1. Introduction
Cardiovascular diseases encompass a range of
conditions that impact the heart and blood vessels, including
coronary artery disease, hypertension and cardiomyopathy. These
diseases have emerged as significant contributors to global
mortality, representing a serious risk to health and life. The
development of cardiovascular disease can be complex and
multifaceted. It involves a number of factors, including genetics,
the environment, lifestyle and age. Research strongly suggests that
epigenetic changes serve a significant role in the development and
progression of cardiovascular disorders, acting as a regulatory
mechanism that can alter gene function/expression/activity without
changing the content of DNA sequences. Epigenetics is widely
considered the primary regulatory mechanism by which cells respond
to environmental changes, as it allows for alterations in gene
expression without changing the underlying DNA sequence, making it
a flexible way for cells to adapt to different conditions (1). Mechanisms associated with epigenetics
including DNA methylation, histone modification and non-coding RNA
activity notably influence the function and expression level of
cardiovascular disease-related genes, thereby participating in the
occurrence and development of these diseases. Related studies have
confirmed that histone modification serves an important role in the
occurrence and development of cardiovascular diseases. Given the
significant role of histone modification in the regulation of the
expression of cardiovascular disease-related genes, it is essential
to investigate the mechanisms underlying histone modification and
to elucidate its critical influence on the progression of
cardiovascular diseases. An improved understanding of the
regulatory mechanisms in the development of cardiovascular disease
may help to identify new therapeutic targets and provide beneficial
effects for patients. On this basis, the mechanism of histone
modification in cardiovascular disease will be reviewed to provide
new ideas for clinical research of cardiovascular disease.
2. Structure and function of histones
Histones are fundamental proteins found in both
eukaryotic and prokaryotic organisms and serve as the primary
protein constituents of chromatin (2). There are six known histones, namely,
H1, H2A, H2B, H3, H4 and archaea histones. Histone octamers are
composed of four types of histone proteins: H2A, H2B, H3 and H4,
with each type contributing two molecules. A total of ~200 base
pairs of DNA wrap around these histone octamers, forming a
structural unit known as a nucleosome. Nucleosomes constitute the
fundamental building blocks of chromatin, formed by DNA wrapped
around a set of eight proteins called histones, known as histone
octamer (3). Within a nucleosome
core, histone proteins consist of a globular domain that forms the
core structure, while their amino-terminal tails extend outwards
and are subject to various modifications such as methylation,
acetylation, ubiquitination, crotonylation and lactylation. The
specific modification mechanism is demonstrated in Fig. 1.
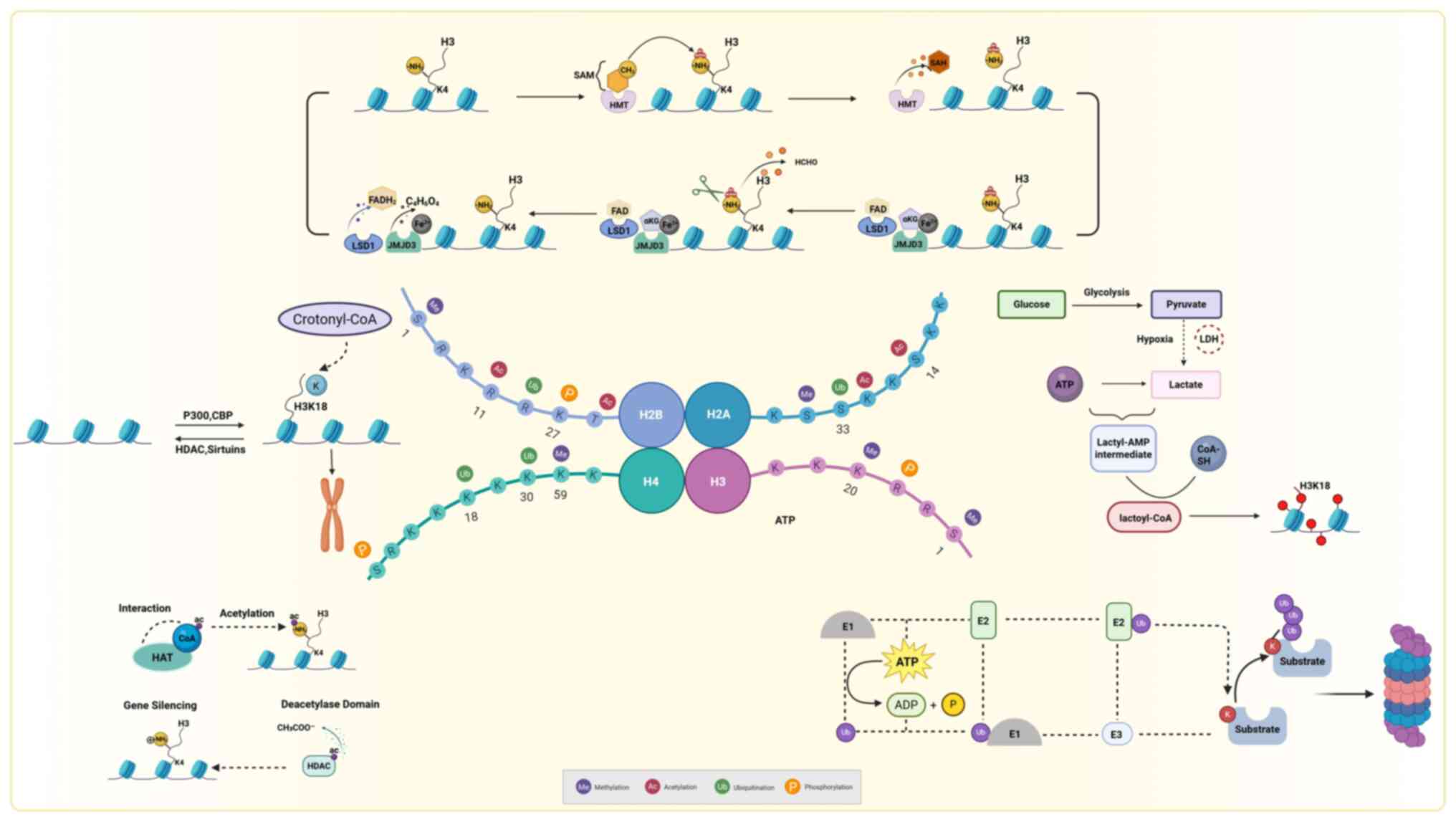 | Figure 1Mechanisms of histone modification.
HAT, histone acetyltransferase; HDAC, histone deacetylase; CBP,
creb binding protein; P300, e1a binding protein p300; SAM,
s-adenosyl methionine; HMT, histone methyltransferase; FAD, flavin
adenine dinucleotide; FADH2, reduced flavin adenine
dinucleotide; LSD1, lysine-specific demethylase 1; JMJD3, jumonji
domain-containing protein 3; SAH, s-adenosyl-l-homocysteine; ATP,
adenosine triphosphate; ADP, adenosine diphosphate. This figure was
created with BioRender.com (https://www.biorender.com/). |
Histone modifications can alter the compactness of
chromatin by altering the affinity between histones and DNA double
strands, essentially controlling whether chromatin is in a loosened
(accessible) or condensed (less accessible) state. Aberrant histone
modifications lead to imbalanced expression of cardiovascular
disease-related genes, resulting in changes in cellular phenotype
and cardiac function. The study of cardiovascular diseases has
largely centered on acetylation, methylation and ubiquitination.
Consequently, the present review comprehensively discussed the
functional roles of these processes and their reversible and
dynamic regulatory mechanisms. Histone methylation usually occurs
on lysine or arginine residues, and its activity is regulated by
histone methyltransferases and demethylases (4). Histone acetylation is mediated by
histone acetyltransferases (HATs). This loosens the chromatin
structure, making DNA more accessible to transcription factors and
thus promoting transcriptional activation. The opposite progression
implies that histone deacetylase (HDAC) removes the acetyl group,
thereby condensing nucleosomes and leading to transcriptional
repression (5). Ubiquitin is a key
covalent modification mechanism, which covalently links ubiquitin
peptide chains containing 76 amino acids to lysine residues of
histones through the continuous action of the E1 activating enzyme,
E2 binding enzyme and E3 ligase (6). Mono-ubiquitination is a type of
protein modification where a single ubiquitin molecule is attached
to a protein, it is most commonly observed on histones H2A and H2B,
where there are two well-defined modification sites (7,8). In
histone acetylation, chromatin is directly loosened by neutralizing
the positive charge on histones, thereby reducing their affinity
for DNA. In methylation such as (H3K4me3 or H3K27me3) and
ubiquitination, the overall charge of histones is not changed,
instead, chromatin architecture is regulated by recruiting effector
proteins such as chromatin remodeling enzymes or transcriptional
repressors (9). During the
ubiquitination process such as H2BK120 ubiquitination, nucleosome
remodeling during processes such as DNA repair or transcription
elongation is facilitated primarily through recruiting
deubiquitinating enzymes and chromatin remodeling complexes
including SWI/SNF (10).
Crotonylation, a post-translational modification
mediated by crotonyl-transferase, involves the enzymatic transfer
of crotonyl groups from crotonyl-coenzyme A to lysine residues on
target proteins (11). In somatic
cells, genomic mapping studies have revealed that histone
crotonylation marks are primarily localized within 200-300 base
pairs flanking transcription start sites, exhibiting symmetrical
distribution around the transcriptional initiation core region
(12). These findings establish
histone crotonylation as an epigenetic signature marking
promoter-specific transcriptional activation and active
transcription hubs. Histone lactylation, another modification,
operates through the covalent conjugation of lactyl moieties to
lysine residues, enabling context-dependent regulation of
transcriptional programs associated with metabolic adaptation
(13). Previous studies focusing
on histone modifications in cardiovascular diseases have found that
histone modifications may affect a wide range of cardiovascular
diseases, including atherosclerosis (As), heart failure, myocardial
infarction (MI), cardiomegaly and myocardial ischemia-reperfusion
injury (MIRI) (14,15). The specific histone modifications
in these cardiovascular diseases and their associated molecular
mechanisms are illustrated in Fig.
2.
