Introduction
According to previously published data, as well as
reports from the World Health Organization, hearing loss of
variable etiology represents a serious health, social and economic
issue worldwide (1-3).
With a prevalence varying between 1-3/1.000(4) and 1/500 newborns (5), congenital hearing loss is relatively
frequent, and presents numerous difficulties in early diagnosis and
treatment that could affect the optimal social integration of the
infant. Genetic etiology has been identified in >50% of all
cases of early onset hearing losses, of which, 75-80% are most
probably autosomal recessive and 70% are non-syndromic (1-3).
Other documented etiologies include clinical and environmental
factors, such as ototoxic medication, prematurity or complications
at birth (6). Due to the
etiological heterogeneity of congenital hearing loss, genetic,
clinical and environmental risk factors often combine to provide a
complex picture that makes genetic evaluation and counselling
difficult, especially for very young children (6).
Heterogenous gene mutations, which often require a
complex diagnosis, can be responsible for complex and serious
neurological conditions (7).
Notably, 30% of congenital hearing loss cases are syndromic
(1-3),
and Radio-Tartaglia syndrome (RATARS) is a rare type, of which
34-45 global cases have been reported thus far (8). RATARS is a neurodevelopmental
disorder caused by a heterozygous mutation in the SPEN gene
localized in chromosome 1p36.21-36.13. The genetic mechanism
involved is represented by autosomal dominant inheritance (8,9). The
most frequent deletions of this chromosome are terminal and present
a phenotype defined by a variety of clinical manifestations
(8), including impaired
intellectual development, speech delay, variable behavioral
abnormalities [aggression, attention-deficit/hyperactivity disorder
(ADHD), autism and impulsivity], ear malformations (hearing
impairment), malformations of the head and neck, and malformations
of the eyes, integumentary, cardiovascular, digestive,
musculoskeletal and endocrine systems. An increase in the number of
copies and mutations in this chromosome have also been documented
in a small number of cases (10).
As well as neurodevelopmental delay, RATARS is characterized by
intellectual disability, language developmental delay and
craniofacial dysmorphism (11).
Information available about the distinctive features of this
disease is scarce, delaying prompt management and the screening of
associated conditions, which, in turn, may lead to reduced life
expectancy.
The SPEN gene is part of the tumor-suppressor gene
family, and has been reported to exert functions as a nuclear
matrix platform, since it organizes and integrates any
transcriptional responses within cells (8). It is hormonally induced through
interactions with other repressor genes, especially those involved
in the remodeling of chromatin, histone deacetylases and the
entrapment of transcriptional activators (8,12).
The importance of this gene in the human genotype has been verified
by the observation that it serves an important role in several
processes in neuronal development (12). Previous research on rodent models
has shown that SPEN gene defects produce a reduction in brain mass,
and a decrease in the thickness of the cerebral cortex and the
hippocampus with subsequent ventriculomegaly (8,11,12).
The present case describes the association between
severe congenital sensorineural hearing loss and a complex
phenotype associated with a variant of the SPEN gene in a
5-year-old male child. The case is unique and important due to the
rarity of this SPEN gene pathology, and to the intricate mechanisms
underlying the effects of genetic and environmental risk factors on
congenital severe sensorineural hypoacusis. To the best our
knowledge, there have been no other reports regarding the
association between SPEN gene and hypoacusis. The medical
literature regarding RATARS is limited due to the global rarity of
this disease.
Case report
The present study describes the case of a 5-year-old
male patient born prematurely at 26 weeks and 6 days (June 2019;
Filantropia Clinical Hospital, Bucharest, Romania), via caesarean
section, after an uncomplicated pregnancy. The child was born with
a very low birth weight (VLBW) of 1,300 g, length of 39 cm,
cephalic perimeter of 28 cm, an Apgar score of 6 in the 1st min and
7 at 5 min after birth, and with difficult postnatal adaptation.
The child required resuscitation and respiratory support
(FiO2 30%; positive end-expiratory pressure, 6 cm
H2O) immediately after birth. They were immediately
admitted to the neonatal intensive care unit (NICU) where they were
diagnosed with neonatal respiratory distress syndrome with a
deficiency of surfactant, premature jaundice, apnea and osteopenia.
The child was kept in a neutral thermal environment in an
incubator, with orotracheal intubation, administered one dose of
intratracheal surfactant (2.5 ml/kg) and mechanically ventilated
for 5 days. Hemodynamic positive inotrope support (dopamine) was
also necessary for the first 48 h. The tracheal tube was removed
after 5 days, and non-invasive respiratory support was administered
for another 5 days via nasal continuous positive airway pressure
and for 18 days via high-flow nasal cannula. Supplementary
free-flow oxygen at FiO2 #x003C;30% was necessary for
the following 4 weeks coupled to the administration of caffeine
intravenously and orally for apnea of prematurity. The patient
slowly evolved towards balanced, normal physiological
parameters.
