Introduction
Schwannomatosis is a rare neurocutaneous disorder
characterized by the development of multiple schwannomas, benign
tumors of the peripheral nerve sheath (1). The pathogenesis of schwannomatosis is
complex and unclear, and it is often misdiagnosed with
Neurofibromatosis type 2 (NF2) due to overlapping phenotypes
(2). Schwannomatosis is distinct
from NF2, which is characterized by bilateral vestibular
schwannomas and is linked to mutations in the NF2 gene
(3). While schwannomas are the
hallmark of schwannomatosis, the clinical presentation is highly
variable, with symptoms ranging from mild to severe, including
chronic pain and neurological deficits (4).
In recent years, studies have shown association
between schwannomatosis and mutations in the LZTR1 and
SMARCB1 genes (5). Both
genes are involved in crucial cellular pathways, including tumor
suppression and chromatin remodeling, and their disruption plays a
role in tumor formation (6,7).
Mutations in SMARCB1 have been implicated less frequently
than LZTR1, but their presence can suggest a more aggressive
tumor phenotype, and in some cases, they are associated with the
risk of malignant transformation (7-9).
This understanding has enabled genetic testing not only for
diagnostic confirmation of schwannomatosis but also for prognostic
evaluation and personalized treatment plans (10).
Intradural, extramedullary schwannomas are rare,
accounting for approximately 2% of all spinal tumors (11). Here, we present the rare case of a
patient with recurrent schwannomas located at the cauda equina and
distal thigh, with mosaic loss of SMARCB1 protein in the
tumor cells revealed on immunohistochemistry. In addition to the
rare location of this schwannoma, the mosaic loss of SMARCB1