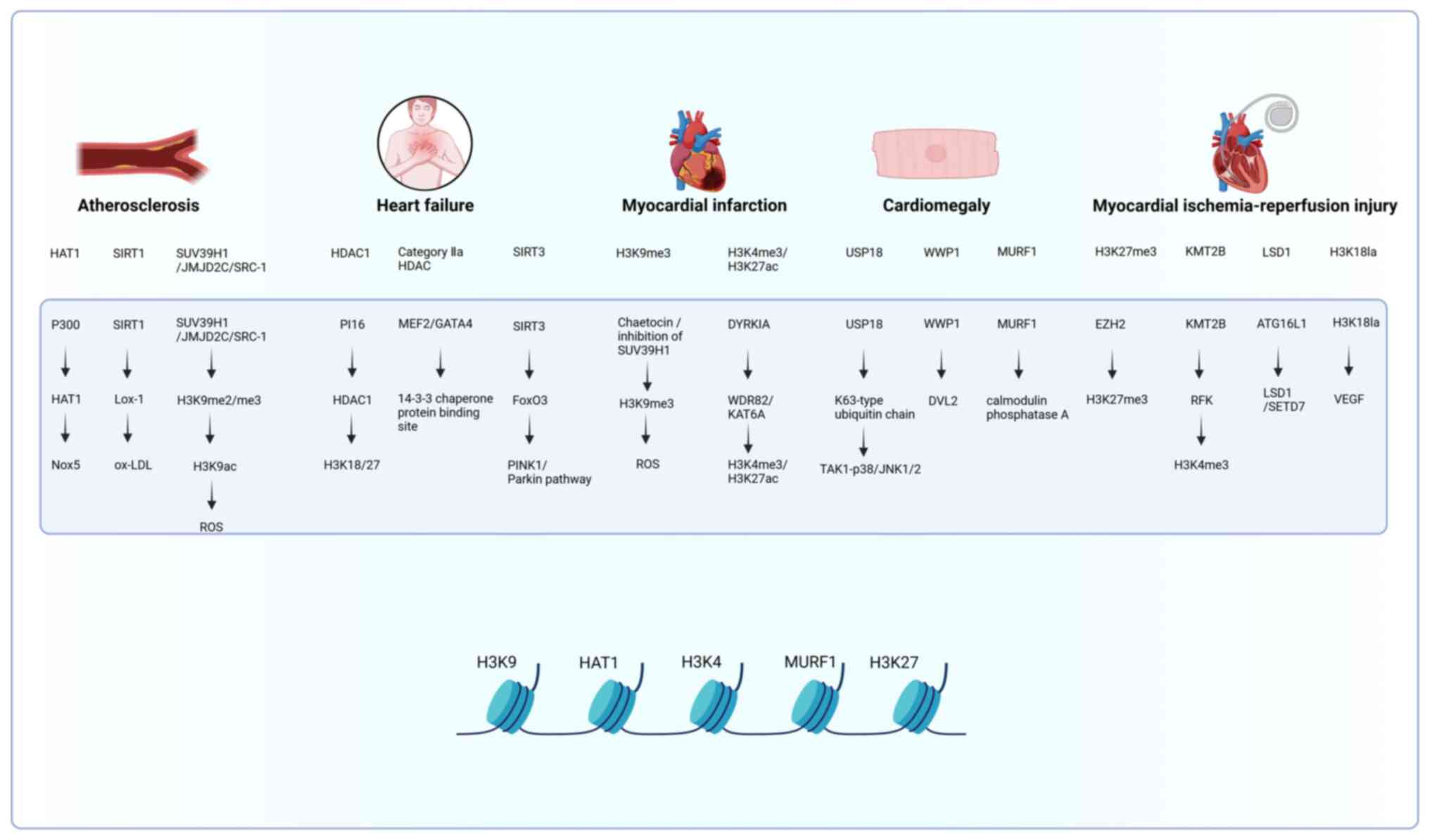 | Figure 2Specific mechanisms studied for the
effects of histone modifications on cardiovascular disease . HAT,
histone acetyltransferase; HDAC, histone deacetylase; ROS, reactive
oxygen species; ox-LDL, oxidized low-density lipoprotein; H3K9me3,
trimethylation of histone H3 at lysine 9; SIRT, silent information
regulator; SUV39H1, suppressor of variegation 3-9 homolog 1;
JMJD2C, jumonji domain-containing protein 2c; SRC-1, steroid
receptor coactivator-1; H3K9ac, histone h3 lysine 9 acetylation;
H3K4me, histone h3 lysine 4 trimethylation; H3K27ac, histone h3
lysine 27 acetylation; H3K27me3, histone h3 lysine 27
trimethylation; H3K18la, histone h3 lysine 18 lactylation; p300,
e1a binding protein p300; KAT6A, lysine acetyltransferase 6a; EZH2,
enhancer of zeste homolog 2; KMT2B, lysine methyltransferase 2b;
LSD1, lysine specific demethylase 1; SETD7, set domain containing
7; TAK1, TGF-β activated kinase 1; PINK1, pten induced kinase 1;
DYRK1A, dual-specificity tyrosine-phosphorylation regulated kinase
1a; FoxO3, forkhead box O3; MEF2, myocyte enhancer factor 2; GATA4,
gata binding protein 4; USP18, ubiquitin specific peptidase 18;
WWP1, ww domain containing e3 ubiquitin protein ligase 1; ATG16L1,
autophagy related 16 like 1; Lox-1, lectin-like oxidized ldl
receptor 1; DVL2, dishevelled segment polarity protein 2; MURF1,
muscle ring finger protein 1; RFK, riboflavin kinase; WDR82, wd
repeat domain 82; PI16, peptidase inhibitor 16; Nox5, NADPH oxidase
5. This figure was created with BioRender.com (https://www.biorender.com/). |
3. Histone modifications in cardiovascular
disease
Histone modifications and As
As is a disease characterized by lipid metabolism
disorders and endothelial dysfunction. It is linked to histone
modifications that regulate inflammatory pathways, with Nox5, a
newly identified NADPH oxidase, serving a key role by generating
superoxide, leading to oxidative stress and promoting As
development. During inflammation, the proteins belonging to the
histone acetylation system (p300 and HAT1) become elevated, leading
to increased acetylation of the Nox5 gene promoter region. This
suggests that histone acetylation is involved in As development
(16). Consequently, regulating
Nox5 expression through epigenetic pathways may be a new approach
to treating As, instead of directly removing reactive oxygen
species (ROS). Silent Information Regulator 1 (SIRT1), a member of
the HDAC family of HDACs, slows the progression of As by inhibiting
macrophage LOX-1 gene expression and decreasing the
phagocytosis of oxidized low-density lipoprotein by macrophages,
thereby leading to a reduction in subendothelial lipid deposition
(17). Krüppel-like factor 2
(KLF2), a transcription factor, serves a crucial role in
maintaining the anti-inflammatory and anti-atherosclerotic
properties of endothelial cells. However, its activity is
negatively regulated by HDAC5, which binds directly to KLF2 and
prevents its proper functioning by downregulating KLF2-dependent
endothelial nitric oxide synthase expression. It ultimately leads
to impaired endothelial cell function and increased risk of As
development (18).
Sabinyl-anilino-hydroxamic acid (SAHA, also known as Vorinostat or
MK0683) is a well-characterized HDAC inhibitor (19). As a pan-inhibitor targeting
multiple HDAC isoforms, SAHA theoretically enhances histone
acetylation levels through broad suppression of HDAC activity,
including HDAC5, thereby theoretically promoting transcriptional
activation of KLF2 to exert anti-atherosclerotic effects. However,
the precise mechanisms underlying SAHA-mediated upregulation of
KLF2 require further elucidation. A previous study has demonstrated
that SAHA is a novel KLF2 activator that prevents endothelial
inflammation in vitro and the development of As in
vivo (20). Based on in
vitro and in vivo experimental data, SAHA reduces
monocyte adhesion primarily by inhibiting tumor necrosis factor α
(TNFα)-stimulated upregulation of vascular cell adhesion molecule 1
(VCAM1). Given that VCAM1 is a target molecule of KLF2(21), the aforementioned study further
investigated whether the downregulation of VCAM1 by SAHA depended
on the KLF2 pathway. Overexpression of KLF2 was found to inhibit
the induction of the TNFα-mediated proinflammatory molecule VCAM1
in endothelial cells (22),
suggesting that the anti-atherosclerotic effect of SAHA may be
partially realized through a KLF2-dependent anti-inflammatory
mechanism. In addition, the upregulation of KLF2 by SAHA may
further exert its anti-atherosclerotic effect by promoting the
expression of a series of genes with antithrombotic,
anti-inflammatory and antiproliferative functions (23-25).
Thus, SAHA may be suitable for targeting the early inflammatory
process of As. Wang et al (26) demonstrated that HDAC3 upregulation
inactivates NF-κB/p65 signaling by enhancing microRNA
(miR)-19b-mediated peroxisome proliferator-activated receptor gamma
(PPARγ) expression. HDAC3 is highlighted as a possible therapeutic
target for treating As in this mechanism, as it inhibits
inflammatory responses and slows the progression of As.
Endothelial-to-mesenchymal transition (EndMT) was
also regarded as a novel therapeutic target for cardiovascular
diseases (27). A previous study
found that the HDAC3 inhibitor RGFP966 inhibited EndMT in the
aortic root of ApoE-/- mice, reducing the development of
As in the aortic root of ApoE-/- mice (28).
SUV39H1, a key enzymatic regulator of histone
methylation, catalyzes trimethylation of histone H3 at lysine 9
(H3K9me3), thereby inducing transcriptional silencing of downstream
genes through chromatin compaction (29). Experimental evidence indicates that
pharmacological suppression of SUV39H1 elevates p21 expression by
attenuating H3K9me3 enrichment at the p21 promoter (30), highlighting its therapeutic
potential in modulating vascular smooth muscle cell (VSMC)
hyperproliferation.
Further investigations reveal context-dependent
regulatory roles of SUV39H1. While it demonstrates a robust
repressive effect on cell cycle inhibitors, such as p21 and
p27Kip1, its regulatory influence on proliferation-associated
genes, such as Id3, is comparatively reduced. This differential
activity may contribute to pathological VSMC activation and
subsequent neointimal hyperplasia, suggesting a complex mechanistic
landscape influenced by cell-specific signaling networks and
experimental conditions.
Collectively, targeted inhibition of SUV39H1
demonstrates efficacy in attenuating post-injury neointima
formation, positioning it as a novel therapeutic candidate for
proliferative vasculopathy including in-stent restenosis (31).
In addition, SUV39H1 is implicated in enhancing the
possibility of As development in obese individuals (32). Free radicals produced by p66Shc in
mitochondria affect energy metabolism and the development of
obesity (33). Obesity-induced
epigenetic modifications, particularly those involving the SUV39H1
methyltransferase, JMJD2C/SRC-1 demethylase and SRC-1
acetyltransferase, have the potential to lower the levels of
H3K9me2/me3 while simultaneously elevating H3K9 acetylation, which
binds to the p66Shc promoter (34). This leads to an overproduction of
mitochondrial ROS in the visceral fat artery of obese individuals.
These epigenetic changes were found not only in humans but also in
the endothelial cells and aorta of obese mice. In particular,
obesity-induced downregulation of SUV39H1 expression is at the
heart of H3K9 modification changes, which may contribute to
ROS-induced endothelial dysfunction and the development of As
(34). Experiments showed that the
SUV39H1, JMJD2C and SRC-1 complexes clustered in the promoter
region of p66Shc. Moreover, it was observed that JMJD2C and SRC-1
did not affect the function of SUV39H1, suggesting that targeting
SUV39H1 may help delay the progression of obesity-induced As. Aging
is also an important risk factor for As. AMPK, known as
AMP-activated protein kinase, serves a crucial role in combating
aging by reducing mitochondrial oxidative stress. It functions as a
cellular energy sensor, activating important proteins such as SIRT1
and PGC-1α. These proteins facilitate mitochondrial biogenesis and
enhance mitochondrial function, thereby improving the cell's
capacity for efficient energy production and decreasing the
generation of harmful ROS. This process ultimately supports an
extended lifespan. Activation of AMPK using drugs such as metformin
or AICAR triggers an increase in DOT1L-mediated H3K79
trimethylation on SIRT1/SIRT3 promoters. This, in turn, results in
higher levels of telomerase reverse transcriptase and the protein
PGC-1α, which are important for cellular longevity and
mitochondrial function. Silencing SIRT3 increases mitochondrial
oxidative stress and prevents age-induced atheromatous plaque
formation in ApoE knockdown mice (35). This finding indicates that the
SIRT-mediated longevity of the vascular system can be promoted
through DOT1L hypermethylation of H3K79. Furthermore, metformin may
be a potential treatment for age-related As.
The study suggests that the development of As may be
linked to specific risk factors that serve a role in influencing
the methylation status of H3K9 and H3K79, as well as the oxidative
stress response of mitochondria. In addition, targeted therapy
against SUV39H1 and DOT1L may help delay the progression of As. It
is important to note that metformin, as a commonly used
anti-diabetic drug, may have a positive effect on diabetes-related
As, especially during aging. In the pathological process of As,
histone methylation may not only trigger the occurrence of As but
also serve as a biomarker to assess the severity of the disease and
participate in its development. For example, the levels of H3K4me2
in smooth muscle cells were elevated during advanced
atherosclerotic plaques in the human carotid artery, compared with
earlier stages. By contrast, H3K9me2 levels were observed to
decline in smooth muscle cells and inflammatory cells, while
H3K27me2 levels decreased specifically in inflammatory cells
(36). In the advanced stages of
As, the expression levels of H3K4 methyltransferase MLL2 and H3K9
methyltransferase G9a are elevated (36). Previous studies have also found
that in human advanced atherosclerotic plaques, SMC and
inflammatory cells lacking H3K9 methyltransferase exhibit reduced
H3K9 and H3K27 methylation levels, while in SMC with elevated
MLL2/4 levels, H3K4 methylation levels are increased. This suggests
that H3K4 methylation may be related to the severity of As
(36,37). Nevertheless, the mechanism of
atherosclerotic plaque formation, disease progression and the
interaction between histone methylation and oxidative stress still
need further study. These areas may represent promising avenues for
future research endeavors.
Histone acetylation (involving regulators such as
HDAC5 and SIRT1) modulates inflammation and endothelial
dysfunction. Pan-HDAC inhibitors such as SAHA enhance KLF2 activity
to suppress VCAM1, while HDAC3-selective agents specifically target
endothelial-mesenchymal transition. Methylation modifiers,
including SUV39H1 and DOT1L, exhibit dual roles: SUV39H1-mediated
H3K9me3 drives vascular smooth muscle cell proliferation and
obesity-linked oxidative stress, whereas DOT1L-driven H3K79me3
activates SIRT1/SIRT3 to mitigate age-related mitochondrial damage.
