously
target triple TAM kinases, we performed in silico screening
using a deep learning-based model (23). By comparing the values predicted
through MT-DTI (predicted IC50 and predicted
EC50), drugs with an inhibitory effect twice that of the
activating effect were selected. Thirty-one drugs with an
IC50 of less than 1 µM in AXL, MERTK, and TYRO3 remained
out of 2,459 FDA drug sets (Table
SI). Of these 31 compounds, 21 were kinase inhibitors,
including multi-kinase inhibitors (e.g. ponatinib, cabozantinib,
lenvatinib), epidermal growth factor receptor (EGFR) inhibitors
(e.g. neratinib, osimertinib, afatinib), and MEK inhibitors (e.g.
selumetinib, trametinib, binimetinib, cobimetinib). In addition to
kinases, 10 drugs included androgen receptor antagonists (e.g.
apalutamide, enzalutamide), an antihypertensive (e.g. sitaxentan),
and antibiotics (e.g. ceforanide, ozenoxacin). To explore the
effects of each drug on TAM signaling, we analyzed the CMap
resource. The CMap database, developed by the Broad Institute, is a
collection of genome-wide gene expression data encompassing 1.5
million gene expression profiles from ~5,000 drug perturbagens and
~3,000 genetic perturbagens (32).
CLUE (https://clue.io/) is a cloud-based software
platform that provides a connectivity score between a query and a
perturbagen and allows users to access publicly available CMap data
(33). We compared the gene
signatures induced by 31 drug treatments with the gene signatures
by knock-down of AXL, MERTK, and TYRO3 using the Touchstone app in
the CLUE platform, which shows connectivity between perturbagens.
Gene signature scores for nine drugs among 31 compounds showed
>40 connectivity with gene knock-down signature scores (Table I). This suggests that the nine
drugs predicted by MT-DTI may act as potential inhibitors of TAM
kinases. According to the FDA labeling information, afatinib,
neratinib, axitinib, and bosutinib have already been approved for
the treatment of various types of tumors, suggesting there is a
limit to the concept of expansion of the drug indication through
repurposing. In addition, ruxolitinib resulted in an additive
effect by combined treatment with the AXL inhibitors, TP-0903 and
bemcentinib (34,35). Because ceforanide and zafirlukast
do not have a kinase inhibitor structure, the possibility of direct
action on the TAM family is relatively low. By contrast, because
fostamatinib, a Syk inhibitor, is already used as a treatment for
ITP, and there are no reports of the effect of fostamatinib on the
TAM family, we sought to verify the effect of fostamatinib on TAM
signaling. The workflow of the current study is illustrated in
Fig. 1.
 | Table IList of drugs with connectivity
scores >40 after knock-down of TAM kinases. |
Table I
List of drugs with connectivity
scores >40 after knock-down of TAM kinases.
| | DTI scores
(predicted IC50, nM) | Connectivity
scores | |
|---|
| Drug | AXL | MERTK | TYRO3 | AXL | MERTK | TYRO3 | MoA | FDA-labeled
indication |
|---|
| Afatinib | 619.3 | 700.7 | 601.5 | 68.6 | - | 94.0 | EGFR inhibitor | Untreated,
metastatic non-small cell lung cancer |
| Neratinib | 156.3 | 349.1 | 158.8 | 67.6 | 45.6 | - | EGFR inhibitor |
HER2-overexpressed/amplified breast
cancer |
| Axitinib | 127.5 | 98.0 | 184.9 | 60.0 | 68.7 | 45.7 | VEGFR
inhibitor | Advanced renal cell
carcinoma |
| Ruxolitinib | 740.3 | 114.9 | 498.8 | 43.6 | 73.6 | 60.8 | JAK inhibitor | Intermediate or
high-risk myelofibrosis, including primary myelofibrosis |
| Bosutinib | 123.6 | 126.4 | 73.9 | 40.7 | 51.9 | 51.9 | Src/Abl
inhibitor | Chronic myelogenous
leukemia |
| Ceforanide | 358.0 | 553.9 | 516.9 | - | 51.9 | - | Antibacterial | Antibacterial |
| Zafirlukast | 643.8 | 623.7 | 748.4 | - | 47.9 | - | Leukotriene D4
receptor antagonist | Asthma |
| Selumetinib | 191.3 | 336.3 | 186.9 | - | - | 46.6 | MEK inhibitor | Neurofibromatosis
type 1 |
| Fostamatinib | 754.4 | 867.8 | 978.4 | - | - | 40.5 | Syk inhibitor | Immune
thrombocytopenia |
Fostamatinib suppresses cell
proliferation via inhibition of TAM kinases
The TAM family (AXL, MERTK, and TYRO3) are known to
play an important role in the development of various cancers. Based
on the results using MT-DTI, fostamatinib was predicted to be an
inhibitor of the TAM family. To determine the effect of the TAM
family on solid cancers, experiments were conducted using the
following cancer cell lines: H1299 cells (lung cancer); SCC4 cells
(head and neck squamous cell carcinoma); and MB231 cells (breast
cancer). We first evaluated the impact of BMS-777607, a known
pan-TAM inhibitor, on cell viability and TAM protein expression in
H1299, SCC4, and MB231 cells. BMS-777607 treatment resulted in a
dose-dependent decrease in cell viability across all three cell
lines (Fig. 2A). Western blot
analysis revealed that BMS-777607 treatment led to reduced
phosphorylation of both AXL and MERTK, indicating successful
inhibition of these kinases (Fig.