suggests a potential genetic alteration that may influence tumor
formation, though this mutation is not yet fully understood in the
context of schwannomatosis. This finding reinforces the complexity
in the genetic mechanisms underlying schwannoma development and
warrants further surveillance. Additionally, the multifocal nature
and recurrence of schwannomatosis in this case highlights the
importance of continued follow-up and comprehensive management for
neuropathic pain and quality of life improvements.
Case presentation
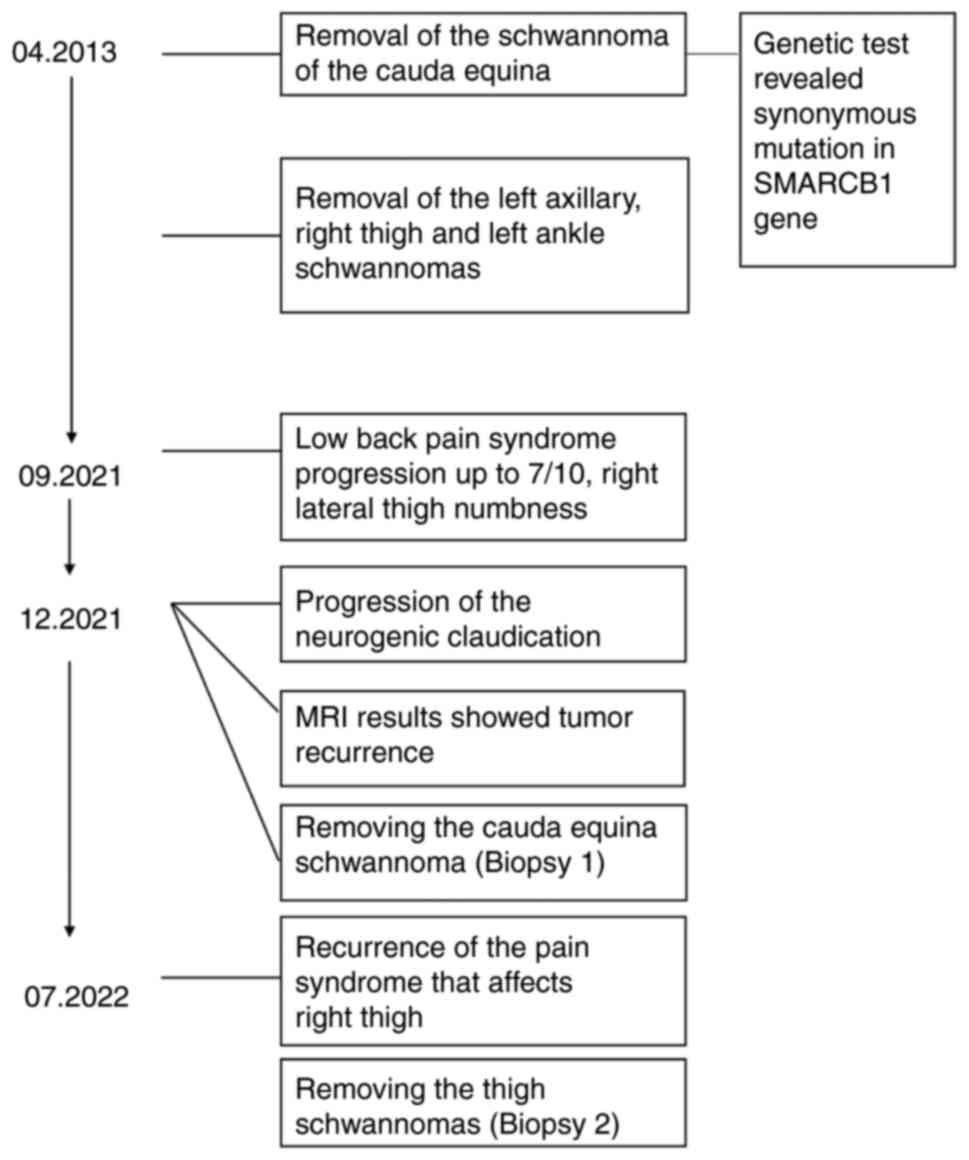
A 53-year-old white female with a history of
schwannomatosis diagnosis at 44 years-old presented to the clinic
in 2021 with increasing symptoms of neurogenic claudication. The
patient had undergone multiple nerve sheath tumor resections in the
past with the first being of the cauda equina in 2013 and
subsequently of the left axilla, right thigh, and left ankle. At
the time, pathology supported schwannoma diagnosis of these tumor
resections (Fig. 1). She had five
cafe-au-lait macules and no family history with features or
symptoms suggestive of neurocutaneous disease.
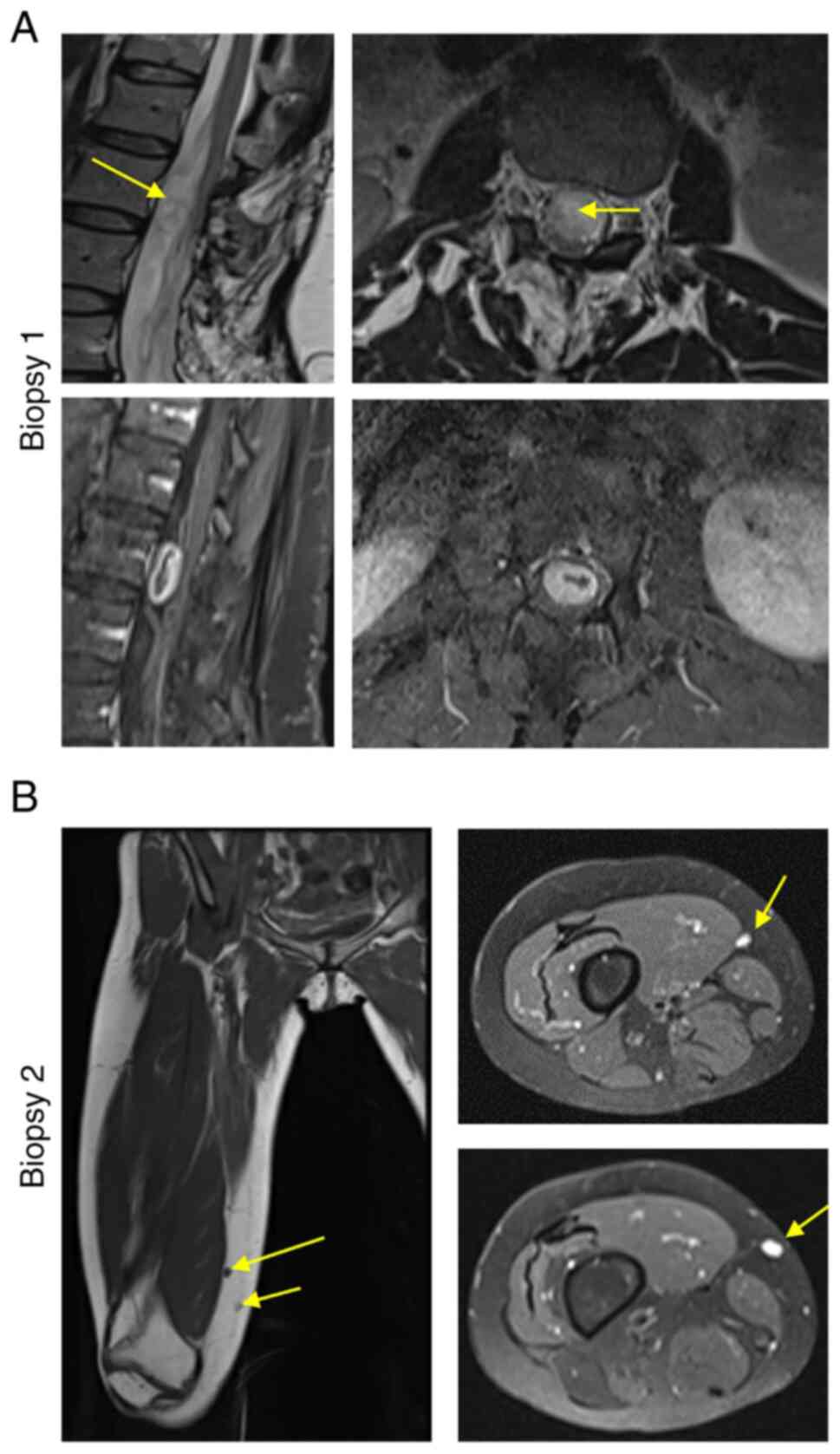
Contrast magnetic resonance imaging (MRI) of the
lumbar spine revealed an intradural extramedullary mass measuring
approximately 11x11x24 mm at the L2-L3 level causing significant
compression of the cauda equina nerve roots (Fig. 2A). A L1-L3 laminectomy was
performed for resection of the intradural mass. Due to the
patient's previous L2-L4 laminoplasty and tumor resection at the
cauda equina in 2013, the procedure was complicated by scarring of
the dorsal nerve roots and dura adherence but concluded with
successful total resection. Pathology confirmed the masses as