Challenges persist in acetylation's isoform specificity and
methylation's contextual risks. Obesity and aging exacerbate
complexity. Combined strategies merging SAHA with SUV39H1
inhibition may synergize anti-inflammatory and antiproliferative
pathways.
Histone modification and heart
failure
Chronic heart failure is a widespread and
potentially fatal condition notably impacting life expectancy and
quality of life. It is characterized by alterations in histone
acetylation, which in turn affects gene expression crucial to the
progression of the disease.
Histone acetylation is a key regulator in
cardiovascular disease, impacting gene expression and disease
progression. This modification influences gene expression by
multiple pathways, some of which are described below. First, the
post-translational modification (PTM) of histone lysine alters the
positive charge of the ε-amino group, which diminishes nucleosome
compaction and impacts DNA-histone or histone-histone interactions.
This leads to a decrease in euchromatin-nucleosome interactions but
enhances the interaction between enhancers and their target
promoters (38). Thus, the PTM of
histones directly affects the structure of chromatin and
nucleosomes. Additionally, the PTMs of lysine residues on histones
can serve as epigenetic signals, either directly or indirectly, by
influencing the recruitment of transcription factors that
facilitate the acetylation of RNA polymerase II. The acetylation
status of lysine residues is governed by the balance between the
activities of HAT and HDAC (38,39).
Studies have demonstrated that HDAC serves a significant regulatory
role in heart failure and is closely connected to the onset and
progression of the condition (40,41).
Group I HDACs may contribute to the development of
heart failure by affecting ventricular remodeling. Studies have
shown that histone H3 lysine trimethylation at position 27 (H3K27),
micro inhibitor of ribonucleic acid-21-3p (MIR-21-3p), and HDAC
3-nuclear receptor co-inhibitory factor 2/thyrotropin receptor
silencer of retinoic acid (Hdac3-NCoR/SMRT) complexes are
associated with ventricular remodeling, suggesting that the
development of ventricular remodeling may be related to histone
acetylation at both the transcriptional and translational levels
(42-44).
The process of ventricular remodeling may be related to histone
acetylation, and group I HDACs may also regulate myocardial
fibrosis, potentially contributing to the development of chronic
heart failure (45). Peptidase
inhibitory protein 16 (PI16), transforming growth factor-β/mother
DPP homolog (TGF-β/Smad) signaling, myofibroblast marker α-smooth
muscle actin (α-SMA), and transforming growth factor-β1/mother DPP
homolog 2/3 (TGF-β1/Smad2/3) were found to be closely related to
myocardial fibrosis. The overexpression of PI16 results in a
decrease in the nuclear level of HDAC1, which subsequently enhances
histone acetylation in K18 and K27 lysine. This process also
reduces myocardial collagen deposition, effectively inhibiting the
proliferation of fibroblasts and fibrotic levels of related
proteins, thereby preventing the occurrence of cardiac hypertrophy
and cardiac fibrosis development (46). When HDAC3 is abnormally expressed,
it actively suppresses the production of Klotho protein, a molecule
that protects against fibrosis in the heart muscle, leading to an
amplified TGF-β/Smad signaling pathway and ultimately causing the
development of myocardial fibrosis (scar tissue buildup), as
observed in mice (47). α-SMA,
regulated by HDAC8, is highly expressed in myocardial fibrosis but
less so in normal myocardium, suggesting an important role of HDAC8
in myocardial fibrosis (48).
Among HDACII, specifically, class IIa HDACs, serve a
key role in suppressing the activity of cardiac-specific
transcription factors such as MEF2 and GATA4 in the heart by a
mechanism involving phosphorylation, which then allows the 14-3-3
chaperonin to bind and shuttle the HDACs out of the nucleus,
effectively inhibiting the transcription of genes related to
cardiac hypertrophy (49). This
action prevents the progression to chronic heart failure, offering
a protective effect on the heart (49). Conversely, HDAC class IIb affects
cardiac function through non-epigenetic mechanisms, including the
pathological remodeling of the ventricles and the modulation of
myogenic fibers, both of which may contribute to the advancement of
chronic heart failure.
Class III HDAC is a family of SIRT proteins, which
are NAD+-dependent HDACs involved in the pathophysiology
of cardiovascular disease (50).
The SIRT family serves a crucial role in managing oxidative stress,
autophagy and apoptosis by lowering ROS levels. This is achieved
through the activation of SIRT2, the deacetylation of hepatic
kinase B1, and the promotion of the protein kinase (AMPK) pathway
activation (51). According to
research, failing hearts of humans, mice and pigs, the expression
of SIRT1, a class III HDAC, is significantly reduced, indicating
its downregulation in these conditions. SIRT1 deficiency leads to
an increase in the acetylation of sarcoplasmic reticulum calcium
ion ATPase (SERCA2a). By contrast, the pharmacological activation
of SIRT1 facilitates the deacetylation of SERCA2a, which in turn
improves cardiac dysfunction in models of heart failure (52). These findings position SIRT1 as a
promising therapeutic target for heart failure. SIRT3's
demethylation impacts the FoxO family by triggering
autophagy-related proteins, including light chain 3 (LC3) of
microtubule-associated protein 1, and the phosphorylation of BNIP3,
a pro-death regulatory protein (53). An upregulation in SIRT3 expression
stimulates the FoxO3/PINK1/Parkin pathway, enhancing mitochondrial
autophagy and thus providing cardiac protection.
Pulmonary arterial hypertension (PAH) is an
important cause of increased right ventricular loading and the
eventual development of right heart failure (54). Valproic acid (VPA), an HDAC
inhibitor targeting Class I enzymes, has demonstrated therapeutic
potential in preclinical models of PHA (55). CS1, an oral sustained-release
formulation of sodium valproate, is currently under investigation
for PAH management. Following oral administration, the compound
undergoes intestinal conversion to its active metabolite, VPA,
which mediates its pharmacological effects. A recent Phase II
randomized clinical trial (NCT05224531) has been designed to
evaluate the safety and exploratory efficacy of CS1, an HDAC
inhibitor, in patients with PAH. This prospective, open-label,
blinded-endpoint (PROBE-design) trial enrolled 30 patients with
PAH, randomized into three dose cohorts: 480, 960 and 1,920 mg/day,
with a target of 10 participants per cohort. CS1 was administered
as controlled-release capsules (160 mg sodium valproate per
capsule), with dose escalation guided by tolerability. The highest
dose (1,920 mg/day) was selected based on effective dose ranges
derived from animal models (equivalent human dose: 1,120-3,430
mg/day) and safety data from a Phase I trial in healthy volunteers
(552 mg/day significantly reduced PAI-1 levels). The study
incorporated continuous ambulatory monitoring of mean pulmonary
arterial pressure via the CardioMEMS HF System, alongside
right heart catheterization, cardiac magnetic resonance imaging
(MRI), and biomarker profiling to enable high-resolution clinical
phenotyping (56). Although
results remain unpublished, this trial provides a scalable model
for evaluating HDAC inhibitors in heart failure through its
innovative integration of dynamic hemodynamic monitoring and
multimodal assessments.
Beyond individual modifications, the functional
antagonism within HDAC subfamilies underscores the complexity of
epigenetic regulation in heart failure. For example, while Class I
HDACs (such as HDAC1/3) promote fibrosis by suppressing
antifibrotic genes, Class III HDACs (SIRT1/3) enhance mitochondrial
autophagy and calcium handling. Such divergence necessitates
subtype-specific interventions. Similarly, methylation dynamics
reveal a paradoxical role: H3K27me3-mediated silencing of
cardioprotective Klotho contrasts with H3K4me3-driven activation of
pro-fibrotic TGF-β/Smad pathways. These findings advocate for
combinatorial strategies [simultaneously inhibiting PRC2 (to
relieve H3K27me3 repression) and enhancing H3K4me3 erasure] to
restore transcriptional balance.
Histone modifications and MI
MI serves as a significant contributor to
cardiovascular disease, exhibiting the highest rates of mortality
and morbidity globally. It is typically induced by the occlusion or
stenosis of coronary arteries, resulting from thrombosis linked to
the degradation of atherosclerotic plaques. This condition is
marked by an inflammatory response, detrimental ventricular
remodeling, fibrosis and oxidative stress. Histone methylation is
important in the pathogenesis of MI. Previous studies have shown
that H3K9 methylation serves a role in cardiac hypertrophy and
fibrosis by influencing free radical production (57,58).
A previous study further confirmed that increased H3K9me2 levels
can exacerbate negative ventricular remodeling after MI (59). Yang et al (60) found that the absence or functional
inhibition of SUV39H1 can reduce the damage caused by myocardial
ischemia, limit the scope of MI, improve the survival rate of mice
with MI, reduce the death of cardiomyocytes, and improve left
ventricular function in a SIRT1-dependent manner. Molecularly,
H3K9me3 is methylated by SUV39H1 and recruited to the promoter
region of SIRT1. By silencing or inhibiting SUV39H1, H3K9me3 levels
on SIRT1 promoters can be reduced, thereby preventing excessive
accumulation of intracellular ROS. Chaetocin is a small molecule
naturally derived from the metabolites of the marine fungus genus
Chaetomium, which primarily functions as an inhibitor of the
enzymes SUV39H1 and G9a. Previous research indicates that Chaetocin
exhibits multiple pharmacological functions in the context of
cancer, bacterial and viral infections, as well as cardiovascular
diseases. It achieves this by inhibiting processes such as
apoptosis, oxidative stress, autophagy and angiogenesis (61), suggesting that Chaetocin may
regulate histone methylation and oxidative stress. It has potential
application prospects in the treatment of MI. Emerging evidence
highlights the therapeutic potential of chaetocin in ischemic
pathologies. Schweizer et al (62) demonstrated that the pharmacological
application of chaetocin confers neuroprotection in cellular models
of cerebral ischemia, concomitant with enhanced histone H3K9
acetylation at BDNF promoter regions, which mechanistically
underlies the upregulation of neurotrophin transcription. This
epigenetic modulation aligns with findings by Yang et al
(60), wherein chaetocin-mediated
cardioprotection against ischemic injury was attributed to SIRT1
transcriptional activation. Collectively, these studies suggest
that chaetocin may functionally mimic SUV39H1 depletion in
ischemia-compromised cells, implicating its role as a multimodal
epigenetic modulator in hypoxic-ischemic disorders. DYRK1A, a
protein kinase that has remained conserved throughout evolution,
can regulate the proliferation of a wide range of cell types,
including neoplastic mouse tumor cells, neural precursor cells,
pancreatic islet β-cells and cardiomyocytes (63). Research indicates that the
overexpression of DYRK1A can disrupt the normal cell cycle of
cardiac myocytes, potentially leading to dilated cardiomyopathy.
This condition is linked to congestive heart failure and may result
in premature mortality in neonatal mice. These results suggest that
DYRK1A may be a potent regulator of cell proliferation, even when
the cell type is resistant to the stimulation of cell division or
prone to excessive proliferation (64).
Recent studies have found that DYRK1A may be a
potential target for promoting the cyclical circulation of
cardiomyocytes and the self-repair of the heart, especially in the
case of MI (65,66). It was revealed that DYRK1A, a
protein kinase, regulates cardiomyocyte cell cycle activation by
inhibiting the deposition of histone modifications (H3K4me3 and
H3K27ac) on the promoters of cell cycle genes; it does this by
phosphorylating two interacting proteins, WDR82 and KAT6A, which
are key players in histone acetylation and methylation, thereby
limiting their transcriptional activity and suppressing cell cycle
gene expression. This finding is significant in the field of
translational medicine because harmaline, a commonly used DYRK1A
inhibitor, has demonstrated the ability to pharmacologically
inhibit DYRK1A. This inhibition subsequently facilitates cardiac
repair following a MI. In addition, complementary experiments were
conducted using inhibitors that phosphorylated WDR82 or
phosphorylated KAT6A to verify the role of phosphorylation of WDR82
and KAT6A in DYRK1A-mediated cardiomyocyte cycle and cardiac
repair. The experimental results confirmed that WDR82 and KAT6A are
key factors in the epigenetic marks H3K4me3 and H3K27ac, which are
essential for the transcriptome that promotes proliferation and
activation of the cardiomyocyte cycle (65).