2B). While we observed a decrease in total TYRO3 protein
levels, we were unable to directly assess TYRO3 phosphorylation
status due to technical limitations of available antibodies. This
limitation stems from the high sequence homology between TYRO3
phosphorylation sites (Y681, Y685) and MERTK phosphorylation sites
(Y749, Y753), which causes commercially available phospho-specific
antibodies to cross-react with phosphorylated MERTK. Despite this
technical constraint, the reduction in cell viability and clear
effects on AXL and MERTK signaling demonstrate the effectiveness of
BMS-777607 as a TAM family inhibitor. Based on these results, the
same experiment was conducted using the cell lines listed above to
confirm the role of fostamatinib in solid cancers (Fig. 2C and D). Cell viability was decreased on
addition of increasing concentrations of fostamatinib, although
there was a difference in degree. Both the phosphorylated and total
forms of AXL and MERTK were decreased following fostamatinib
treatment. These results suggest that fostamatinib reduces cell
viability through the inhibition of TAM family proteins.
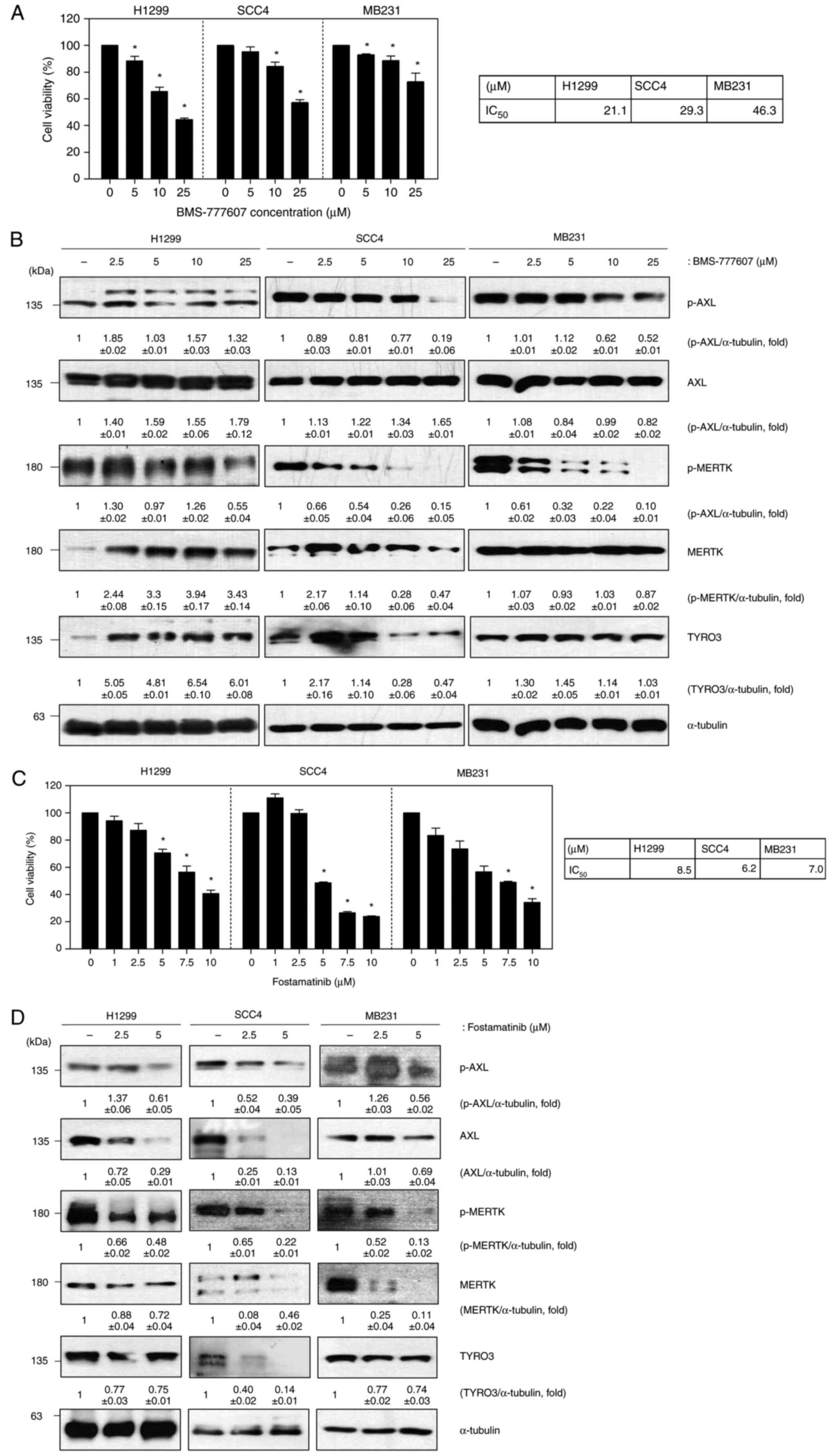 | Figure 2Fostamatinib suppresses cell
proliferation via inhibition of TAM kinases. (A) Cell viability
assay of H1299, SCC4 and MB231 cells after BMS-777607 treatment for
48 h; (B) Western blot of p-AXL, AXL, p-MERTK, MERTK and TYRO3 in
BMS-777607-treated cells (α-tubulin was used as a loading control);
(C) Cell viability assay of H1299, SCC4 and MB231 cells after
fostamatinib treatment for 48 h; (D) Western blot of p-AXL, AXL,
p-MERTK, MERTK and TYRO3 in fostamatinib-treated cells (α-tubulin