At 6 weeks the child was transferred to the
premature unit (July 2019; Filantropia Clinical Hospital,
Bucharest, Romania) where they continued to develop slowly until
they were discharged with a good general status; at discharge, they
were in a cardiorespiratory and hemodynamically balanced state in
atmospheric air pressure, had normal stethacoustic signs and were
breast-fed. Health problems, a notable family history or
consanguinity were not issues for either parent. During the first 4
years of life, the child was extensively investigated and closely
monitored for the evolution of neuromotor development in various
clinics until, in November 2023, the child was referred to the
‘Marie Sklodowska Curie’ Emergency Children's Hospital (Bucharest,
Romania) where they were investigated by a multidisciplinary team,
including ophthalmology (1st degree retinopathy with spontaneous
remission); otorhinolaryngology with complete audiological testing
[auditory brainstem response (ABR), behavioral observation
audiometry that suggested severe bilateral hearing loss, middle-ear
immittance audiometry, distortion product otoacoustic emissions
absent from 1,500-4,000 Hz bilaterally); neurology; neurosurgery
(normal exam); electroencephalogram; brain MRI; and whole exome
sequencing (October 2023; myLifeGenome, Arcensus GmbH).
The milestones of the patient included hand grip at
4 months, sitting at 8 months, crawling at 12 months and
independent walking at 23 months, although with precarious balance
and coordination. By the age of 3 years, the patient was able to
use a fork, pick up object with two fingers, chew and eat on their