The functional significance of histone lactylation
during MI recovery remains to be fully characterized. Emerging
evidence from murine MI models indicates that circulating monocytes
exhibit rapid induction of tissue-reparative transcriptional
programs, accompanied by elevated histone lactylation levels during
the acute injury phase, a modification that directly coordinates
the expression of reparative mediators including LRG1, VEGF-A and
IL-10. These lactylation-driven molecular cascades establish a
cardioprotective microenvironment by exhibiting both
anti-inflammatory and angiogenic properties. Notably, this
epigenetic modification attenuated pathological inflammatory
responses while enhancing cardiac functional recovery in post-MI
hearts. Mechanistically, monocyte metabolic rewiring characterized
by glycolytic flux dysregulation and MCT1-dependent lactate
shuttling was identified as a critical driver of lactylation
dynamics. Furthermore, IL-1β signaling was found to orchestrate
lactylation patterns through GCN5 recruitment, partially mediating
the activation of tissue-restorative genetic networks.
Collectively, these insights position histone lactylation as both a
biomarker and a tunable epigenetic mechanism governing
post-ischemic myocardial repair, thereby offering translational
opportunities for targeted cardiac regeneration strategies
(67).
Trans-differentiation of cardiac fibroblasts into
functional cardiomyocytes has previously been recognized as an
innovative therapeutic strategy to repair and rejuvenate damaged
myocardial tissue following injury. Previous studies have revealed
a functional interplay between H3K27me3 dynamics and a miR
combination (comprising miR-1, miR-133, miR-208 and miR-499) in
driving direct reprogramming of cardiac fibroblasts into
cardiomyocytes, offering a novel therapeutic avenue for MI
(68). The miR combo remodels
chromatin states through dual epigenetic modulation: It
downregulates the H3K27 methyltransferase Ezh2 (a PRC2 complex
component), reducing the deposition of the repressive H3K27me3 mark
at cardiac gene promoters, while simultaneously upregulating the
expression of demethylases Kdm6A/B to further erase H3K27me3 and
activate cardiomyocyte-specific genes (such as those regulated by
enhancer binding and transcription factor interactions) (69,70).
Experiments demonstrated that knockdown of Kdm6A/B or
pharmacological inhibition of Ezh2 activity (for instance, using
3-Deazaneplanocin A) could either reverse or mimic the
reprogramming effects of miR combo, underscoring the central role
of H3K27me3 homeostasis (70,71).
This ‘epigenetic-miR synergy’ bypasses transcription
factor-dependent genetic manipulation, significantly enhancing
reprogramming efficiency and safety, and highlights a promising
strategy for in situ cardiac repair and functional recovery
post-MI.
During cardiac repair after MI, there are critical
regulatory interactions between miRs and histone demethylases. For
instance, Kdm3a, an epigenetic modifier that specifically removes
H3K9 monomethylation/dimethylation (H3K9me1/2), is directly
suppressed by miR-22-3p. Intriguingly, the long non-coding RNA H19
competitively binds miR-22-3p, thereby indirectly upregulating
Kdm3a expression. Experimental evidence from animal models
demonstrates that overexpression of H19 via adenoviral vectors
prior to MI significantly reduces infarct size, attenuates fibrosis
and inflammatory responses, and improves cardiac contractile
function (72). Further
investigations confirm that either knockdown of miR-22-3p using
gene editing techniques or direct overexpression of Kdm3a enhances
cardiomyocyte survival and ameliorates post-injury cardiac
phenotypes (72). This regulatory
network underscores the synergistic mechanisms by which non-coding
RNAs and histone-modifying enzymes coordinate multidimensional
epigenetic regulation, offering novel therapeutic insights for
targeted MI intervention.
Histone modifications exhibit distinct roles in
cardiovascular pathologies: H3K9me2/3 exacerbates ventricular
remodeling through SUV39H1-driven ROS accumulation, whereas
H3K27me3 dynamics, regulated by Ezh2/Kdm6A/B alongside miR combos,
enable fibroblast-to-cardiomyocyte reprogramming. Lactylation
promotes reparative angiogenesis by upregulating VEGF-A expression.
The DYRK1A-WDR82/KAT6A axis drives cardiomyocyte regeneration by
dynamically balancing H3K4me3 and H3K27ac levels.
Pharmacologically, Chaetocin, a selective SUV39H1 inhibitor, and
harmaline, targeting DYRK1A, demonstrate therapeutic potential, but
studies on specificity and off-target risk remain scarce. These
mechanisms underscore context-dependent interplay among oxidative
stress, cell cycle control and inflammation resolution, positioning
epigenetic modifiers as precision targets for myocardial
repair.
Histone modifications and cardiac
hypertrophy
Cardiac hypertrophy, often stemming from
hypertension or valve diseases, is associated with specific histone
modifications that impact cardiomyocyte growth. This condition
ultimately leads to decreased cardiac output, increased heart
failure risk, and the potential for heart failure to develop
(73).
Ubiquitination, regulated by ligases and DUBs, is a
critical modifier in the pathogenesis of cardiac hypertrophy
(74). This process is tightly
controlled by specific enzymes called E3 ubiquitin ligases and DUBs
within the ubiquitin-proteasome pathway which are implicated in
cardiovascular diseases (6). For
example, ubiquitin-specific protease 18 (USP18) exerts a mitigating
effect on cardiac hypertrophy by exclusively removing the K63-type
ubiquitin chain on TAK1, which leads to inhibition of the
TAK1-p38/JNK1/2 signaling pathway (75,76).
In addition, calmodulin phosphatase is a protein associated with
promoting cardiac hypertrophy, while atrogin-1 acts as a
countermeasure by forming a complex called ‘SCFatrogin-1’ which,
through its E3 ubiquitin ligase activity (by interacting with Skp1,
Cul1 and Roc1), can target and degrade proteins such as calmodulin
phosphatase, thereby inhibiting cardiac hypertrophy. When the
expression level of atrogin-1 is reduced, it enhances
agonist-triggered calmodulin phosphatase activity, which in turn
leads to cardiomyocyte hypertrophy. It has been shown that the
SCFatrogin-1 complex serves a crucial role in the regulation and
inhibition of calmodulin phosphatase activity through the
ubiquitination-dependent protein degradation pathway. Thus,
atrogin-1 acts as a negative regulator of calmodulin phosphatase
and ultimately inhibits the cardiac response to pathological
stimuli, thereby attenuating symptoms of cardiac hypertrophy.
The WW structural domain E3 ubiquitin ligase 1
(WWP1) serves a key role in a variety of age-related diseases,
including cardiovascular disease and cancer. According to research,
the expression level of WWP1 is notably elevated in cardiac tissue
samples from patients with heart failure, as well as in animal
models where cardiac hypertrophy is induced by transverse aortic
constriction (TAC), indicating a potential role of WWP1 in the
development of pathological cardiac remodeling associated with
heart failure. Notably, TAC-induced cardiac hypertrophy could be
inhibited by interfering with the interaction of WWP1 with DVL2
protein. Thus, WWP1 may be a potential therapeutic target for the
treatment of cardiac hypertrophy and heart failure (77). In addition, previous studies have
shown that muscle-specific ring finger protein 1 (MuRF1), an E3
ubiquitin ligase, can attenuate pathological cardiac hypertrophy by
promoting the degradation of calmodulin phosphatase A (6,78).
These observations underscore the significant role of
ubiquitination in the progression of cardiac hypertrophy and
highlight the potential for developing anti-hypertrophic
medications that target E3 ligases and deubiquitinating
enzymes.
Sumoylation is a PTM where small ubiquitin-like
modifier (SUMO) proteins are attached to lysine residues on target
proteins, including histones, with the help of a cascade involving
E1 activating, E2 conjugating and E3 ligase enzymes, effectively
altering the function of the modified protein. This process is
closely related to ubiquitination and has a decisive impact on the
regulation of cardiac gene expression and cardiac development,
including the differentiation process of cardiomyocytes. Previous
studies have revealed the key role of SUMOisation in the
maintenance of cardiac function, in particular its protective role
in the heart's response to stress. UBC9 upregulation of the SUMO E2
enzyme enhances protein quality control in the heart and promotes
higher levels of autophagy, suggesting that increasing
UBC9-mediated SUMO may be a novel strategy to treat heart disease,
improve heart function, and increase survival (79). In addition, SUMO-1-mediated
SUMOylation of heat shock factor 2 (HSF2) has been shown to
attenuate cardiac hypertrophy. In the presence of Ang II, increased
expression of MEL-18 leads to de-SUMOisation of HSF2, which in turn
increases the expression of IGF-IIR, thereby triggering cardiac
hypertrophy (80). The COP9
signaling complex (CSN) modulates the function of cullin-RING
ligases, with CSN8 being a crucial subunit of the CSN complex. This
complex is integral to numerous biological processes, including the
process of demethylation. Deletion of CSN8 not only disrupts the
assembly of the CSN complex but also leads to an increase in the
level of ubiquitylation of the cullin proteins, which in turn
induces cardiac hypertrophy. In addition, CSN8 deficiency affects
the function of the ubiquitin-proteasome system, leading to
myocardial necrosis. Elevated activity of ZAK, a kinase with mixed
specificity, may facilitate the development of cardiac hypertrophy.
Estrogen receptor β has the capacity to interact with ZAK,
inhibiting its accumulation within the nucleus by blocking SUMO-1
modification. This interaction results in a reduction of ZAK
protein levels, thereby producing an antihypertrophic effect
(81).
Previous findings indicate that the re-expression of
the Scn5a gene in denervated skeletal muscle shares similar
molecular mechanisms with those that drive Scn5a transcription in
cardiac tissue. Experiments using ChIP-qPCR have demonstrated that
the re-expression of Scn5a correlates with increased levels of
H3K4me3 and H3K27ac histone marks, as well as the binding of the
transcription factor Gata4 to the gene's promoter region. Notably,
ChIP-seq analysis of H3K27ac has revealed that denervation
activates a super enhancer previously identified as regulating
Scn5a expression in cardiac tissue. The aforementioned data suggest
that Gata4 serves a significant role in the transcriptional
activation of the Scn5a gene in denervated muscle, as evidenced by
a substantial increase in Gata4 expression observed through RNA-seq
analysis (82). Gata4, a
zinc-finger transcription factor belonging to the Gata family, is
extensively expressed in both developing and mature cardiac tissue.
It serves as a key regulator of transcriptional networks and is
essential for cardiac differentiation and morphogenesis (83). Studies have shown that mice lacking
Gata4 exhibit severe cardiac defects leading to embryonic lethality
(84) while genetic mutations
affecting GATA4 activity are linked to various cardiac
abnormalities, such as right ventricular hypoplasia and
cardiomyopathy (85). Moreover,
Gata4+/− mice have been reported to exhibit shortened PR
intervals, highlighting the critical role of Gata4 in the
development of the atrioventricular cardiac conduction system
(86). Mechanistically, Gata4
interacts synergistically with other transcription factors,
including Nkx2-5, TBX5, and MEF2, to regulate gene expression.
ChIP-seq studies have identified the co-localization of these
factors with the HAT KAT3B at cardiac enhancers, revealing that
Gata4 promotes gene activation through the deposition of H3K27ac
(87-89).
Hypertrophic cardiomyopathy (HCM) is a genetically
diverse condition primarily linked to mutations in
sarcomere-related genes, leading to pathological thickening of the
left ventricular wall, tissue scarring, excessive contractile
activity and impaired diastolic relaxation (90,91).
Previous studies have identified suppressed expression of
short-chain enoyl-CoA hydratase 1 (ECHS1) and elevated histone
crotonylation modifications at H3K18 and H3K12 sites in cardiac
tissues of patients with HCM. Mechanistic investigations
demonstrated that ECHS1 modulates crotonyl-CoA metabolic levels,
orchestrates histone crotonylation dynamics, and regulates the
NFATc3 signaling pathway, thereby contributing to cardiomyocyte
maturation and homeostatic stability. These findings suggest that
targeting epigenetic regulation of histone crotonylation may
represent a promising therapeutic approach for pediatric patients
harboring ECHS1 mutations associated with HCM (92,93).
Ubiquitination is not the only epigenetic driver of
cardiac hypertrophy. Comparative studies highlight SUMOylation as a
counterregulatory mechanism: SUMO1 conjugation to HSF2 attenuates
hypertrophy by blocking IGF-IIR signaling, whereas ubiquitination,
exemplified by the WWP1-DVL2 axis, accelerates pathological
remodeling. This antagonistic interplay underscores the imperative
for selective modulation of distinct PTM pathways. Additionally,
non-canonical modifications such as H3K12/H3K18 crotonylation,
which are regulated by ECHS1 in HCM, reveal metabolic-epigenetic
crosstalk distinct from acetylation. Unlike H3K27ac, which broadly
enhances transcription, crotonylation fine-tunes NFATc3 activation,
urging a re-evaluation of metabolic interventions in
hypertrophy.