schwannomatosis. Proceeding surgery, the patient's recovery was
without complications with no leaking from the incision, foley
removed two days post-surgery with passed voiding trial, and pain
was controlled. While recovering in the hospital, the patient was
given acetaminophen (325 mg, oral) for pain, buspirone (10 mg,
oral) for generalized anxiety, and gabapentin (300 mg, oral) for
nerve-associated pain.
The patient returned home with family three days
after surgery with no prescribed discharge medications. At
discharge, the patient had bilateral thigh numbness and significant
right lower extremity weakness. At the one-month post-operation
appointment, the patient presented with no fever, no infection,
well healed incision site. Seven months after the surgery, she
regained significant function in her right lower extremity. At this
appointment, the patient presented again with burning pain in her
distal thigh.
Following this appointment, in 2022, subsequent MRI
of the femur revealed enhancing lesions in the distal thigh in
proximity to the right sciatic and right common peroneal nerve
(Fig. 2B). The pain was determined
to be caused by these masses found in the distal thigh in proximity
to the right sciatic and right common peroneal nerve. After
discussion with neurosurgery, the lesions in the distal thigh
illustrated by MRI of the femur were removed with blunt dissection.
Pathology confirmed the masses as schwannomatosis. The patient
returned home the day of surgery with no complications. Tissue
samples from both surgeries, laminectomy and distal thigh
dissection, as well as control nerve samples from the ulnar nerve
from a consenting patient with no genetic changes were sent for
genetic sequencing, histopathological examination, and
SMARCB1 protein level quantification.