own, socialize but not interact in play groups and use ~10
meaningful words. Communication was primarily through gestures and
facial expressions, and complex words were used only in specific
contexts. Autism spectrum disorder was also diagnosticated.
The child did not have specific facies, and there
were no craniofacial abnormalities or any other system affliction
(e.g. cardiovascular, digestive, endocrine) reported thus far. The
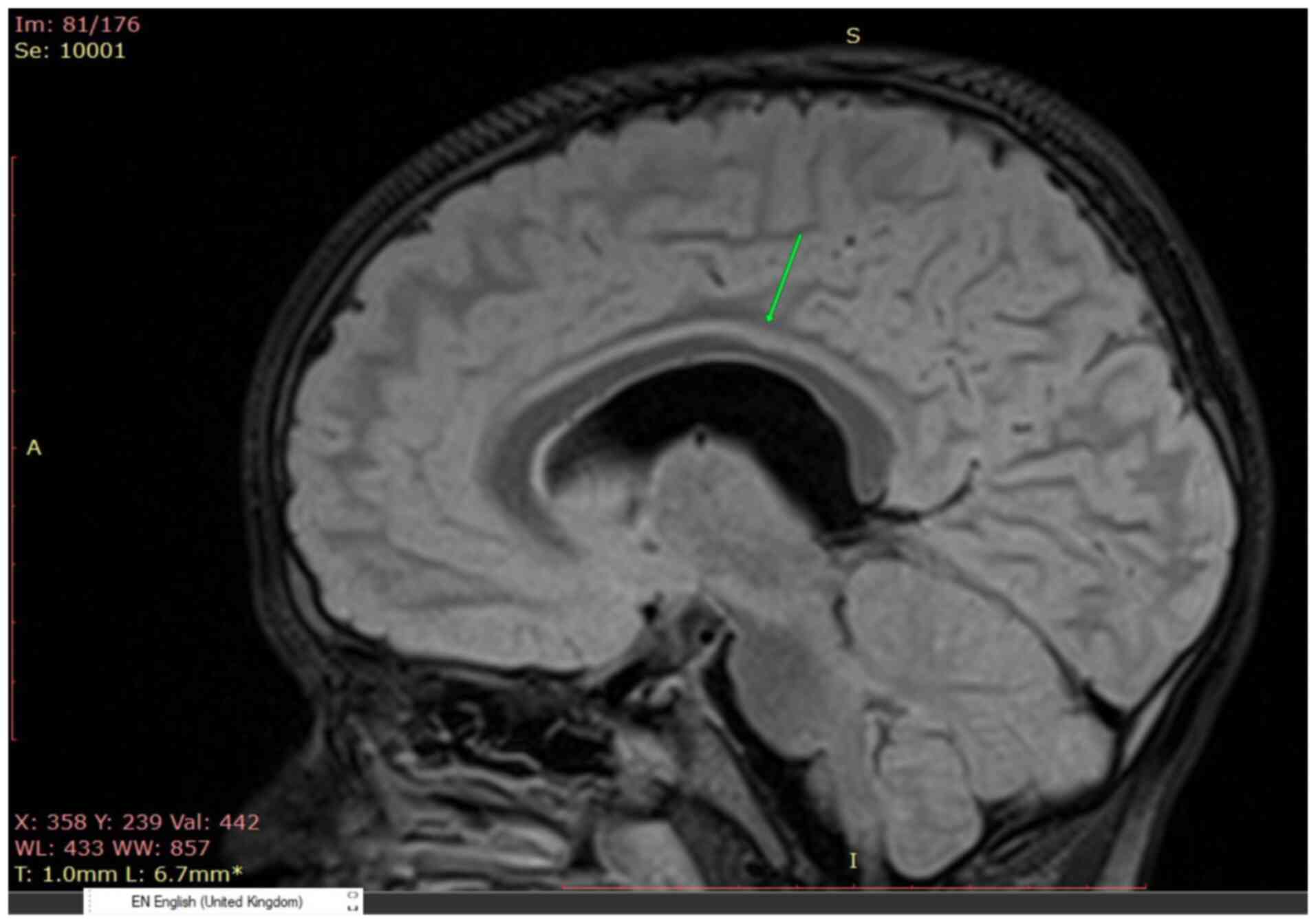
MRI showed nonspecific demyelinating lesions of the white matter
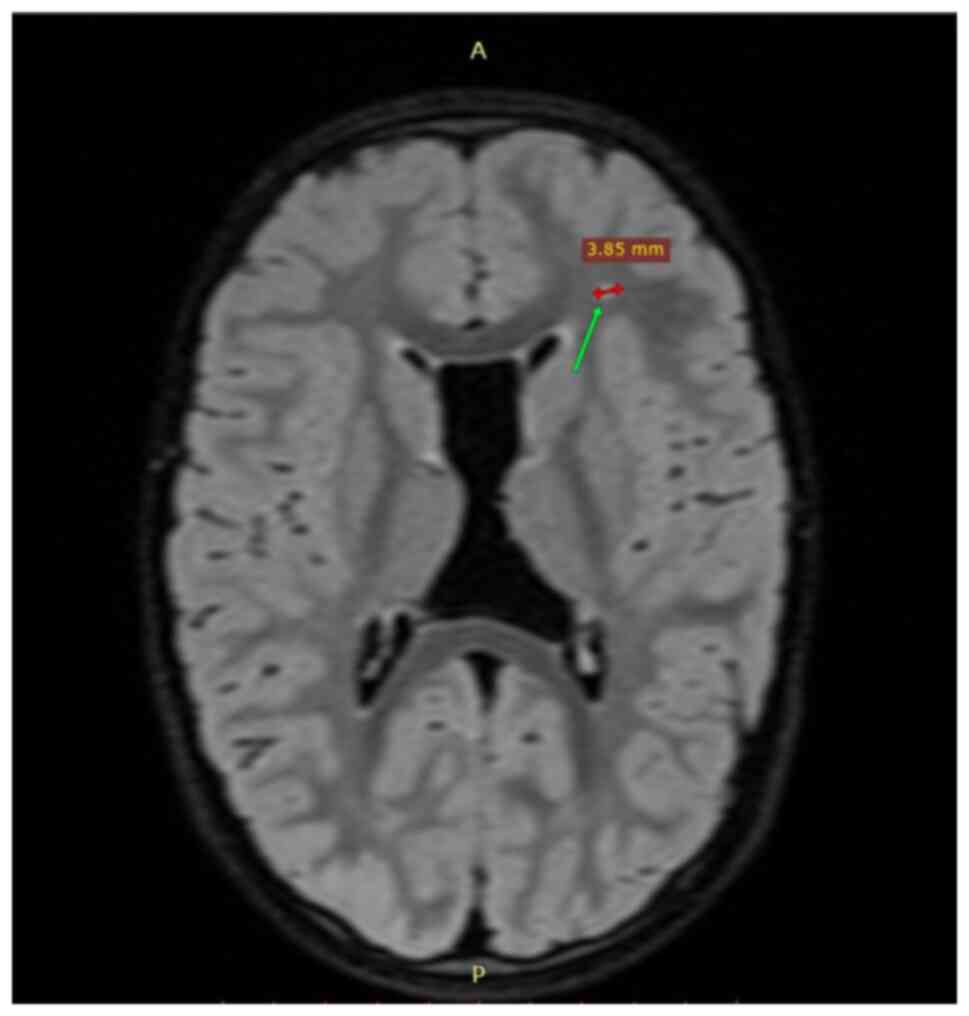
(Fig. 1), a thin corpus callosum
with irregular contour (Fig. 2)
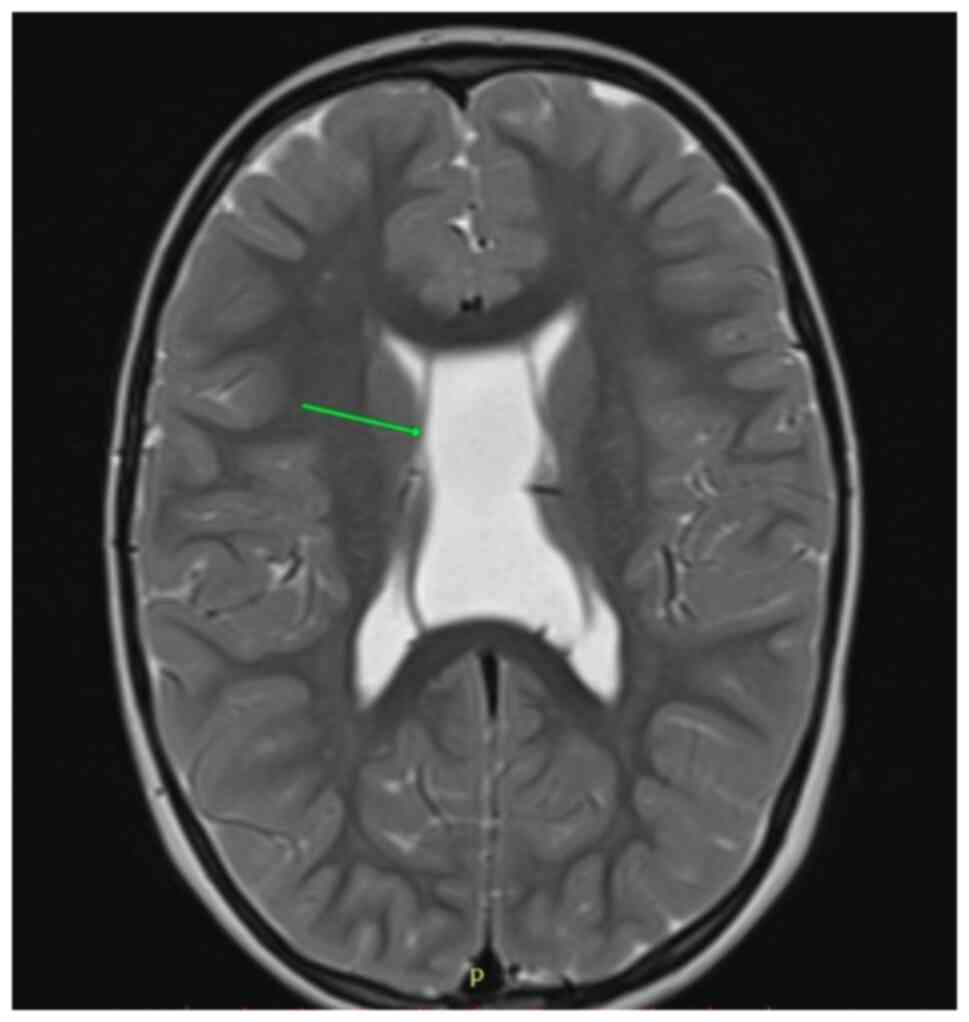
and a cyst of the septum pellucidum with no apparent clinical
significance (Fig. 3). The eye