Histone modifications and MIRI
Ischemic heart disease (IHD) is considered the most
prevalent form of cardiovascular disease, and it is significantly
linked to the highest rates of morbidity and mortality globally
(94). While reperfusion therapy,
which aims to restore blood flow to the heart during a heart attack
(IHD), is the standard treatment, the process of re-establishing
blood flow can paradoxically cause further damage to the heart
muscle, known as MIRI, due to cellular dysfunction and tissue
damage that occurs when blood flow returns after a period of
ischemia (95). MIRI contributes
to cardiomyocyte apoptosis, myocardial damage, the no-reflow
phenomenon and microvascular endothelial injury, significantly
impacting patient prognosis and increasing follow-up care costs
(96).
Histone modifications significantly impact MIRI
pathology by controlling the expression of genes related to cardiac
damage and the repair mechanisms that follow ischemia/reperfusion
(I/R), essentially acting as an epigenetic regulator that
influences the cellular response to injury and recovery process. A
research investigation has identified epigenetic alterations in
cardiac cells following I/R injury by integrating transcriptomic
and epigenetic information regarding histone modifications.
Particularly at 24 and 48 h after the onset of I/R injury, the
investigators observed disease-associated histone marker changes in
the regions of genes modified by H3K27me3, H3K27ac and H3K4me1.
These differentially modified genes are involved in biological
processes such as immune function, cardiac electrophysiology or
muscle contraction, cytoskeletal structure and vascular neogenesis.
After I/R injury, there is an observed increase in the expression
of H3K27me3 and its associated methyltransferase complex, PRC2,
within the myocardium. However, selective inhibition of EZH2, the
principal catalytic subunit of PRC2, leads to a decrease in
H3K27me3 levels. This intervention results in the activation of
protective gene expression, enhancement of cardiac function,
promotion of neovascularization, and a reduction in fibrosis in
mice. Further studies showed that EZH2 inhibition enhanced
angiogenesis in vitro and in vivo by modulating the
modification of multiple angiogenesis-related genes by
H3K27me3(97). In I/R injury,
lactate accumulation drives pathological angiogenesis through H3K18
lactylation (H3K18la)-mediated activation of VEGF transcriptional
programs. Concurrently, H3K27 trimethylation (H3K27me3) works in
conjunction with DNA hypermethylation to inhibit antioxidant
defense mechanisms, such as the silencing of SOD2, thereby
exacerbating oxidative damage. Dual therapeutic strategies
targeting both lactate metabolic pathways (via LDH-A inhibition)
and epigenetic modifiers such as EZH2 inhibitor may synergistically
mitigate these maladaptive responses (98,99).
However, research has indicated that the use of GSK126, an EZH2
inhibitor, is associated with elevated arterial rigidity and
breakdown of elastin fibers, suggesting potential negative impacts
on cardiovascular health (100).
Lysine-specific methyltransferase 2B (KMT2B),
commonly referred to as MLL2, is a substantial protein composed of
2,715 amino acids. Its primary role is to facilitate the
trimethylation modification (specifically, H3K4me3) of the lysine 4
residue on histone H3 within the promoter regions of specific genes
(101). The KMT2 family
encompasses the H3K4 methyltransferases found in six mammalian
species, including KMT2A,KMT2B, KMT2C, KMT2D, KMT2F and KMT2G.
These KMT2 family members regulate the transcriptional activity of
target genes through H3K4 methylation. Nonetheless, the role of
KMT2B in regulating histone modifications, especially in terms of
its association with abnormal responses in iron metabolism, has
been relatively limited.
Iron death, also known as ferroptosis, is a specific
form of cell death heavily reliant on iron and characterized by
excessive lipid peroxidation, which is considered a key contributor
to I/R injury and subsequent organ failure due to the uncontrolled
oxidative damage it causes when blood flow is restored to
previously ischemic tissue; this process involves the release of
free iron, which can trigger a chain reaction of lipid peroxidation
leading to cell death. Iron death, through the modulation of
glutathione peroxidase activity, whether directly or indirectly,
results in the peroxidation of cellular membrane lipids. This
phenomenon is attributed to a disturbance in intracellular redox
homeostasis and an excessive generation of ROS, ultimately
culminating in the disruption of the cell membrane (102). This finding provides a new
strategy for the prevention of MIRI. The aforementioned study
demonstrated that reducing the expression of KMT2B can attenuate
the injury of cardiomyocytes under hypoxia/reoxygenation (H/R)
conditions and reduce the occurrence of iron death, which can
reduce the area of MI in MIRI rats. In addition, the study revealed
the potential mechanism by which KMT2B affects iron death. KMT2B
was able to recruit H3K4me3 in the promoter region of the RFK gene
to promote the transcription of the RFK gene. The RFK protein
further interacted with the p22 subunit of the NOX2 complex and
with FOX and TRADD (103).
In addition, KMT2B has been recognized as an
activator of the RFK gene through H3K4me3. Studies have noted
increased RFK levels in individuals experiencing acute MI (AMI).
The overexpression of RFK counteracts the suppressive impact of
KMT2B reduction on ROS generation and iron death during H/R. RFK
can interact with TRADD and p22phox. Moreover, blocking TNF-α has
been observed to enhance MIRI by elevating mouse lipocalin levels
(104). The TNF-α antagonist ETA
mitigates MIRI, reduces infarct size, and enhances cardiac
performance by inhibiting the overproduction of gp91phox, another
NOX2 subunit, and superoxide (105). A decrease in NOX2 or NOX4 levels
significantly reverses TNF-α-induced ROS overproduction and
decreases IL-1β and IL-6 accumulation in cardiomyocytes (106). Consistent with multiple studies,
the NOX2 inhibitor apocynin alleviates H/R-induced ROS accumulation
and the release of pro-inflammatory cytokines, thus preventing iron
death (107,108). In vivo studies have shown
that NOX2 inhibitors reduce MIRI and infarct size when KMT2B is
present, which is partially consistent with the results presented
by Wang et al (109).
Their study indicated that the NOX2 inhibitor Vas2870 or NOX2
silencing via small interfering RNA improved MIRI in diabetic rats
by inhibiting oxidative stress, apoptosis, pyroptosis and iron
death in cardiomyocytes under high-glucose conditions. Another
study found that NOX2 expression is significantly increased in
cardiomyocytes following AMI and its downregulation reduces
superoxide production and apoptosis in these cells. Pharmacological
interventions that partially inhibit ROS production due to NOX2
deficiency have shown potential for post-I/R recovery, suggesting a
therapeutic strategy for MIRI (110,111). These results suggest that KMT2B
may promote iron death by activating the TNF-α/RFK/NOX2 signaling
pathway.
Research suggests that Lysine-specific demethylase 1
(LSD1) can interact with autophagy-related 16-like 1 protein,
serving a role in the cellular response to H/R conditions, which
can lead to apoptosis (programmed cell death). This interaction can
directly influence SETD7 and LSD1, reversing the apoptotic process
and mitigating myocardial damage caused by H/R (112). In a mouse cardiomyocyte cell line
that underwent H/R treatment, a decrease in LSD1 protein expression
was observed. The study revealed that elevated levels of LSD1
expression can suppress the transcriptional activity of the SOX9
gene by decreasing the H3K4 trimethylation (H3K4me3) in the
promoter region of the SOX9 gene. This mechanism serves a role in
mitigating apoptosis triggered by H/R conditions in cardiomyocytes
(113). Furthermore, other key
members of the KDM family closely associated with cardiovascular
functions predominantly belong to the JmjC domain family (114). Research has shown that KDM6A
expression is upregulated in rat models of AMI and hypoxia-induced
cardiomyocytes. The lack of KDM6A results in elevated H3K27
trimethylation at the Ncx gene promoter, which subsequently results
in decreased Ncx expression and a decrease in calcium influx into
cardiac muscle cells (115). This
indicates that members of the JmjC family may serve as new
regulatory factors in the protection of ischemic
cardiomyopathy.
In addition to methylation and lactylation,
acetylation is also strongly associated with cardiac I/R injury.
Whether HDAC inhibitors exert cardioprotective effects by limiting
infarct size in cardiac IR injury has previously been investigated.
Available studies suggest that the core therapeutic window for the
selective HDAC6 inhibitor ACY1215 corresponds to the reperfusion
phase, particularly during the peak oxidative stress and
inflammatory response in the early phase of reperfusion. Modulation
of ROS-related pathways, peroxiredoxin 1, and hypoxia-inducible
factor-1α (HIF-1α) signaling through HDAC6 inhibition may represent
a potential strategy to attenuate reperfusion injury. However, the
specific role of HIF-1α and the optimal timing for administering
HDAC6 inhibitors still require thorough validation (116).
The competing roles of methylation marks in MIRI
underscore the challenge of selective targeting. EZH2 inhibitors,
such as GSK126 enhance angiogenesis but risk vascular stiffness,
whereas KMT2B knockdown reduces iron death but may impair other
H3K4me3-dependent pathways. By contrast, lactylation and
acetylation modifiers operate within metabolic constraints:
Lactate-driven H3K18la requires glycolytic activity, while HDAC6
inhibitors like ACY1215 depend on HIF-1α signaling. This metabolic
contextuality necessitates patient stratification; for instance,
diabetic patients with impaired glycolysis may benefit less from
lactylation-focused therapies.
Histone modifications as biomarkers in
cardiovascular diseases
The dynamic and disease-specific nature of histone
modifications has shown great potential as biomarkers in the
diagnosis, prognostic assessment and efficacy monitoring of
cardiovascular diseases. For instance, elevated plasma homocysteine
levels have been shown to enhance abnormal DNA methylation and
upregulate the expression of N-methyl-D-aspartate receptor-1
(NMDAR1), DNA (cytosine-5)-methyltransferase-1 (DNMT1), and matrix
metalloproteinase-9 (MMP-9). This regulatory mechanism is driven by
H3K9 acetylation and suppression of HDAC1. These proteins (NMDAR1,
DNMT1 and MMP-9) serve as indicative markers for heart failure,
with experimental evidence from cardiomyocyte studies confirming
their direct association with disease pathogenesis (117).
Previous studies have identified plasma levels of
S-adenosylhomocysteine (SAH) as a key biomarker of As, and its
concentration is significantly and positively associated with
atherosclerotic plaque volume and the pathological course of
hyper-homo-cysteinemia. Research reveals that excessive SAH
accumulation disrupts epigenetic regulation by suppressing histone
H3 lysine 9 trimethylation (H3K9me3), thereby markedly upregulating
endoplasmic reticulum (ER) stress markers, including
glucose-regulated protein 78 and C/EBP homologous protein. This
mechanistic pathway underscores the pivotal role of SAH in driving
As development through ER dysfunction, solidifying its utility as a
biomarker for disease diagnosis and progression monitoring
(118).
Super enhancers (SEs) are emerging as pivotal
biomarkers for cardiac remodeling following MI. Integrative
analysis of SEs and RNA sequencing (RNA-seq) data identified 76
differentially expressed genes (DEGs) linked to H3K27ac-enriched
SEs in MI, with functional enrichment primarily in
angiogenesis-related pathways. Notably, SEs unassociated with DEGs
showed significant involvement in actin filament dynamics and cell
migration. These SE-regulated genes may maintain a
transcriptionally poised state during early MI and contribute to
pathological cardiac remodeling in later disease stages (119).
Similarly, ATP2A2/SERCA2a, a cardiomyocyte-specific
calcium transporter critical for regulating myocardial contraction
and relaxation, has emerged as a potential biomarker for HCM.
Integrated multi-omics analyses, encompassing ChIP-seq, RNA-seq and
proteomics, revealed significantly reduced mRNA and protein
expression levels of ATP2A2/SERCA2a in HCM cardiac tissues compared
with controls. This suppression was further validated in
hiPSC-derived cardiomyocyte models. The observed downregulation
strongly correlates with hallmark HCM pathological features, such
as diastolic dysfunction, specifically impaired myocardial
relaxation, indicating its role as a molecular indicator of disease
progression (120).
In summary, methylation, acetylation,
ubiquitination, crotonylation and lactylation modifications of
histones can regulate a wide range of genes in patients with
cardiovascular disease, with different modifications or levels of
modification exhibiting different associated effects.
4. Conclusions and prospects
Cardiovascular diseases are predominantly chronic
conditions requiring long-term management, such as hypertension,
coronary heart disease and heart failure. Patients are often
required to adhere to prolonged medication regimens, as
pharmacological treatments can only manage the disease's
progression and have limited efficacy and numerous adverse effects.