Comprehensive germline genetic analysis was
conducted of the tissue using whole exome sequencing (WES) and
clinical exome sequencing (CES) with Next-generation sequencing
(NGS) technology, including all genes associated with
schwannomatosis, NF2, and LZTR1 (Table I). The analysis did not identify
any pathological variants (PVs) or other gene mutations in
NF2, and LZTR1. Although, a single nucleotide
polymorphism (SNP) c.1032 C>T [p.Gly344Gly (GGC>GGT)]
in exon 8 of the SMARCB1 gene was identified as a likely
benign variant of unknown significance.
 | Table ISummary of variants detected by WES in
schwannomatosis-associated genes. |
Table I
Summary of variants detected by WES in
schwannomatosis-associated genes.
| Gene | Disease | Mode of
inheritance | Variant | Classification |
|---|
| NF2 | Schwannomatosis | Autosomal
dominant | None detected | Normal/benign |
| LZTR1 | Schwannomatosis | Autosomal
dominant | None detected | Normal/benign |
| SMARCB1 | Schwannomatosis | Autosomal
dominant | c.1032 C>T | Variant of unknown
significance/likely benign |
H&E staining of both samples displayed a
prominent myxoid background with the extensive deposition of
stromal mucin, supporting the pathological diagnosis of schwannoma
(Fig. 3A). The histologic sections
showed an encapsulated tumor made up of spindle cells with a bland
appearance, arranged in short bundles. The tumor exhibited areas
with dense cellularity and nuclear palisading (Antoni A) alongside
regions that were hypocellular (Antoni B). No histologic features
of malignancy were identified.
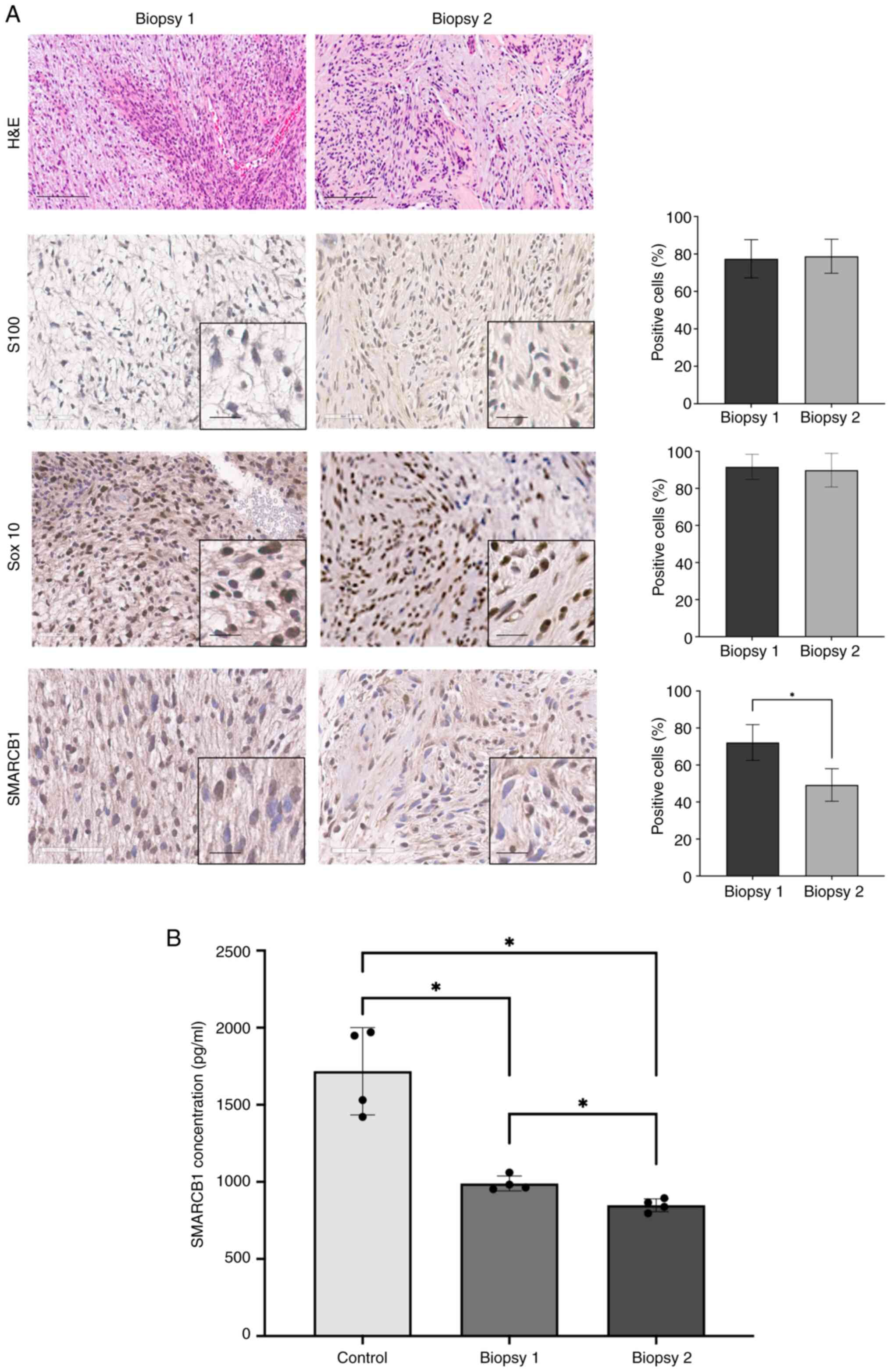 | Figure 3(A) (Top to bottom) High-power
H&E-stained section images of removed schwannomas during first
and second surgeries. The recurrent neoplasm, intradural mass
attached to the cauda equina roots. Image showing spindle cell
neoplasm with hypercellular Antoni A and myxoid hypocellular Antoni
B areas. Blood vessels have thickened hyalinized walls. Nuclear
palisading around fibrillary process (Verocay bodies). Scale bar,
100 μm. S100 immunohistochemistry and quantification
demonstrating intense diffuse labeling of nuclei and cytoplasm of
the tumor cells. Sox10 immunohistochemistry and quantification
representing positive nuclear staining: Elongated nuclei, no