was used as a loading control). Data are representative of three
independent experiments and are presented as the mean ± SD. The
statistical significance was analyzed via Student's t-test;
*P<0.05 vs. untreated group. TAM, TYRO3, AXL, MERTK;
p-, phosphorylated. |
Other Syk inhibitors do not affect TAM
signaling
Fostamatinib was previously known as a Syk
inhibitor; therefore, we investigated whether other Syk inhibitors
had the same effect on TAM signaling. TAK-659 is a potent and
selective inhibitor of Syk. H1299, SCC4, and MB231 cells were
assessed for cell viability and TAM family protein levels after
TAK-659 treatment. H1299 and SCC4 cells did not show a change in
cell viability on exposure to TAK-659, but MB231 cell viability was
decreased slightly in a dose-dependent manner (Fig. 3A). Unlike the results after
fostamatinib treatment, TAM family protein expression levels did
not change after TAK-659 treatment in any of the cell lines tested
(Fig. 3B). Basal levels of Syk
protein expression were undetectable in H1299, SCC4, and MB231
cells, so we confirmed the effect of TAK-659 on TAM signaling using
CAL27 cells, a cell line with high Syk expression. TAK-659
significantly reduced the Syk protein levels in CAL27 cells,
whereas it did not affect the activity of AXL, MERTK, and TYRO3
(Fig. S1). These results suggest
that the effect of fostamatinib on TAM signaling is a unique
feature not found in other Syk inhibitors.
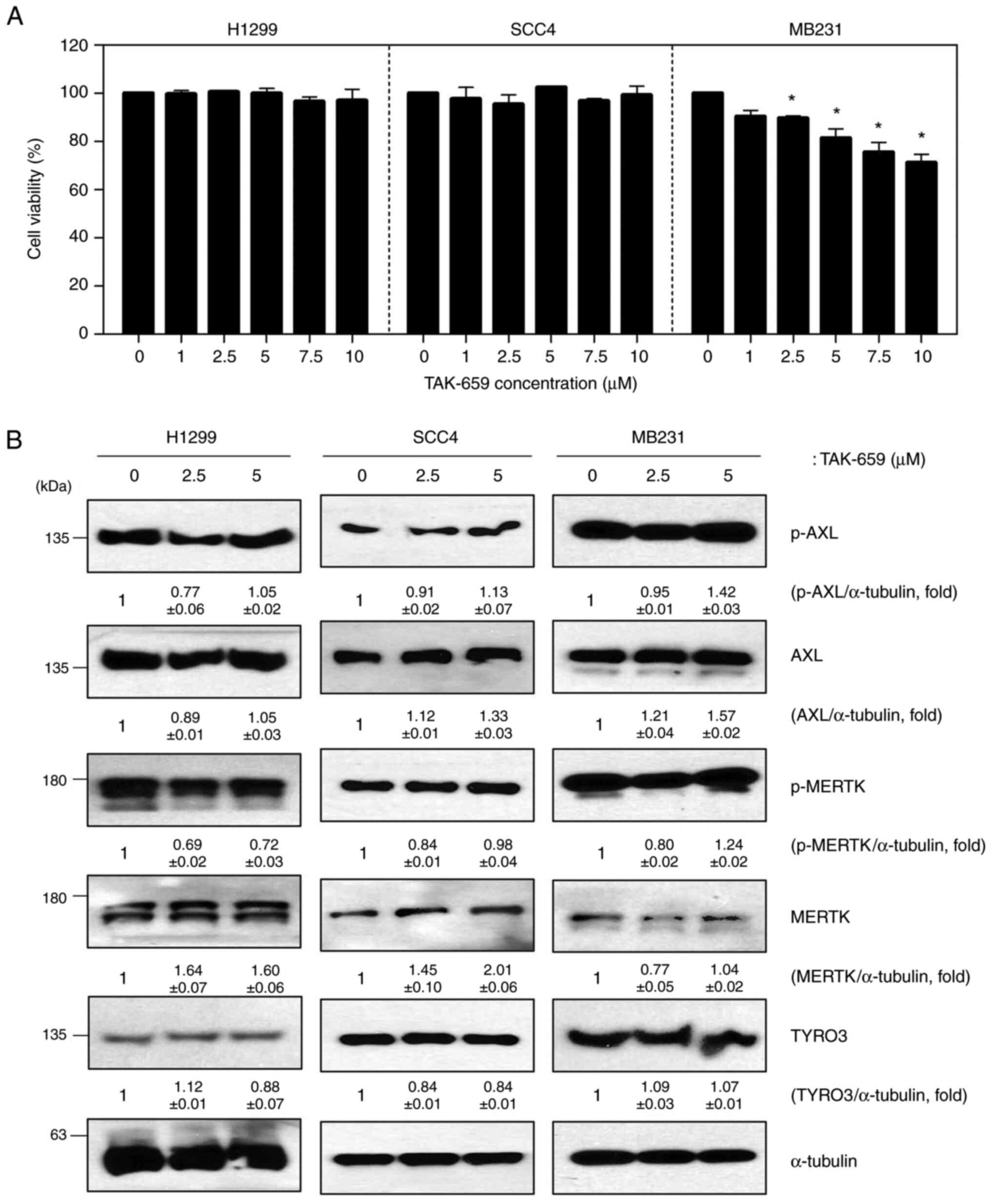 | Figure 3TAK-659, a Syk inhibitor, does not
affect TAM signaling. (A) Cell viability assay of H1299, SCC4 and
MB231 cells after TAK-659 treatment for 48 h; (B) Western blot of