However, advancements in high-throughput sequencing have
revolutionized traditional disease research paradigms through the
field of epigenetics. New research has uncovered a substantial link
between cardiovascular ailments and their associated risk factors
with alterations in histone proteins.
Histone modifications serve a critical role in the
development and progression of various cardiovascular diseases,
particularly histone acetylation and methylation, making them
potential therapeutic targets due to their ability to regulate gene
expression and cellular function within the cardiovascular system.
These modifications regulate gene expression, influencing processes
such as cardiomyocyte apoptosis, proliferation, inflammatory
responses, oxidative stress and fibrosis. They are also intricately
associated with cardiac remodeling, inflammation and vascular
function. Notably, the modification states of histones H3K9 and
H3K27 correlate significantly with the onset and progression of
cardiovascular diseases. These findings offer new insights into the
molecular mechanisms underlying cardiovascular conditions.
Despite the progress made in understanding the
relationship between histone modifications and cardiovascular
diseases, several limitations persist. First, most studies have
primarily focused on specific histone modifications, such as
acetylation and methylation, while research on other crucial
changes, including phosphorylation and ubiquitination, remains
limited. This narrow focus hampers a comprehensive understanding of
the overall network of histone modifications. Moreover, there is a
scarcity of relevant literature providing detailed statistics on
the prevalence and incidence of specific histone modifications in
cardiovascular diseases, which limits our understanding of their
epidemiological significance. Second, current research
predominantly relies on animal models or in vitro cellular
experiments, with few clinical studies validating histone-modifying
drugs' therapeutic efficacy and safety in human cardiovascular
diseases. For instance, while preclinical studies highlight the
potential of HDAC inhibitors such as SAHA, EZH2 inhibitors such as
GSK126, and SUV39H1 inhibitors such as Chaetocin in attenuating As,
MI, or hypertrophy, these findings have yet to be translated into
clinical trials targeting cardiovascular indications (100,121). However, the Phase II trial of CS1
(NCT05224531), a sustained-release HDAC inhibitor for PAH, has
completed enrollment and preliminary evaluations (56), but clinical results are still not
available. This lack of transparency hampers the translation of
preclinical epigenetic discoveries into validated therapeutic
strategies. Furthermore, the intricate dynamics of histone
modifications and their multifaceted interactions with other
epigenetic mechanisms remain inadequately investigated, which
complicates the accurate evaluation of their specific contributions
to cardiovascular diseases.
Future research should focus on the dynamic changes
in histone modifications and their interactions with other
epigenetic mechanisms, such as non-coding RNAs and DNA methylation.
This focus holds vast potential for applications in diagnosing and
treating cardiovascular diseases. For example, the hematopoietic
DNA demethylase TET2 has been shown by Walsh's and Ebert's
laboratories (122,123), respectively, to prevent As by
inhibiting the increased levels of gene expression of
pro-inflammatory cytokines and chemokines as well as the activation
of inflammatory vesicles. In addition, the expression level of
miR-1 in non-coding RNAs directly affects cardiac function. Studies
have shown that miR-1 can mitigate the pathological effects of
cardiomyopathy by regulating cardiomyocyte transformation while
inhibiting cell reproduction and growth (124,125). In addition, circRNA CDR1as could
enhance HDAC4 expression and promote cardiac hypertrophy by
adsorbing miR-7, whereas lactate-mediated lactylation could modify
circRNA-binding proteins to form a metabolic-epistatic-noncoding
RNA interaction axis (98,126). A critical yet understudied aspect
is the therapeutic time window of histone-modifying agents. For
example, while SAHA's anti-inflammatory effects may be most
effective in early As, Chaetocin's ability to suppress H3K9me3
could target advanced ischemic injury. Systematic evaluation of
these agents across distinct disease stages-including acute injury,
chronic progression, and tissue repair phases-is imperative to
establish precision therapeutic regimens. Emerging technologies,
such as optical imaging, are revolutionizing our ability to
visualize epigenetic dynamics in vivo. For example,
activatable two-photon fluorescent probes (127) applied in HeLa cells were able to
dynamically monitor HDAC activity at cellular resolution. This
finding provides a potential technological crossover and future
research direction for cardiovascular disease research.
Advancements in these areas could significantly reduce mortality
rates among cardiovascular patients, enhance the quality of life
for a lot of individuals, and propel research and clinical
applications in the field forward. Additionally, the gap in
clinical research underscores a critical bottleneck in
translational research. Off-target effects and long-term safety
profiles of epigenetic drugs, which are well-documented in oncology
(128), require rigorous
evaluation in cardiovascular cohorts. For example, EZH2 inhibitors
such as GSK126, though promising in preclinical models for
enhancing post-ischemic angiogenesis, may pose risks such as
vascular stiffness, as observed in experimental settings (100). Thus, future clinical studies must
prioritize dose optimization, patient stratification, and
comprehensive monitoring of adverse effects to ensure therapeutic
viability. By integrating clinical studies with foundational
science, the authors anticipate uncovering the potential
applications of histone modifications in cardiovascular diseases.
This approach aims to offer new perspectives for early diagnosis
and personalized treatment, ultimately contributing significantly
to the understanding and management of human cardiovascular
conditions.
Acknowledgements
Not applicable.
Funding
Funding: The present project was supported by the National
Natural Science Foundation of China (grant nos. 82305092,
82274411), the National Natural Science Foundation of Hunan (grant
no. 2023JJ30453), Hunan Youth Science and Technology Innovation
Talent Project (grant no. 2024RC3199). Excellent Young Research
Fund Project of Hunan University of Chinese Medicine (grant no.
Z2023XJYQ03), Undergraduate Research Innovation Foundation of Hunan
University of Chinese Medicine (grant no. 2023BKS017), Hunan
University of Chinese Medicine Entrepreneurship Training Program
(grant no. 186) and the Research Foundation of Hunan Provincial
Department of Education (grant no. 23C0162).
Availability of data and materials
Not applicable.
Authors' contributions
QQ revised the manuscript regarding intellectual
content. YDZ performed literature research. LL performed data
analysis. QQ and HL were responsible for the editing of the
manuscript. YDZ revised and validated the manuscript. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Feinberg AP: The key role of epigenetics
in human disease prevention and mitigation. N Engl J Med.
378:1323–1334. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhou BR and Bai Y: Chromatin structures
condensed by linker histones. Essays Biochem. 63:75–87.
2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ke L: Single molecule study of DNA and
nucleosome complexes. Journal 2022.
|
|
4
|
Hyun K, Jeon J, Park K and Kim J: Writing,
erasing and reading histone lysine methylations. Exp Mol Med.
49(e324)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wang K, Li Y, Qiang T, Chen J and Wang X:
Role of epigenetic regulation in myocardial ischemia/reperfusion
injury. Pharmacol Res. 170(105743)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gupta I, Varshney NK and Khan S: Emergence
of members of TRAF and DUB of ubiquitin proteasome system in the
regulation of hypertrophic cardiomyopathy. Front Genet.
9(336)2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
He B, Zhao YC, Gao LC, Ying XY, Xu LW, Su
YY, Ji QQ, Lin N and Pu J: Ubiquitin-specific protease 4 is an
endogenous negative regulator of pathological cardiac hypertrophy.
Hypertension. 67:1237–1248. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yan K, Ponnusamy M, Xin Y, Wang Q, Li P
and Wang K: The role of K63-linked polyubiquitination in cardiac
hypertrophy. J Cell Mol Med. 22:4558–4567. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kim U and Lee DS: Epigenetic regulations
in mammalian cells: Roles and profiling techniques. Mol Cells.
46:86–98. 2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bannister AJ and Kouzarides T: Regulation
of chromatin by histone modifications. Cell Res. 21:381–395.
2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cheng X and Wang K, Zhao Y and Wang K:
Research progress on post-translational modification of proteins
and cardiovascular diseases. Cell Death Discov.
9(275)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tan M, Luo H, Lee S, Jin F, Yang JS,
Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al:
Identification of 67 histone marks and histone lysine crotonylation
as a new type of histone modification. Cell. 146:1016–1028.
2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Soler-Botija C, Gálvez-Montón C and
Bayés-Genís A: Epigenetic biomarkers in cardiovascular diseases.
Front Genet. 10(950)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shu F, Xiao H, Li QN, Ren XS, Liu ZG, Hu
BW, Wang HS, Wang H and Jiang GM: Epigenetic and post-translational
modifications in autophagy: biological functions and therapeutic
targets. Signal Transduct Target Ther. 8(32)2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Vlad ML, Manea SA, Iazar AG, Raicu M,
Muresian H, Simionescu M and Manea A: Histone
acetyltransferase-dependent pathways mediate upregulation of nadph
oxidase 5 in human macrophages under inflammatory conditions: A
potential mechanism of reactive oxygen species overproduction in
atherosclerosis. Oxid Med Cell Longev. 2019(3201062)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chan SH, Hung CH, Shih JY, Chu PM, Cheng
YH, Lin HC, Hsieh PL and Tsai KL: Exercise intervention attenuates
hyperhomocysteinemia-induced aortic endothelial oxidative injury by
regulating SIRT1 through mitigating NADPH oxidase/LOX-1 signaling.
Redox Biol. 14:116–125. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lijuan L: Discovery of microbially derived
KLF2 small molecule up-regulation Study on the effect of
anti-atherosclerosis. Journal 2023.
|
|
19
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: Development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Xu Y, Xu S, Liu P, Koroleva M, Zhang S, Si
S and Jin ZG: Suberanilohydroxamic acid as a pharmacological
kruppel-like factor 2 activator that represses vascular
inflammation and atherosclerosis. J Am Heart Assoc.
6(e007134)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
SenBanerjee S, Lin Z, Atkins GB, Greif DM,
Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, et
al: KLF2 is a novel transcriptional regulator of endothelial
proinflammatory activation. J Exp Med. 199:1305–1315.
2004.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu M, Kluger MS, D'Alessio A,
García-Cardeña G and Pober JS: Regulation of arterial-venous
differences in tumor necrosis factor responsiveness of endothelial
cells by anatomic context. Am J Pathol. 172:1088–1099.
2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bu DX, Griffin G and Lichtman AH:
Mechanisms for the anti-inflammatory effects of statins. Curr Opin
Lipidol. 22:165–170. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ridker PM and Lüscher TF:
Anti-inflammatory therapies for cardiovascular disease. Eur Heart
J. 35:1782–1791. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sen-Banerjee S, Mir S, Lin Z, Hamik A,
Atkins GB, Das H, Banerjee P, Kumar A and Jain MK: Kruppel-like
factor 2 as a novel mediator of statin effects in endothelial
cells. Circulation. 112:720–726. 2005.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang J, Xu X, Li P, Zhang B and Zhang J:
HDAC3 protects against atherosclerosis through inhibition of
inflammation via the microRNA-19b/PPARγ/NF-κB axis.
Atherosclerosis. 323:1–12. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jackson AO, Zhang J, Jiang Z and Yin K:
Endothelial-to-mesenchymal transition: A novel therapeutic target
for cardiovascular diseases. Trends Cardiovasc Med. 27:383–393.
2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chen L, Shang C, Wang B, Wang G, Jin Z,
Yao F, Yue Z, Bai L, Wang R, Zhao S, et al: HDAC3 inhibitor
suppresses endothelial-to-mesenchymal transition via modulating
inflammatory response in atherosclerosis. Biochem Pharmacol.
192(114716)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cai L, Ma X, Huang Y, Zou Y and Chen X:
Aberrant histone methylation and the effect of SUV39H1 siRNA on
gastric carcinoma. Oncol Rep. 31:2593–2600. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cherrier T, Suzanne S, Redel L, Calao M,
Marban C, Samah B, Mukerjee R, Schwartz C, Gras G, Sawaya BE, et
al: p21(WAF1) gene promoter is epigenetically silenced by CTIP2 and
SUV39H1. Oncogene. 28:3380–3389. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhang J, Chen J, Yang J, Xu C, Hu Q, Wu H,
Cai W, Guo Q, Gao W, He C, et al: SUV39H1 downregulation inhibits
neointimal hyperplasia after vascular injury. Atherosclerosis.
288:76–84. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Masi S, Ambrosini S, Mohammed SA,
Sciarretta S, Luescher TF, Paneni F and Costantino S: Epigenetic
remodeling in obesity-related vascular disease. Antioxid Redox
Signal. 34:1165–1199. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Costantino S, Paneni F and Cosentino F:
Ageing, metabolism and cardiovascular disease. J Physiol.