mitotic figures. Immunohistochemical staining with SMARCB1
antibody. Mosaic nuclear staining for SMARCB1 protein in the cauda
equina roots and distal thigh schwannomas with the c.1032 C>T
mutation. There is notable statistical loss in SMARCB1 expression
in the distal thigh schwannoma. Original scale bar, 60 μm.
Magnified scale bar, 20 μm. (B) ELISA SMARCB1 Concentrations
for Biopsy 1, Biopsy 2 from SMARCB1 mutation patient and the
control nerve sample from the SMARCB1 wild-type patient. Samples
were split into four different areas to conduct four replicates of
the ELISA assay (n=4). *P<0.05 (one-way ANOVA; data
are shown as means ± SEM). SEM, standard error of the mean;
H&E, hematoxylin and eosin; SMARCB1, SWI/SNF related, matrix
associated, actin dependent regulator of chromatin subfamily B
member 1. |
IHC indicated positive strong expression of
S100 and Sox10 (~78% and 90% respectively), markers
commonly associated with schwannoma (Fig. 3A). ELISA demonstrated mosaic loss
of nuclear SMARCB1 protein was present in both samples
ranging from 10 to 60% with significant loss of SMARCB1
expression in the distal thigh sample. Additionally, ELISA
indicated statistically significant lower concentrations of
SMARCB1 in both biopsies compared to the control nerve
sample (P<0.05) (Fig. 3B).
Discussion
This case is an unusual presentation of recurrent
schwannomas, mosaic loss of SMARCB1, and no identifiable
direct genetic alteration that is pathogenic. The only identified
alteration is SMARCB1:c.1032C>T, p.Gly344Gly which is a
synonymous change and has been reported in ClinVar five times as a
likely benign or benign variant (12). Based on NHLBI Exome Sequencing
Project (phs000422.v1.p1), this mutation is not observed at any
significant frequency with 6,500 individuals being of European and
African ancestry. According to ACMG guidelines, this variant should
be classified as a likely benign variant and not considered the
cause of the patient's disease, whether it is of de novo or
inherited origin (13). While no
clear pathogenic SMARCB1 mutation was identified, the observed
mosaic loss of SMARCB1 suggests a potential role in disease
pathogenesis by impairing tumor suppressor mechanisms and
deregulation of gene expression and cell cycle control.