exam noted 1st degree retinopathy with spontaneous remission.
By the age of 4 years, the patient was referred to
genetic counseling due to abnormal corpus callosum morphology,
global developmental delay, hearing impairment, premature birth and
short attention span (older brother affected with global
developmental delay). Subsequently, whole exome sequencing was
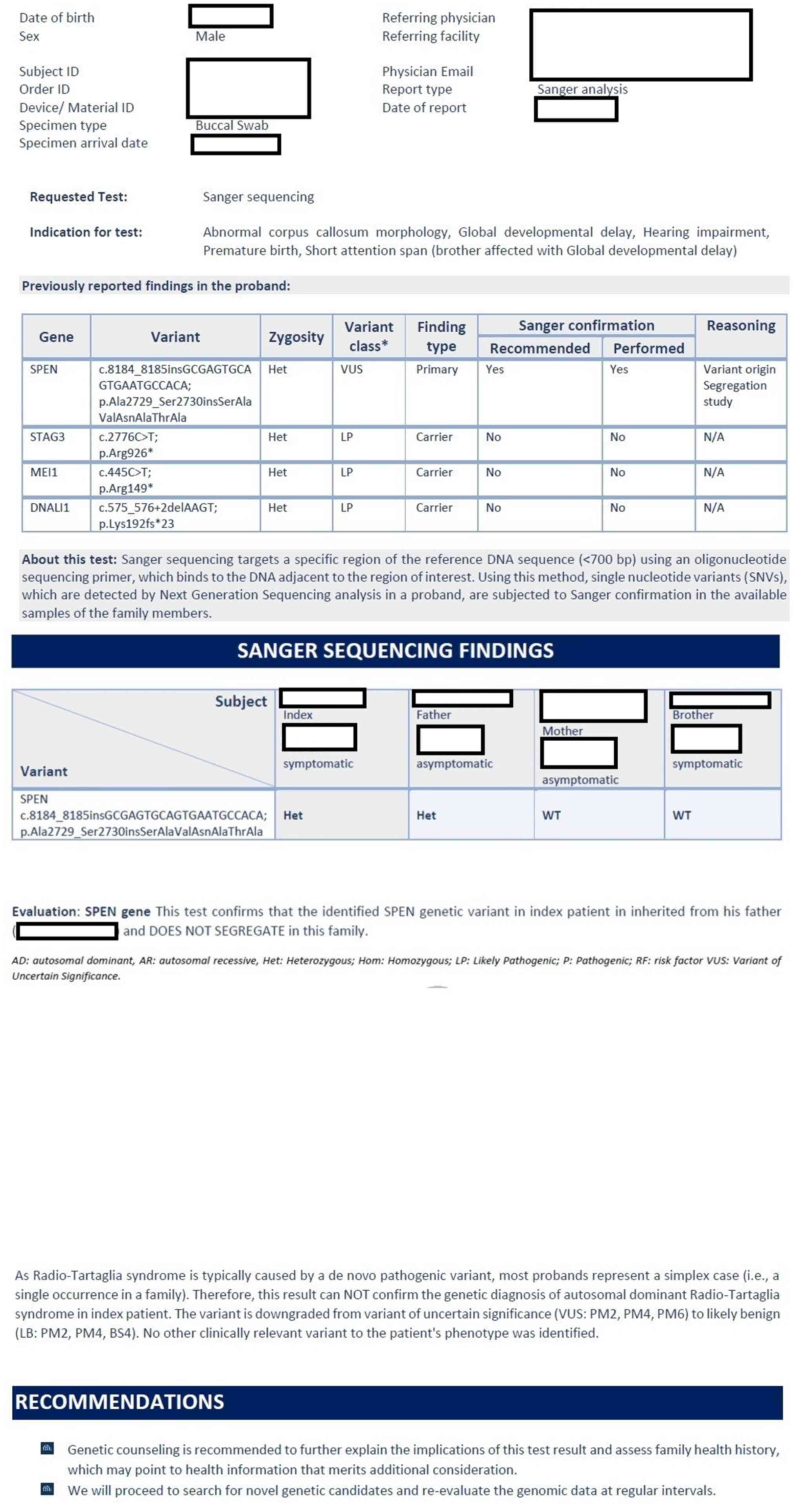
performed by myLifeGenome, Arcensus GmbH. The result revealed the
presence of a variant of uncertain significance in heterozygous
state in the SPEN gene, (c.8184_8185insGCGAGTGCAGTGAATGCCACA,
p.Ala2729_Ser2730insSerAlaValAsnAlaThrAla); this variant was
confirmed by Sanger analysis (Fig.
4). The child was also a carrier for other heterozygous
variants without phenotypic effects in STAG3, MEI1 and DNALI1