p-AXL, AXL, p-MERTK, MERTK, and TYRO3 in TAK-659-treated cells
(α-tubulin was used as a loading control). Data are representative
of three independent experiments and presented as the mean ± SD.
The statistical significance was analyzed via Student's t-test.
*P<0.05 vs. untreated group. TAM, TYRO3, AXL, MERTK;
p-, phosphorylated; Syk, spleen tyrosine kinase. |
Discussion
Syk is a non-RTK and is known to play a crucial role
in autoimmune diseases and hematological malignancies (1). Syk mediates diverse biological
functions, including innate immune recognition, platelet
activation, cellular adhesion, osteoclast maturation, and vascular
development (1,2). Fostamatinib is a prodrug that is
converted to the active metabolite R406, which is a specific
inhibitor of Syk-dependent Fcγ receptors-mediated signaling in a
wide variety of tissues (36). In
phase III randomized double-blind placebo-controlled FIT1 and FIT2
trials, the efficacy and safety of fostamatinib were demonstrated
in patients with chronic ITP (37). Most adverse events were mild to
moderate, and manageable. Based on these results, in 2018, the FDA
approved fostamatinib for the treatment of chronic ITP in adults
who had insufficient response to prior therapy (36,37).
Although the clinical activity of fostamatinib has been reported in
some hematological and solid cancers (8,9), the
mechanism of action of fostamatinib, other than as a Syk kinase
inhibitor, has not yet been described, and it has not been approved
as an anticancer treatment. In this study, we discovered a novel
function of fostamatinib by employing a drug repositioning approach
via a machine learning algorithm.