While exact recurrence rates for spinal schwannomas
can vary depending on the series, studies show spinal schwannoma
recurs after initial surgery at a rate of 4-6% (14). Known risk factors include subtotal
resection, tumor size and location, histopathology characteristics,
and follow-up (15). The
relationship between SMARCB1 loss and schwannoma recurrence
is complex. While SMARCB1 loss is associated with the
development of certain schwannomas, its role in recurrence is not
entirely clear and needs further research to be fully understood.
For instance, a study on epithelioid schwannomas discovered that
while most tumors followed a benign clinical course, some with
notable cytologic atypia showed recurrence or malignant
transformation (16). Additional
research on epithelioid malignant peripheral nerve sheath tumors,
which can arise from pre-existing schwannomas, revealed that
SMARCB1 inactivation is a recurrent event (17).
This report illustrates that effective management of
recurrent schwannomas hinges on early detection, regular
monitoring, and a comprehensive, multidisciplinary approach even
when genetic testing shows no clear mutative cause. Early detection
of recurrence may allow for timely intervention, minimizing the
need for extensive treatment and preserving the patient's quality
of life. Long term, regular follow-ups with physical assessments
help in identifying new neurological symptoms, such as pain,
weakness, or sensory deficits, which might indicate tumor
progression. Monitoring also facilitates the stratification of
tumors by risk and guides decisions about the urgency and type of
intervention needed. By integrating surgical expertise, advanced
imaging, pain management, and genetic counseling, clinicians can
tailor individualized care plans that address both the immediate
and long-term needs of patients with recurrent schwannomas.
Despite the comprehensive genetic analysis of WES
and CES conducted in this report, a limitation is the possibility
that SMARCB1 pathogenic variants may escape detection. WES
primarily captures exonic regions and may miss pathogenic variants
in intronic regions, which could affect gene expression without
being detected by standard exome sequencing approaches. There are
also limitations of mosaicism detection, as WES has reduced
sensitivity for low-frequency mosaic variants. Because of this
limitation, promoter analysis was also not conducted in this study.
Future investigations could incorporate tumor genetic testing to
provide a more comprehensive understanding of potential somatic
mutations and the promoters contributing to the disease.
In conclusion, in this case report, a patient with
mosaic loss of SMARCB1 protein exhibits recurrence of
schwannomas impacting her quality of life. A better understanding
of the role of SMARCB1 loss in schwannomas and a
multi-leveled approach to diagnosis and treatment may improve
diagnostic accuracy, prognostication, and treatment strategies,
offering hope for more personalized approaches to managing this
challenging condition.
Acknowledgements
Not applicable.
Funding
Funding: This research is financially supported by the
Children's Tumor Foundation Contract Award (grant no. 2022-04-007).
The publication also acknowledges support from Georgia Clinical and
Translational Science Alliance UL1 (grant no. UL1TR002378) and KL2
(grant no. KL2TR002381).
Availability of data and materials
Exome sequencing was provided by GeneDx. GeneDx data
cannot be shared publicly due to consent restrictions tied to
clinical testing. Patients referred to GeneDx consent to
deidentified, aggregate research use under HIPAA privacy
protections. As such, patient-level exome sequencing files, which
may be identifiable, cannot be shared without a HIPAA Business
Associate Agreement or other legally required contract. Requestors
must meet all HIPAA requirements for data access, use, disclosure
and storage. Once all documentation is in place, patient-level data
may be shared per the terms of the agreement. Deidentified
aggregate data from this analysis are available upon request to
GeneDx (support@genedx.com), with
typical fulfillment within 60 days. Data was shared in accordance
with patient consent guidelines to support improved clinical
interpretation.
Authors' contributions
NMB and KL conceived the project. YL and MGY
designed and performed most of the experiments. YL, MAL and MGY
critically analyzed the data. MC handled patient materials. MST,
YD, MAL and SK aided with the interpretation of data and critically
read the manuscript. Manuscript drafting and figure preparation
were performed by YL, MAL and MGY. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Emory University IRB
(approval no. STUDY00002544). Informed consent was obtained from
the patient in accordance with the ethical principles of the
Declaration of Helsinki. The patient has provided written consent
for the publication of the data.