594:2061–2073. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Costantino S, Paneni F, Virdis A, Hussain
S, Mohammed SA, Capretti G, Akhmedov A, Dalgaard K, Chiandotto S,
Pospisilik JA, et al: Interplay among H3K9-editing enzymes SUV39H1,
JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative
stress in obesity. Eur Heart J. 40:383–391. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Karnewar S, Neeli PK, Panuganti D,
Kotagiri S, Mallappa S, Jain N, Jerald MK and Kotamraju S:
Metformin regulates mitochondrial biogenesis and senescence through
AMPK mediated H3K79 methylation: Relevance in age-associated
vascular dysfunction. Biochim Biophys Acta. 1864:1115–1128.
2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Greissel A, Culmes M, Napieralski R,
Wagner E, Gebhard H, Schmitt M, Zimmermann A, Eckstein HH, Zernecke
A and Pelisek J: Alternation of histone and DNA methylation in
human atherosclerotic carotid plaques. Thromb Haemost. 114:390–402.
2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Greissel A, Culmes M, Burgkart R,
Zimmermann A, Eckstein HH, Zernecke A and Pelisek J: Histone
acetylation and methylation significantly change with severity of
atherosclerosis in human carotid plaques. Cardiovasc Pathol.
25:79–86. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Huang Z, Song S, Zhang X, Zeng L, Sun A
and Ge J: Metabolic substrates, histone modifications, and heart
failure. Biochim Biophys Acta. 1866(194898)2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jing Y, Li X, Liu Z and Li XD: Roles of
negatively charged histone lysine acylations in regulating
nucleosome structure and dynamics. Front Mol Biosci.
9(899013)2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Papait R and Condorelli G: Epigenetics in
heart failure. Ann N Y Acad Sci. 1188:159–164. 2010.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Chelladurai P, Boucherat O, Stenmark K,
Kracht M, Seeger W, Bauer UM, Bonnet S and Pullamsetti SS:
Targeting histone acetylation in pulmonary hypertension and right
ventricular hypertrophy. Br J Pharmacol. 178:54–71. 2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yan M, Chen C, Gong W, Yin Z, Zhou L,
Chaugai S and Wang DW: miR-21-3p regulates cardiac hypertrophic
response by targeting histone deacetylase-8. Cardiovasc Res.
105:340–352. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Xiao-mei L, Chang P, Shu-qi W, Huan-ting Z
and Xiao-chun T: Role of histone deacetylase 2-mediated histone
acetylation imbalance in myocardial remodeling induced by pressure
overload. Chinese Journal of Pathophysiology. 38:584–591. 2022.
|
|
44
|
Yuhang C, Rui H, Yujun S and Li S:
Myocardial-specific Hdac3 deletion induces by ventricular
remodeling in mice. J Army Med Univ. 40:1205–1212. 2018.(In
Chinese).
|
|
45
|
Wang B, Zhang LD, Zhao QF, Zhu MJ and Wang
XL: Research progress of histone acetylation in prevention and
treatment of heart failure and new ideas based on traditional
Chinese medicine. China J Chinese Materia Medica. 48:2010–2019.
2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mengqing D: Mechanism of peptidase
inhibitory protein PI16 inhibiting angiotensin-ⅱinduced cardiac
hypertrophy and cardiac fibrosis by down-regulating HDAC1. Journal
2019.
|
|
47
|
Jia-pei X and Yu-hua L: Role and mechanism
of histone deacetylase 3 in cardiac fibrosis in mice. Hainan
Medical Journal. 32:2998–3002. 2021.
|
|
48
|
Min Z, Hui T and Zewen C: The role of HDAC
8 in isoprenaline-induced myocardial fibrosis of rat. Acta Univ Med
Anhui. 50:950–953. 2015.(In Chinese).
|
|
49
|
Han Y, Nie J, Wang DW and Ni L: Mechanism
of histone deacetylases in cardiac hypertrophy and its therapeutic
inhibitors. Front Cardiovasc Med. 9(931475)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhang HN, Dai Y, Zhang CH, Omondi AM,
Ghosh A, Khanra I, Chakraborty M, Yu XB and Liang J: Sirtuins
family as a target in endothelial cell dysfunction: Implications
for vascular ageing. Biogerontology. 21:495–516. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wu B, You S, Qian H, Wu S, Lu S, Zhang Y,
Sun Y and Zhang N: The role of SIRT2 in vascular-related and
heart-related diseases: A review. J Cell Mol Med. 25:6470–6478.
2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Gorski PA, Jang SP, Jeong D, Lee A, Lee P,
Oh JG, Chepurko V, Yang DK, Kwak TH, Eom SH, et al: Role of SIRT1
in modulating acetylation of the sarco-endoplasmic reticulum
Ca(2+)-ATPase in heart failure. Circ Res. 124:e63–e80.
2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Li J, Chen T, Xiao M, Li N, Wang S, Su H,
Guo X, Liu H, Yan F, Yang Y, et al: Mouse Sirt3 promotes autophagy
in AngII-induced myocardial hypertrophy through the deacetylation
of FoxO1. Oncotarget. 7:86648–86659. 2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Mehra MR, Park MH, Landzberg MJ, Lala A
and Waxman AB: Right heart failure: Toward a common language. J
Heart Lung Transplant. 33:123–126. 2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Tomson T, Battino D and Perucca E: The
remarkable story of valproic acid. Lancet Neurol.
15(141)2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Benza RL, Adamson PB, Bhatt DL, Frick F,
Olsson G, Bergh N and Dahlöf B: CS1, a controlled-release
formulation of valproic acid, for the treatment of patients with
pulmonary arterial hypertension: Rationale and design of a Phase 2
clinical trial. Pulm Circ. 14(e12323)2024.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Pang M, Li Y, Gu W, Sun Z, Wang Z and Li
L: Recent advances in epigenetics of macrovascular complications in
diabetes mellitus. Heart Lung Circ. 30:186–196. 2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Zang R, Tan Q, Zeng F, Wang D, Yu S and
Wang Q: JMJD1A represses the development of cardiomyocyte
hypertrophy by regulating the expression of catalase. Biomed Res
Int. 2020(5081323)2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Liu X, Chen J, Zhang B, Liu G, Zhao H and
Hu Q: KDM3A inhibition modulates macrophage polarization to
aggravate post-MI injuries and accelerates adverse ventricular
remodeling via an IRF4 signaling pathway. Cell Signal.
64(109415)2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Yang G, Weng X, Zhao Y, Zhang X, Hu Y, Dai
X, Liang P, Wang P, Ma L, Sun X, et al: The histone H3K9
methyltransferase SUV39H links SIRT1 repression to myocardial
infarction. Nat Commun. 8(14941)2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Jiang H, Li Y, Xiang X, Tang Z, Liu K, Su
Q, Zhang X and Li L: Chaetocin: A review of its anticancer
potentials and mechanisms. Eur J Pharmacol.
910(174459)2021.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Schweizer S, Harms C, Lerch H, Flynn J,
Hecht J, Yildirim F, Meisel A and Märschenz S: Inhibition of
histone methyltransferases SUV39H1 and G9a leads to neuroprotection
in an in vitro model of cerebral ischemia. J Cereb Blood Flow
Metab. 35:1640–1647. 2015.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Wang P, Alvarez-Perez JC, Felsenfeld DP,
Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK,
Garcia-Ocaña A and Stewart AF: A high-throughput chemical screen
reveals that harmine-mediated inhibition of DYRK1A increases human
pancreatic beta cell replication. Nat Med. 21:383–388.
2015.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Hille S, Dierck F, Kuehl C, Sosna J,
Adam-Klages S, Adam D, Luellmann-Rauch R, Frey N and Kuhn C: Dyrk1a
regulates the cardiomyocyte cell cycle via D-cyclin-dependent
Rb/E2f-signalling. Cardiovasc Res. 110:381–394. 2016.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Lan C, Chen C, Qu S, Cao N, Luo H, Yu C,
Wang N, Xue Y, Xia X, Fan C, et al: Inhibition of DYRK1A, via
histone modification, promotes cardiomyocyte cell cycle activation
and cardiac repair after myocardial infarction. EBioMedicine.
82(104139)2022.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Young A, Bradley LA, Farrar E, Bilcheck
HO, Tkachenko S, Saucerman JJ, Bekiranov S and Wolf MJ: Inhibition
of DYRK1a enhances cardiomyocyte cycling after myocardial
infarction. Circ Res. 130:1345–1361. 2022.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Wang N, Wang W, Wang X, Mang G, Chen J,
Yan X, Tong Z, Yang Q, Wang M, Chen L, et al: Histone lactylation
boosts reparative gene activation post-myocardial infarction. Circ
Res. 131:893–908. 2022.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Dal-Pra S, Hodgkinson CP, Mirotsou M,
Kirste I and Dzau VJ: Demethylation of H3K27 is essential for the
induction of direct cardiac reprogramming by miR combo. Circ Res.
120:1403–1413. 2017.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Lee S, Lee JW and Lee SK: UTX, a histone
H3-lysine 27 demethylase, acts as a critical switch to activate the
cardiac developmental program. Dev Cell. 22:25–37. 2012.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Tan J, Yang X, Zhuang L, Jiang X, Chen W,
Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q, et al: Pharmacologic
disruption of Polycomb-repressive complex 2-mediated gene
repression selectively induces apoptosis in cancer cells. Genes
Dev. 21:1050–1063. 2007.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Hou P, Li Y, Zhang X, Liu C, Guan J, Li H,
Zhao T, Ye J, Yang W, Liu K, et al: Pluripotent stem cells induced
from mouse somatic cells by small-molecule compounds. Science.
341:651–654. 2013.PubMed/NCBI View Article : Google Scholar
|
|
72
|
LncRNA H19 ameliorates myocardial
infarction-induced myocardial injury and maladaptive cardiac
remodelling by regulating KDM3A. J Cell Mol Med. 27:1757–1760.
2023.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Repetti GG, Toepfer CN, Seidman JG and
Seidman CE: Novel therapies for prevention and early treatment of
cardiomyopathies now and in the future. Circ Res. 124:1536–1550.
2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Xu J, Liang S, Wang Q, Zheng Q, Wang M,
Qian J, Yu T, Lou S, Luo W, Zhou H and Liang G: JOSD2 mediates
isoprenaline-induced heart failure by deubiquitinating CaMKIIδ in
cardiomyocytes. Cell Mol Life Sci. 81(18)2024.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Ying X, Zhao Y, Yao T, Yuan A, Xu L, Gao
L, Ding S, Ding H, Pu J and He B: Novel protective role for
ubiquitin-specific protease 18 in pathological cardiac remodeling.
Hypertension. 68:1160–1170. 2016.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Liu N, Chai R, Liu B, Zhang Z, Zhang S,
Zhang J, Liao Y, Cai J, Xia X, Li A, et al: Ubiquitin-specific
protease 14 regulates cardiac hypertrophy progression by increasing
GSK-3β phosphorylation. Biochem Biophys Res Commun. 478:1236–1241.
2016.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Zhao D, Zhong G, Li J, Pan J, Zhao Y, Song
H, Sun W, Jin X, Li Y, Du R, et al: Targeting E3 ubiquitin ligase
WWP1 prevents cardiac hypertrophy through destabilizing DVL2 via
inhibition of K27-linked ubiquitination. Circulation. 144:694–711.
2021.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Maejima Y, Usui S, Zhai P, Takamura M,
Kaneko S, Zablocki D, Yokota M, Isobe M and Sadoshima J:
Muscle-specific RING finger 1 negatively regulates pathological
cardiac hypertrophy through downregulation of calcineurin A. Circ
Heart Fail. 7:479–490. 2014.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Gupta MK, McLendon PM, Gulick J, James J,
Khalili K and Robbins J: UBC9-mediated sumoylation favorably
impacts cardiac function in compromised hearts. Circ Res.
118:1894–1905. 2016.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Huang CY, Kuo CH, Pai PY, Ho TJ, Lin YM,
Chen RJ, Tsai FJ, Padma VV, Kuo WW and Huang CY: Data supporting
the angiotensin II activates MEL18 to deSUMOylate HSF2 for
hypertension-related heart failure. Data Brief. 16:521–526.