An AI-driven drug repurposing approach, such as
MT-DTI, and computational pharmacogenomics using large-scale
perturbation databases, provide effective drug discovery and drug
repurposing/repositioning methodologies. As a result, we predicted
fostamatinib to be a drug that can inhibit TAM RTKs. The inhibitory
effect of fostamatinib on TAM kinases was verified with three
different types of cancer cell lines: lung cancer (H1299), head and
neck cancer (SCC4), and breast cancer (MB231). Fostamatinib
significantly decreased the expression levels of AXL, MERTK, and
TYRO3 equally in all three different types of cancer cell lines,
and reduced cell viability in a dose-dependent manner. For each
cell line, the IC50 values of fostamatinib were between
5 and 10 µM, and the cytotoxic effect of fostamatinib was superior
to that of the pan-TAM kinase inhibitor, BMS-777607. Regan-Fendt
et al (38) investigated
the possibility of dasatinib and fostamatinib as a treatment for
hepatocellular carcinoma using a transcriptomics-based drug
repurposing method. They showed that fostamatinib was able to
inhibit the growth of hepatocellular carcinoma cells with
IC50 values between 20 and 35 µM (38). In the experiment using another
well-known Syk inhibitor, the inhibitory effect on TAM kinases was
not shown, indicating it is a unique characteristic of
fostamatinib. Regarding the mechanism of action by which
fostamatinib inhibits TAM receptors, we investigated the potential
involvement of proteasomal degradation pathway. Our preliminary
experiment with the proteasome inhibitor, MG132, showed that MG132
pretreatment could attenuate the fostamatinib-induced decrease in
both phosphorylated and total protein levels of AXL and MERTK (data
not shown). While these initial findings suggested that
fostamatinib might promote TAM receptor degradation through a
proteasome-dependent mechanism, we faced significant technical
challenges in fully validating this hypothesis. Specifically, when
we attempted to optimize the MG132 and fostamatinib co-treatment
conditions using H1299 cells, the combination showed considerable
cellular toxicity that prevented us from obtaining reliable protein
expression data, particularly for TYRO3 blots. This technical
limitation, combined with the complex nature of receptor tyrosine
kinase regulation, suggests that additional studies using
alternative approaches would be needed to fully elucidate the
precise mechanism by which fostamatinib regulates TAM receptor
stability and function. Further investigation into the role of
fostamatinib as a potential molecular glue degrader (MGD) might
provide additional insights into its mechanism of action, as MGDs
destabilize target proteins by bridging them near E3 ubiquitin
ligases leading to polyubiquitylation and proteasomal degradation
of the target protein.
The TAM family of RTKs, including AXL, MERTK, and
TYRO3, is expressed in hematopoietic-derived cells that play
important roles in efferocytosis, and in the balancing of immune
responses and inflammation (10).
TAM kinases are also overexpressed in a variety of cancers that are
associated with chemoresistance, metastasis, and poor survival
outcomes (10,11). As well as the promotion of tumor
cell survival, proliferation, and invasion through activation of
downstream oncogenic signaling pathways, TAM receptors contribute
to tumor progression through an immunosuppressive tumor
microenvironment and tumor immune escape (12-14).
The key roles of TAM kinases as both oncogenic drivers and immune
system regulators have led to a growing interest in TAM receptor
inhibition, especially in the era of cancer immunotherapy. Of note,
the TAM receptors, in particular MERTK, have been shown to
upregulate the immune checkpoint molecule programmed cell death
ligand 1 (PD-L1) in tumor cells (39). These data suggest that TAM tyrosine
kinase inhibitors, in combination with immune checkpoint inhibitors
(ICIs), could enhance treatment efficacy. In support of this
hypothesis, Kasikara et al (40) reported that combined administration
of an ICI with the pan-TAM tyrosine kinase inhibitor BMS-777607
enhanced tumor-infiltrating lymphocytes and T-cell-mediated
immunity, with improved antitumor activity in a murine model of
triple-negative breast cancer.
Several multitargeted tyrosine kinase inhibitors
show the inhibitory effects of TAM kinases, but few have been
specifically developed as TAM tyrosine kinase inhibitors. Several
TAM-selective inhibitors are currently in development, and
promising agents undergoing clinical trials include bemcentinib and
BMS-777607 (10,13). Bemcentinib (also known as BGB324 or
R428) is an orally available, selective inhibitor of AXL RTK that
showed antitumor effects in preclinical models with multiple cancer
cell lines (1,3,10).
Bemcentinib also demonstrated synergistic effects when combined
with ICIs, targeted therapies, and chemotherapeutic agents. As a
result, this drug is now in phase I/II clinical trials for a
variety of cancers, either alone or in combination with other
chemotherapy regimens including ICIs (7,10,39).
BMS-777607 was initially designed as a selective and orally
available MET kinase inhibitor, but it was found to have a role as
a potent pan-TAM inhibitor. BMS-777607 also demonstrated antitumor
activity in in vivo models of several cancers, and it is now
undergoing phase I/II clinical trials in patients with advanced
tumors (7,8,35,40).