genes. The genetic testing of both parents and older brother
(November 2023; myLifeGenome; Arcensus GmbH) confirmed that the
identified SPEN genetic variant in the index patient was inherited
from the father (asymptomatic) and did not segregate within the
family.
At the age of 4 years, extensive audiological and
vestibular evaluations were performed as preliminary steps for a
cochlear implant. The vestibular examination revealed a
satisfactory labyrinthine function with a slight hypovalent
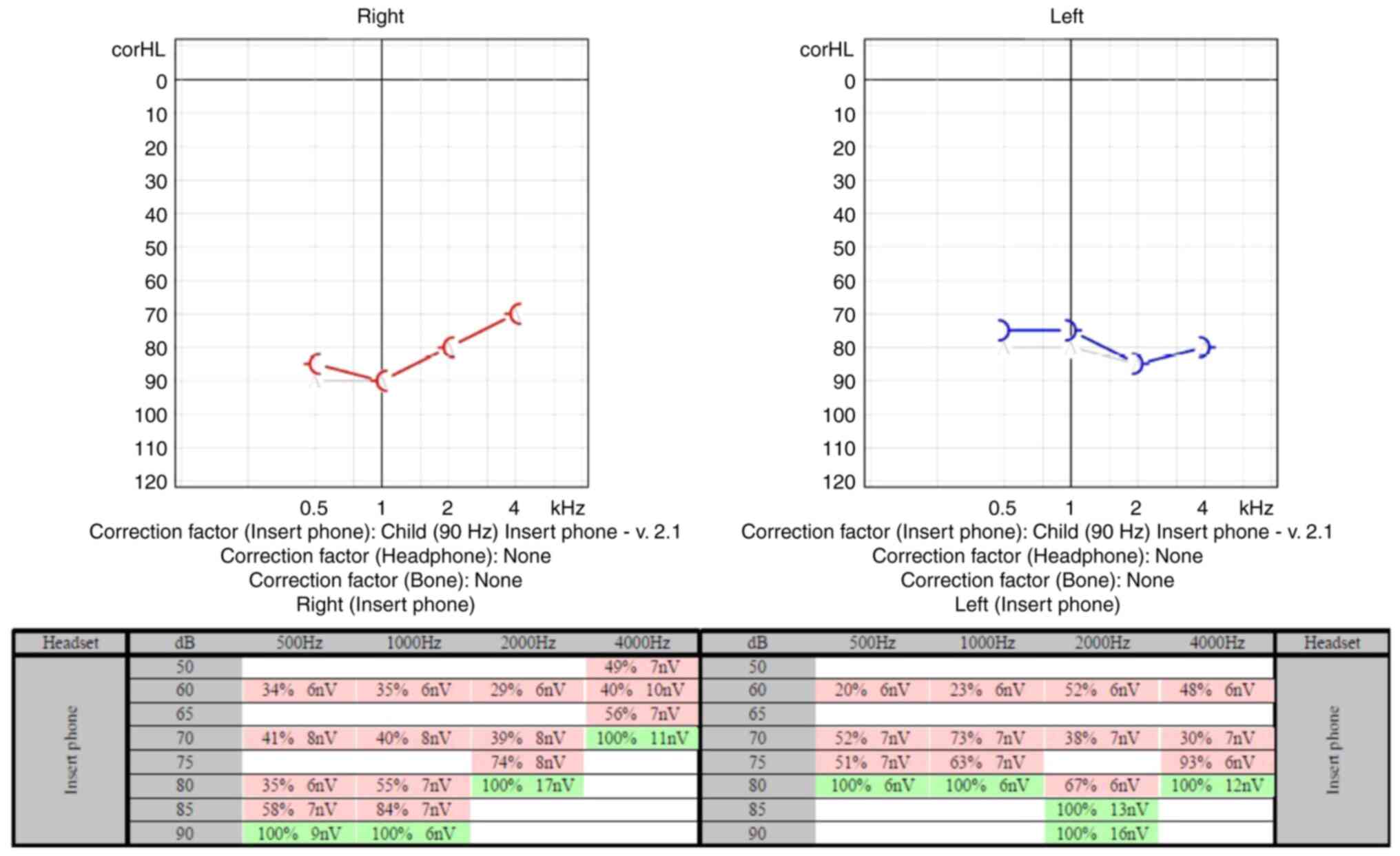
labyrinth on the right side. The audiological examination included
hearing assessment in the free field for specific frequencies and
speech using behavioral observation audiometry, auditory steady
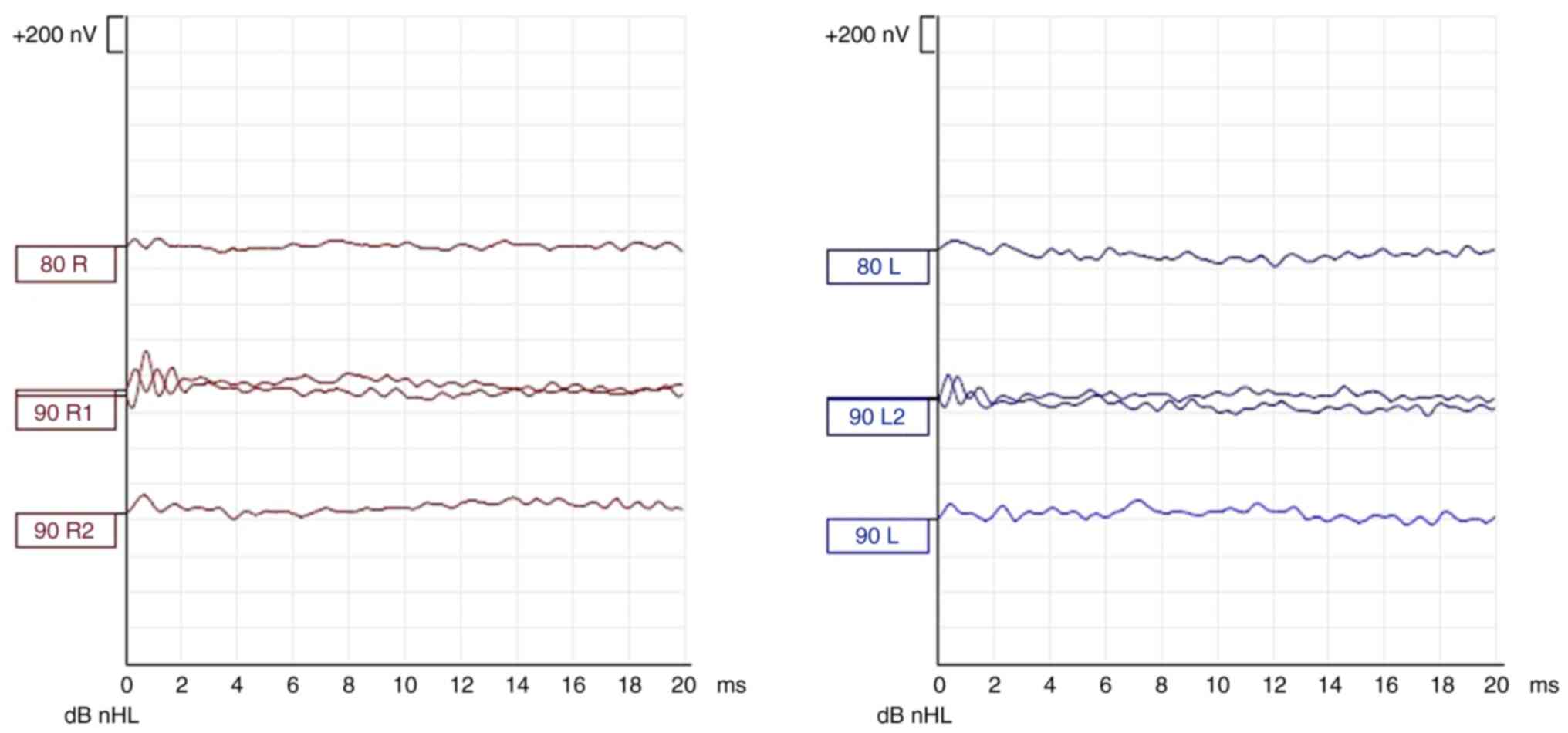
state response audiometry (Fig.