2018.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Pai P, Shibu MA, Chang RL, Yang JJ, Su CC,
Lai CH, Liao HE, Viswanadha VP, Kuo WW and Huang CY: ERβ targets
ZAK and attenuates cellular hypertrophy via SUMO-1 modification in
H9c2 cells. J Cell Biochem. 119:7855–7864. 2018.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Carreras D, Martinez-Moreno R,
Pinsach-Abuin ML, Santafe MM, Gomà P, Brugada R, Scornik FS, Pérez
GJ and Pagans S: Epigenetic changes governing scn5a expression in
denervated skeletal muscle. Int J Mol Sci. 22(2755)2021.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Peterkin T, Gibson A and Patient R:
Redundancy and evolution of GATA factor requirements in development
of the myocardium. Dev Biol. 311:623–635. 2007.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Kuo CT, Morrisey EE, Anandappa R, Sigrist
K, Lu MM, Parmacek MS, Soudais C and Leiden JM: GATA4 transcription
factor is required for ventral morphogenesis and heart tube
formation. Genes Dev. 11:1048–1060. 1997.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Rajagopal SK, Ma Q, Obler D, Shen J,
Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V,
Srivastava D, et al: Spectrum of heart disease associated with
murine and human GATA4 mutation. J Mol Cell Cardiol. 43:677–685.
2007.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Munshi NV, McAnally J, Bezprozvannaya S,
Berry JM, Richardson JA, Hill JA and Olson EN: Cx30.2 enhancer
analysis identifies Gata4 as a novel regulator of atrioventricular
delay. Development. 136:2665–2674. 2009.PubMed/NCBI View Article : Google Scholar
|
|
87
|
He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA,
Visel A, Pennacchio LA and Pu WT: Dynamic GATA4 enhancers shape the
chromatin landscape central to heart development and disease. Nat
Commun. 5(4907)2014.PubMed/NCBI View Article : Google Scholar
|
|
88
|
He A, Kong SW, Ma Q and Pu WT:
Co-occupancy by multiple cardiac transcription factors identifies
transcriptional enhancers active in heart. Proc Natl Acad Sci USA.
108:5632–5637. 2011.PubMed/NCBI View Article : Google Scholar
|
|
89
|
van den Boogaard M, Wong LY, Tessadori F,
Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, Hoen PA, Bakkers
J, Barnett P and Christoffels VM: Genetic variation in T-box
binding element functionally affects SCN5A/SCN10A enhancer. J Clin
Invest. 122:2519–2530. 2012.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Maron BJ: Clinical course and management
of hypertrophic cardiomyopathy. N Engl J Med. 379:655–668.
2018.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Spudich JA: Three perspectives on the
molecular basis of hypercontractility caused by hypertrophic
cardiomyopathy mutations. Pflugers Arch. 471:701–717.
2019.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Tang X, Chen XF, Sun X, Xu P, Zhao X, Tong
Y, Wang XM, Yang K, Zhu YT, Hao DL, et al: Short-Chain Enoyl-CoA
hydratase mediates histone crotonylation and contributes to cardiac
homeostasis. Circulation. 143:1066–1069. 2021.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Liu S, Yu H, Liu Y, Liu X, Zhang Y, Bu C,
Yuan S, Chen Z, Xie G, Li W, et al: Chromodomain protein CDYL acts
as a crotonyl-CoA hydratase to regulate histone crotonylation and
spermatogenesis. Mol Cell. 67:853–866.e855. 2017.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Nussbaum SS, Henry S, Yong CM, Daugherty
SL, Mehran R and Poppas A: Sex-specific considerations in the
presentation, diagnosis, and management of ischemic heart disease:
JACC focus seminar 2/7. J Am Coll Cardiol. 79:1398–1406.
2022.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Li Y, Chen B, Yang X, Zhang C, Jiao Y, Li
P, Liu Y, Li Z, Qiao B, Lau WB, et al: S100a8/a9 signaling causes
mitochondrial dysfunction and cardiomyocyte death in response to
ischemic/reperfusion injury. Circulation. 140:751–764.
2019.PubMed/NCBI View Article : Google Scholar
|
|
96
|
He J, Liu D, Zhao L, Zhou D, Rong J, Zhang
L and Xia Z: Myocardial ischemia/reperfusion injury: Mechanisms of
injury and implications for management (Review). Exp Ther Med.
23(430)2022.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Ni L, Lin B, Zhang Y, Hu L, Lin J, Fu F,
Shen M, Li C, Chen L, Yang J, et al: Histone modification landscape
and the key significance of H3K27me3 in myocardial
ischaemia/reperfusion injury. Sci China Life Sci. 66:1264–1279.
2023.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Wang G, Zou X, Chen Q, Nong W, Miao W, Luo
H and Qu S: The relationship and clinical significance of
lactylation modification in digestive system tumors. Cancer Cell
Int. 24(246)2024.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Xu Y and Fang F: Histone methylation and
transcriptional regulation in cardiovascular disease. Cardiovasc
Hematol Disord Drug Targets. 14:89–97. 2014.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Ibarrola J, Xiang RR, Sun Z, Lu Q, Hill MA
and Jaffe IZ: Inhibition of the histone methyltransferase EZH2
induces vascular stiffness. Clin Sci (Lond). 138:251–268.
2024.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Klonou A, Chlamydas S and Piperi C:
Structure, activity and function of the MLL2 (KMT2B) protein lysine
methyltransferase. Life (Basel). 11(823)2021.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Zhao WK, Zhou YT and Wu Q: Ferroptosis:
Opportunities and challenges in myocardial ischemia-reperfusion
injury. Oxid Med Cell Longev. 2021(9929687)2021.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Cao Y, Luo F, Peng J, Fang Z, Liu Q and
Zhou S: KMT2B-dependent RFK transcription activates the TNF-α/NOX2
pathway and enhances ferroptosis caused by myocardial
ischemia-reperfusion. J Mol Cell Cardiol. 173:75–91.
2022.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Gao C, Liu Y, Yu Q, Yang Q, Li B, Sun L,
Yan W, Cai X, Gao E, Xiong L, et al: TNF-α antagonism ameliorates
myocardial ischemia-reperfusion injury in mice by upregulating
adiponectin. Am J Physiol Heart Circ Physiol. 308:H1583–H1591.
2015.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Pei H, Song X, Peng C, Tan Y, Li Y, Li X,
Ma S, Wang Q, Huang R, Yang D, et al: TNF-α inhibitor protects
against myocardial ischemia/reperfusion injury via Notch1-mediated
suppression of oxidative/nitrative stress. Free Radic Biol Med.
82:114–121. 2015.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Moe KT, Yin NO, Naylynn TM, Khairunnisa K,
Wutyi MA, Gu Y, Atan MS, Wong MC, Koh TH and Wong P: Nox2 and Nox4
mediate tumour necrosis factor-α-induced ventricular remodelling in
mice. J Cell Mol Med. 15:2601–2613. 2011.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Bravo-Sánchez E, Peña-Montes D,
Sánchez-Duarte S, Saavedra-Molina A, Sánchez-Duarte E and
Montoya-Pérez R: Effects of apocynin on heart muscle oxidative
stress of rats with experimental diabetes: Implications for
mitochondria. Antioxidants (Basel). 10(335)2021.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Du ZD, Yu S, Qi Y, Qu TF, He L, Wei W, Liu
K and Gong SS: NADPH oxidase inhibitor apocynin decreases
mitochondrial dysfunction and apoptosis in the ventral cochlear
nucleus of D-galactose-induced aging model in rats. Neurochem Int.
124:31–40. 2019.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang
Z and Yao W: Diabetes aggravates myocardial ischaemia reperfusion
injury via activating Nox2-related programmed cell death in an
AMPK-dependent manner. J Cell Mol Med. 24:6670–6679.
2020.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Szekeres FLM, Walum E, Wikström P and
Arner A: A small molecule inhibitor of Nox2 and Nox4 improves
contractile function after ischemia-reperfusion in the mouse heart.
Sci Rep. 11(11970)2021.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Yu B, Meng F, Yang Y, Liu D and Shi K:
NOX2 antisense attenuates hypoxia-induced oxidative stress and
apoptosis in cardiomyocyte. Int J Med Sci. 13:646–652.
2016.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Song H, Feng X, Zhang M, Jin X, Xu X, Wang
L, Ding X, Luo Y, Lin F, Wu Q, et al: Crosstalk between lysine
methylation and phosphorylation of ATG16L1 dictates the apoptosis
of hypoxia/reoxygenation-induced cardiomyocytes. Autophagy.
14:825–844. 2018.PubMed/NCBI View Article : Google Scholar
|
|
113
|
He L, Wang Y and Luo J: Epigenetic
modification mechanism of histone demethylase KDM1A in regulating
cardiomyocyte apoptosis after myocardial ischemia-reperfusion
injury. PeerJ. 10(e13823)2022.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Thinnes CC, England KS, Kawamura A,
Chowdhury R, Schofield CJ and Hopkinson RJ: Targeting histone
lysine demethylases-progress, challenges, and the future. Biochim
Biophys Acta. 1839:1416–1432. 2014.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Li Y, Quan X, Li X, Pan Y, Zhang T, Liang
Z and Wang Y: Kdm6A protects against hypoxia-induced cardiomyocyte
apoptosis via H3K27me3 demethylation of Ncx gene. J Cardiovasc
Transl Res. 12:488–495. 2019.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Lin CF, Hsu KC, HuangFu WC, Lin TE, Huang
HL and Pan SL: Investigating the potential effects of selective
histone deacetylase 6 inhibitor ACY1215 on infarct size in rats
with cardiac ischemia-reperfusion injury. BMC Pharmacol Toxicol.
21(21)2020.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Chaturvedi P, Kalani A, Givvimani S, Kamat
PK, Familtseva A and Tyagi SC: Differential regulation of DNA
methylation versus histone acetylation in cardiomyocytes during
HHcy in vitro and in vivo: An epigenetic mechanism. Physiol
Genomics. 46:245–255. 2014.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Xiao Y, Huang W, Zhang J, Peng C, Xia M
and Ling W: Increased plasma S-adenosylhomocysteine-accelerated
atherosclerosis is associated with epigenetic regulation of
endoplasmic reticulum stress in apoE-/- mice. Arterioscler Thromb
Vasc Biol. 35:60–70. 2015.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Wang J, Lin B, Zhang Y, Ni L, Hu L, Yang
J, Xu L, Shi D and Chen YH: The regulatory role of histone
modification on gene expression in the early stage of myocardial
infarction. Front Cardiovasc Med. 7(594325)2020.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Pei J, Schuldt M, Nagyova E, Gu Z, El
Bouhaddani S, Yiangou L, Jansen M, Calis JJA, Dorsch LM, Blok CS,
et al: Multi-omics integration identifies key upstream regulators
of pathomechanisms in hypertrophic cardiomyopathy due to truncating
MYBPC3 mutations. Clin Epigenetics. 13(61)2021.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Shi Y, Zhang H, Huang S, Yin L, Wang F,
Luo P and Huang H: Epigenetic regulation in cardiovascular disease:
Mechanisms and advances in clinical trials. Signal Transduct Target
Ther. 7(200)2022.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Fuster JJ, MacLauchlan S, Zuriaga MA,
Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S,
Muralidharan S, Rius C, et al: Clonal hematopoiesis associated with
TET2 deficiency accelerates atherosclerosis development in mice.
Science. 355:842–847. 2017.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Jaiswal S, Natarajan P, Silver AJ, Gibson
CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino
D, et al: Clonal hematopoiesis and risk of atherosclerotic
cardiovascular disease. N Engl J Med. 377:111–121. 2017.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Souidi A, Nakamori M, Zmojdzian M, Jagla
T, Renaud Y and Jagla K: Deregulations of miR-1 and its target
Multiplexin promote dilated cardiomyopathy associated with myotonic
dystrophy type 1. EMBO Rep. 24(e56616)2023.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Kura B, Kalocayova B, Devaux Y and
Bartekova M: Potential clinical implications of miR-1 and miR-21 in
heart disease and cardioprotection. Int J Mol Sci.
21(700)2020.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Lazar IM, Hoeschele I, de Morais J and
Tenga MJ: Cell cycle model system for advancing cancer biomarker
research. Sci Rep. 7(17989)2017.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Liu X, Xiang M, Tong Z, Luo F, Chen W, Liu
F, Wang F, Yu RQ and Jiang JH: Activatable fluorescence probe via
self-immolative intramolecular cyclization for histone deacetylase
imaging in live cells and tissues. Anal Chem. 90:5534–5539.
2018.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Hussain S, Tulsyan S, Dar SA, Sisodiya S,
Abiha U, Kumar R, Mishra BN and Haque S: Role of epigenetics in
carcinogenesis: Recent advancements in anticancer therapy. Semin
Cancer Biol. 83:441–451. 2022.PubMed/NCBI View Article : Google Scholar
|