Despite some of these promising drugs, there are still unmet needs
for more potent TAM kinase inhibitors with good safety profiles as
new anticancer agents. The first consideration is the relative
potency of the drug against the target. Previous studies indicate
that inhibiting only one of three TAM receptors may not be
effective because of the induction of compensatory activation of
the other subunits (11,16). In preclinical models, MERTK
inhibition alone was not sufficient to impact tumor growth, and
some small-molecule inhibitors of AXL or MERTK showed low
inhibitory potency against targets (7). In addition, some studies showed that
AXL expression was increased after treatment with bemcentinib
(41). This increase is caused by
the inhibition of ubiquitination and degradation of AXL receptors
by AXL phosphorylation blocking with small-molecule AXL inhibitors
(42). Further research is needed
to determine whether the paradoxical upregulation of AXL protein
could have the potential to reverse any desired effect of
treatment. Based on these previous data, we chose BMS-777607, the
same pan-TAM inhibitor, instead of bemcentinib, a selective AXL
inhibitor, to compare the efficacy of fostamatinib. Second, the
potential concern of TAM kinase inhibition is about safety issues.
TAM receptors are expressed in a wide range of tissues and have
crucial roles in the maintenance of an organ's normal functions.
Furthermore, because of their role in diminishing the innate immune
response, any sustained TAM kinase inhibition may lead to adverse
immune or inflammatory reactions, such as autoimmunity; thus,
careful monitoring is required (39).
Taken together, TAM family kinases are distinct
oncogenic drivers of novel anticancer targets through their
promotion of tumor cell survival, proliferation, invasion, and
chemoresistance, as well as their suppression of antitumor
immunity. This study suggests that fostamatinib has the potential
to be a novel anticancer agent through its inhibition of TAM
tyrosine kinase. In addition, its efficacy is expected to be better
than other previous TAM kinase inhibitors. An AI-driven drug-target
interaction prediction model, such as MT-DTI, can contribute to the
development of targeted therapy and be a powerful tool for drug
repurposing. Further validation is warranted in animal models and
clinical trials of patients with various types of cancer.
Additional investigation into the role of fostamatinib in immune
modulation in the tumor microenvironment, and in combination
therapy with ICI, may provide clinically useful information for a
new therapeutic strategy.
Supplementary Material
TAK-659 inhibits the Syk expression
but does not affect TAM signaling in CAL27 cells with high Syk
expression. Western blot of p-Syk, Syk, p-AXL, AXL, p-MERTK, MERTK,
and TYRO3 in TAK-659-treated cells (α-tubulin was used as a loading
control). TAM, TYRO3, AXL, MERTK; p-, phosphorylated.
MT-DTI prediction results of Food and
Drug Administration-approved drugs against the TAM receptor
kinases.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by a grant from the National
R&D Program for Cancer Control, Ministry of Health &
Welfare, Republic of Korea (grant no. 1720100). This study was also
supported by National Research Foundation of Korea grants funded by
the Korean government (MSIT) (grant nos. NRF-2020R1A5A2019210 and
NRF-2022R1A2C1011772).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YC, HSW and YHK conceptualized the study. The
machine learning was performed by BL. BRB, HL, JHL, SK and SHC
performed experiments and analyzed data. YC, HL, HSW and YHK wrote
the original draft. YC, HSW and YHK reviewed and edited the
manuscript. HSW and YHK acquired funding. YC and HSW confirm the
authenticity of all the raw data. All authors have read approved
the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
YC, BRB and BL were employed by the company Deargen
Inc., who developed the deep DTI prediction model called Molecule
Transformer (MT)-DTI employed in this study. The remaining authors
declare that the research was conducted in the absence of any
commercial or financial relationships that could be construed as a
potential conflict of interest. All other authors do not have any
financial and non-financial conflict of interest and declare that
they have no competing interests.
References
|
1
|
Graham DK, DeRyckere D, Davies KD and Earp
HS: The TAM family: Phosphatidylserine sensing receptor tyrosine
kinases gone awry in cancer. Nat Rev Cancer. 14:769–785.
2014.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang
S, Li T, Chen F and Yang Y: Targeting Gas6/TAM in cancer cells and
tumor microenvironment. Mol Cancer. 17(20)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lemke G and Rothlin CV: Immunobiology of
the TAM receptors. Nat Rev Immunol. 8:327–336. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Cook RS, Jacobsen KM, Wofford AM,
DeRyckere D, Stanford J, Prieto AL, Redente E, Sandahl M, Hunter
DM, Strunk KE, et al: MerTK inhibition in tumor leukocytes
decreases tumor growth and metastasis. J Clin Invest.
123:3231–3242. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Paolino M, Choidas A, Wallner S, Pranjic
B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa
JP, et al: The E3 ligase Cbl-b and TAM receptors regulate cancer
metastasis via natural killer cells. Nature. 507:508–512.
2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Werfel TA and Cook RS: Efferocytosis in
the tumor microenvironment. Semin Immunopathol. 40:545–554.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Axelrod H and Pienta KJ: Axl as a mediator
of cellular growth and survival. Oncotarget. 5:8818–8852.