5), ABR evaluation (Figs. 6
and 7), assessment of middle ear
function using immittance audiometry and of cochlear outer hair
cell function using otoacoustic emissions. Behavioral test results
were obtained with fair reliability for the speech stimuli.
Responses to speech stimuli were obtained at 70 dB HL in the sound
field condition. No reliable responses to frequency-specific
stimuli in the free field could be obtained; however, no noticeable
behavioral responses to stimuli at the limits of the equipment were
observed. Tympanometry revealed type A tympanograms bilaterally.
Ipsilateral acoustic reflexes were absent at 1,000 Hz bilaterally.
Distortion product otoacoustic emissions were absent from
1,500-4,000 Hz bilaterally. ABR showed no evoked response until 90
dBnHL bilaterally, with the presence of cochlear microphonics,
indicating an auditory neuropathy. The auditory steady state
response results indicated severe and bilateral hearing loss. The
results of the hearing tests concluded severe hearing loss to sound
stimuli in a sound field. No reliable responses to
frequency-specific stimuli could be obtained. Middle ear pressure
and mobility were normal. Acoustic reflexes were absent
bilaterally. Otoacoustic emissions suggested abnormal cochlear
outer hair cell function bilaterally.
The young patient's hypoacusis was initially treated
with hearing aids and speech therapy with poor results.
Subsequently they underwent cochlear implantation in September 2024
at the ‘Marie Sklodowska Curie’ Emergency Children's Hospital. The
implant was activated in December 2024 and showed excellent results
within the first month of use. At present, the child refuses to
wear the classic hearing aid on the contralateral ear and has
suggested that they desire a similar implant for the other ear.
Discussion
Congenital sensorineural hypoacusis has different
etiologies, both genetic and non-genetic. Notably, prematurity is
known to produce hypoacusis, often in association with
developmental delay, as in the present patient. A particularity of
the present case was the association of a novel variant of the SPEN
gene. This variant was inherited from the healthy father and
classified as a variant with unknown significance. Pathogenic
variants of the SPEN gene have been reported to be associated with
RATARS, a rare genetic syndrome with only ~45 previously reported
cases (8), which is characterized
by global developmental delay, hypotonia, brain malformations
(including polymicrogiria, heterotopia, thin corpus callosum,
cerebellar atrophy, periventricular white matter anomalies),
behavioral problems (autism, hyperkinesia, aggression), heart
malformations and dysmorphic facial features. The current patient
also presented with some of these features (global developmental
delay, autistic behavior, abnormal corpus callosum) (11). The role of this SPEN gene variant
in the phenotype, together with the effect of prematurity on
development, was taken into consideration in the present case. The
SPEN gene encodes a protein that is involved in neurogenesis and
acts as a transcriptional repressor. It has an important role in a
number of cellular processes, including cell division, adhesion and
migration, and neuronal development, with implications in learning
and memory processes (11). To the
best of our knowledge, the variant described in the present case
has not been reported before, both in the general population and in
patient databases; therefore, it is difficult to predict the effect
of this variant on the patient phenotype, and a definitive
connection between the patient phenotype and the SPEN gene variant
could not be made. Functional studies or reports of other cases
with this variant could contribute to a better classification of
this gene variant.
In the present case, the presence of prematurity and
numerous other risk factors associated with the early postnatal
state (VLBW, low Apgar score, NICU admission, respiratory distress
syndrome and the need for respiratory support) cannot be ignored as
a likely cause for congenital hypoacusis. Notably, we can only
speculate on the association of the SPEN gene mutation with this
patient due to its role in neural development.
The presence of congenital sensorineural hypoacusis,
especially when associated with neurodevelopmental delay and
intellectual disability at an early age, should be investigated by
genetic sequencing, as heterogeneity between most genetic
pathologies is present (12).
Congenital hypoacusis of syndromic origin (30%) is far more rare
than that of non-syndromic origin (70%) (13) and the presence of the SPEN mutation
in the 1p36 chromosome, associated with RATARS, represents an
extremely rare find. The presence of only ~45 reported cases of
RATARS worldwide makes the diagnosis in this case challenging
(8).
The SPEN gene serves an important role in neuronal
development. In vivo rodent experiments have shown that gene
defects produce a reduction in brain mass, as well thinning of the
cortex and hippocampus (11,12,14-16).
These results have been verified by clinical observations of
patients with RATARS. In 64% of cases, important neurological
abnormalities have been reported (8). Of all the abnormalities identified by
imaging in patients with RATARS (white matter abnormalities,
agenesis of the corpus callosum, cerebellar atrophy and
polymicrogyria), the present case exhibited minimal white matter
abnormalities and a thin, irregular corpus callosum. Such cerebral
malformations are associated with obvious clinical neurological
effects including: Motor and developmental delay, cerebral ataxia,
seizures, intellectual disability, ADHD, psychiatric and behavioral
abnormalities (impulsivity, aggressive behavior, reduced eye
contact, motor stereotypes) and autism spectrum disorder, some of
which have also been reported in the present patient (17).
In 2021, Radio et al (12), reported on 34 patients with RATARS.