Patient consent for publication
Written consent was obtained from the patient for
publication of clinical data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
MacCollin M, Chiocca EA, Evans DG,
Friedman JM, Horvitz R, Jaramillo D, Lev M, Mautner VF, Niimura M,
Plotkin SR, et al: Diagnostic criteria for schwannomatosis.
Neurology. 64:1838–1845. 2005.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tamura R, Yo M and Toda M: Historical
development of diagnostic criteria for NF2-related schwannomatosis.
Neurol Med Chir (Tokyo). 64:299–308. 2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kresak JL and Walsh M: Neurofibromatosis:
A review of NF1, NF2, and schwannomatosis. J Pediatr Genet.
5:98–104. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Merker VL, Esparza S, Smith MJ,
Stemmer-Rachamimov A and Plotkin SR: Clinical features of
schwannomatosis: A retrospective analysis of 87 patients.
Oncologist. 17:1317–1322. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Min BJ, Kang YK, Chung YG, Seo ME, Chang
KB and Joo MW: Germline mutations for novel candidate
predisposition genes in sporadic schwannomatosis. Clin Orthop Relat
Res. 478:2442–2450. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kohashi K and Oda Y: Oncogenic roles of
SMARCB1/INI1 and its deficient tumors. Cancer Sci. 108:547–552.
2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dhamija R, Plotkin S, Gomes A and
Babovic-Vuksanovic D: LZTR1- and SMARCB1-Related Schwannomatosis.
In: GeneReviews® [Internet]. Adam MP, Feldman J, Mirzaa
GM, Pagon RA, Wallace SE and Amemiya A (eds). Seattle (WA):
University of Washington, Seattle, 1993.
|
|
8
|
Smith MJ, Wallace AJ, Bowers NL, Eaton H
and Evans DG: SMARCB1 mutations in schwannomatosis and genotype
correlations with rhabdoid tumors. Cancer Genet. 207:373–378.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Cooper GW and Hong AL: SMARCB1-deficient
cancers: Novel molecular insights and therapeutic vulnerabilities.
Cancers (Basel). 14(3645)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Loh J, Ong PY, Goh DLM, Puhaindran ME,
Vellayappan BA, Ow SGW, Chan G and Lee SC: Clinical characteristics
and genetic testing outcome of suspected hereditary peripheral
nerve sheath tumours in a tertiary cancer institution in Singapore.
Hered Cancer Clin Pract. 20(23)2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Koeller KK and Shih RY: Intradural
extramedullary spinal neoplasms: Radiologic-pathologic correlation.
Radiographics. 39:468–490. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
National Library of Medicine: ClinVar.
https://www.ncbi.nlm.nih.gov/clinvar/.
|
|
13
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Takahashi T, Hirai T, Yoshii T, Inose H,
Yuasa M, Matsukura Y, Morishita S, Kobayashi Y, Utagawa K, Kawabata
A, et al: Risk factors for recurrence and regrowth of spinal
schwannoma. J Orthop Sci. 28:554–559. 2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fehlings MG, Nater A, Zamorano JJ,
Tetreault LA, Varga PP, Gokaslan ZL, Boriani S, Fisher CG, Rhines
L, Bettegowda C, et al: Risk factors for recurrence of surgically
treated conventional spinal schwannomas: Analysis of 169 patients
from a multicenter international database. Spine (Phila Pa 1976).
41:390–398. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jo VY and Fletcher CDM: SMARCB1/INI1 loss
in epithelioid schwannoma: A clinicopathologic and
immunohistochemical study of 65 cases. Am J Surg Pathol.
41:1013–1022. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Schaefer IM, Dong F, Garcia EP, Fletcher
CDM and Jo VY: Recurrent SMARCB1 inactivation in epithelioid
malignant peripheral nerve sheath tumors. Am J Surg Pathol.
43:835–843. 2019.PubMed/NCBI View Article : Google Scholar
|