2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Holland SJ, Pan A, Franci C, Hu Y, Chang
B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, et al: R428, a
selective small molecule inhibitor of Axl kinase, blocks tumor
spread and prolongs survival in models of metastatic breast cancer.
Cancer Res. 70:1544–1554. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Crittenden MR, Baird J, Friedman D, Savage
T, Uhde L, Alice A, Cottam B, Young K, Newell P, Nguyen C, et al:
Mertk on tumor macrophages is a therapeutic target to prevent tumor
recurrence following radiation therapy. Oncotarget. 7:78653–78666.
2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ludwig KF, Du W, Sorrelle NB,
Wnuk-Lipinska K, Topalovski M, Toombs JE, Cruz VH, Yabuuchi S,
Rajeshkumar NV, Maitra A, et al: Small-molecule inhibition of Axl
targets tumor immune suppression and enhances chemotherapy in
pancreatic cancer. Cancer Res. 78:246–255. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kim JE, Kim Y, Li G, Kim ST, Kim K, Park
SH, Park JO, Park YS, Lim HY, Lee H, et al: MerTK inhibition by
RXDX-106 in MerTK activated gastric cancer cell lines. Oncotarget.
8:105727–105734. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cummings CT, Zhang W, Davies KD,
Kirkpatrick GD, Zhang D, DeRyckere D, Wang X, Frye SV, Earp HS and
Graham DK: Small molecule inhibition of MERTK is efficacious in
non-small cell lung cancer models independent of driver oncogene
status. Mol Cancer Ther. 14:2014–2022. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gay CM, Balaji K and Byers LA: Giving AXL
the axe: Targeting AXL in human malignancy. Br J Cancer.
116:415–423. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang W, DeRyckere D, Hunter D, Liu J,
Stashko MA, Minson KA, Cummings CT, Lee M, Glaros TG, Newton DL, et
al: UNC2025, a potent and orally bioavailable MER/FLT3 dual
inhibitor. J Med Chem. 57:7031–7041. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
DeRyckere D, Lee-Sherick AB, Huey MG, Hill
AA, Tyner JW, Jacobsen KM, Page LS, Kirkpatrick GG, Eryildiz F,
Montgomery SA, et al: UNC2025, a MERTK small-molecule inhibitor, is
therapeutically effective alone and in combination with
methotrexate in leukemia models. Clin Cancer Res. 23:1481–1492.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
McDaniel NK, Cummings CT, Iida M, Hülse J,
Pearson HE, Vasileiadi E, Parker RE, Orbuch RA, Ondracek OJ, Welke
NB, et al: MERTK mediates intrinsic and adaptive resistance to
AXL-targeting agents. Mol Cancer Ther. 17:2297–2308.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rios-Doria J, Favata M, Lasky K, Feldman
P, Lo Y, Yang G, Stevens C, Wen X, Sehra S, Katiyar K, et al: A
potent and selective dual inhibitor of AXL and MERTK possesses both
immunomodulatory and tumor-targeted activity. Front Oncol.
10(598477)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ekins S, Puhl AC, Zorn KM, Lane TR, Russo
DP, Klein JJ, Hickey AJ and Clark AM: Exploiting machine learning
for end-to-end drug discovery and development. Nat Mater.
18:435–441. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Issa NT, Stathias V, Schurer S and
Dakshanamurthy S: Machine and deep learning approaches for cancer
drug repurposing. Semin Cancer Biol. 68:132–142. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gilson MK, Liu T, Baitaluk M, Nicola G,
Hwang L and Chong J: BindingDB in 2015: A public database for
medicinal chemistry, computational chemistry and systems
pharmacology. Nucleic Acids Res. 44 (D1):D1045–D1053.
2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kim S, Chen J, Cheng T, Gindulyte A, He J,
He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, et al: PubChem 2019
update: Improved access to chemical data. Nucleic Acids Res. 47
(D1):D1102–D1109. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gaulton A, Bellis LJ, Bento AP, Chambers
J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D,
Al-Lazikani B and Overington JP: ChEMBL: A large-scale bioactivity
database for drug discovery. Nucleic Acids Res. 40 (Database
Issue):D1100–D1107. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Beck BR, Shin B, Choi Y, Park S and Kang
K: Predicting commercially available antiviral drugs that may act
on the novel coronavirus (SARS-CoV-2) through a drug-target
interaction deep learning model. Comput Struct Biotechnol J.
18:784–790. 2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Choi Y, Shin B, Kang K, Park S and Beck
BR: Target-centered drug repurposing predictions of human
angiotensin-converting enzyme 2 (ACE2) and transmembrane protease
serine subtype 2 (TMPRSS2) interacting approved drugs for
coronavirus disease 2019 (COVID-19) treatment through a drug-target
interaction deep learning model. Viruses. 12(1325)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Connell NT and Berliner N: Fostamatinib
for the treatment of chronic immune thrombocytopenia. Blood.
133:2027–2030. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tanoli Z, Alam Z, Vähä-Koskela M,
Ravikumar B, Malyutina A, Jaiswal A, Tang J, Wennerberg K and
Aittokallio T: Drug target commons 2.0: A community platform for
systematic analysis of drug-target interaction profiles. Database
(Oxford). 2018:1–13. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu T, Lin Y, Wen X, Jorissen RN and
Gilson MK: BindingDB: A web-accessible database of experimentally
determined protein-ligand binding affinities. Nucleic Acids Res. 35
(Database Issue):D198–D201. 2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhou J, Cui G, Hu S, Zhang Z, Yang C, Liu
Z, Wang L, Li C and Sun M: Graph neural networks: A review of
methods and applications. AI Open. 1:57–81. 2020.
|
|
29
|
Elnaggar A, Heinzinger M, Dallago C,
Rihawi G, Wang Y, Jones L, Gibbs T, Feher T, Angerer C, Steinegger
M, et al: ProtTrans: Towards cracking the language of life's code
through self-supervised deep learning and high performance
computing. arXiv preprint arXiv: 200706225, 2020.
|
|
30
|
Ruder S: An overview of multi-task
learning in deep neural networks. arXiv preprint arXiv: 170605098,
2017.
|
|
31
|
Duan Q, Flynn C, Niepel M, Hafner M,
Muhlich JL, Fernandez NF, Rouillard AD, Tan CM, Chen EY, Golub TR,
et al: LINCS canvas browser: Interactive web app to query, browse
and interrogate LINCS L1000 gene expression signatures. Nucleic
Acids Res. 42 (Web Server Issue):W449–W460. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lamb J, Crawford ED, Peck D, Modell JW,
Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et
al: The connectivity map: Using gene-expression signatures to
connect small molecules, genes, and disease. Science.
313:1929–1935. 2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Subramanian A, Narayan R, Corsello SM,
Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK,
et al: A next generation connectivity map: L1000 platform and the
first 1,000,000 profiles. Cell. 171:1437–1452.e17. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Beitzen-Heineke A, Berenbrok N,
Waizenegger J, Paesler S, Gensch V, Udonta F, Vargas Delgado ME,
Engelmann J, Hoffmann F, Schafhausen P, et al: AXL inhibition
represents a novel therapeutic approach in BCR-ABL negative
myeloproliferative neoplasms. Hemasphere. 5(e630)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yan D, Earp HS, DeRyckere D and Graham DK:
Targeting MERTK and AXL in EGFR mutant non-small cell lung cancer.
Cancers (Basel). 13(5639)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Markham A: Fostamatinib: First global
approval. Drugs. 78:959–963. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Bussel J, Arnold DM, Grossbard E, Mayer J,
Treliński J, Homenda W, Hellmann A, Windyga J, Sivcheva L,
Khalafallah AA, et al: Fostamatinib for the treatment of adult
persistent and chronic immune thrombocytopenia: Results of two
phase 3, randomized, placebo-controlled trials. Am J Hematol.
93:921–930. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Regan-Fendt K, Li D, Reyes R, Yu L, Wani
NA, Hu P, Jacob ST, Ghoshal K, Payne PRO and Motiwala T:
Transcriptomics-based drug repurposing approach identifies novel
drugs against sorafenib-resistant hepatocellular carcinoma. Cancers
(Basel). 12(2730)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Aehnlich P, Powell RM, Peeters MJW,
Rahbech A and Thor Straten P: TAM receptor inhibition-implications
for cancer and the immune system. Cancers (Basel).
13(1195)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kasikara C, Davra V, Calianese D, Geng K,
Spires TE, Quigley M, Wichroski M, Sriram G, Suarez-Lopez L, Yaffe
MB, et al: Pan-TAM tyrosine kinase inhibitor BMS-777607 enhances
anti-PD-1 mAb efficacy in a murine model of triple-negative breast
cancer. Cancer Res. 79:2669–2683. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Chen F, Song Q and Yu Q: Axl inhibitor
R428 induces apoptosis of cancer cells by blocking lysosomal
acidification and recycling independent of Axl inhibition. Am J
Cancer Res. 8:1466–1482. 2018.PubMed/NCBI
|
|
42
|
Lauter M, Weber A and Torka R: Targeting
of the AXL receptor tyrosine kinase by small molecule inhibitor
leads to AXL cell surface accumulation by impairing the
ubiquitin-dependent receptor degradation. Cell Commun Signal.
17(59)2019.PubMed/NCBI View Article : Google Scholar
|