These patients were diagnosed through genetic and Sanger sequencing
in which heterozygous mutations of the SPEN gene
(613484.0001-613484.0005) were reported (8). These mutations have not yet been
described in the gnomAD database, and the patients were verified
through the GeneMatcher program (18) and DECIPHER database (19). In most cases there was no
association between genotype and phenotype, and the mutations
occurred de novo. The methylation profile of the genome did
not show much difference between cells in the normal population
compared with in the patients in a clinical trial conducted by
Radio et al in 2021(12).
Williams syndrome (WS) is very similar to RATARS and
is defined as an autosomal dominant multisystemic disorder caused
by a microdeletion on chromosome 7q11.23, a region that contains
26-28 genes including the ELN gene (20,21).
It is also associated with intellectual disability, facial
abnormalities and arteriopathy, which makes a clinical differential
diagnosis unreliable. Both pathologies present reduced cerebral
volume, which leads to developmental and intellectual delay
associated with psychiatric disorders (anxiety, phobias, ADHD,
impulsivity, aggression, motor/behavioral stereotypes, such as
thumb sucking, nail or lip biting, hair twirling, body rocking,
self-biting, teeth clenching or grinding and head banging).
However, RATARS may express more neurological features such as:
Polymicrogyria, cerebellar atrophy, corpus callosum agenesis or
tethered cord syndrome (12,20).
There are some clinical features that are present both in RATARS
and WS, such as bitemporal narrowing, prominent bulbous nasal tip,
long nasal philtrum, muscle hypotonia, global developmental delay,
and mild to moderate intellectual disability. Regarding facial
dysmorphisms, a broad forehead, dysplastic ears, arched elongated
eyebrows, epicanthus and synophrys are reported as common for
patients with RATARS (12).
Although both syndromes are associated with facies, these are not
distinctive but rather suggestive of a genetic pathology. This is
not the case for the present patient, as they did not present with
any specific or distinctive facies. RATARS may present with frontal
bossing, telecanthus, arched elongated eyebrows, a pointed chin and
dental abnormalities, whereas WS typically presents with ‘elf-like’
features with epicanthic folds, puffiness around the eyes, small
widely spaced teeth, a small jaw and full cheeks (20-22).
Psychiatric conditions, including ADHD, autism, aggressive behavior
and anxiety, are very common in RATARS compared with in WS
(12). In addition, congenital
heart defects have been reported in both syndromes, but in RATARS
they are represented by ventricular and atrial septal defects,
whereas in WS they are represented by supravalvular aortic stenosis
or pulmonary stenosis. Regarding the genetic defect, WS is caused
by a microdeletion on chromosome 7q11.23, whereas in patients with
RATARS heterozygous mutations of the SPEN gene are the cause
(8).
In conclusion, the present findings indicated that a
patient with no craniofacial dysmorphism or specific facies, with
neurodevelopmental delay but normal laboratory analyses and
slightly modified brain images, associated with severe congenital
sensorineural hypoacusis should be considered as a possible RATARS
case and should undergo exome sequencing for a definitive
diagnosis.
The novel variant of the SPEN gene described in the
present case was inherited by the patient from their healthy father
and was classified as a variant with unknown significance. Since
the patient also had severe pre- and postnatal issues, such as
VLBW, low Apgar score, NICU admission, respiratory distress
syndrome and the need for respiratory support, which are documented
causes of congenital hearing loss, it is difficult to predict the
effect of this gene variant on the patient phenotype. Functional
studies or reports of other cases with this variant could
contribute to a better classification of this gene variant.
RATARS represents an extremely rare genetic syndrome
and possible cause of congenital sensorineural hearing loss
associated with neurodevelopmental delay, intellectual disability
and multiple systemic manifestations that share several
characteristics with other genetic pathologies. The number of
reported cases varies from 34 to 45 worldwide. The lack of
information about the distinctive features of RATARS leads to
delayed diagnosis and management which, in turn, affects the
screening of associated conditions and life expectancy. The
clinical complexity of patients with RATARS, including the presence
of hearing loss and precarious language development, indicates the
need to further establish more clear parameters of the disease so
that it does not go undiagnosed.
Taking into account the global scarcity of reported
cases of RATARS with pathogenic variants of the SPEN gene, the
publication of this case may increase the interest for further
research studies into this gene. A more accurate diagnosis could be
provided through future research directions, including functional
cell or animal model experiments, to observe the impact of this
variant on relevant cell processes and phenotypes, thereby
clarifying the causal relationship between the variant and
disease.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
ACN and HM conceptualized the study. ACN, HM and MB
devised and approved the methodology. AIM, MED, DMP and RN were
responsible for using software and graphical representations. ACN,
MB and HM performed validation. AIM, MED, DMP, RN and DCG performed
formal analysis. ACN, HM and MED were in charge of running the
investigation. AIM, MED, RN and DMP were responsible for resources
(provision of study materials, medical materials, patients,
laboratory samples and results, computing resources, or other
analysis tools). HM, MB, AIM and RN provided data curation (data
management, maintaining research data for initial use and later
reuse). MB and HM were responsible for writing the original draft.
ACN and MED were responsible for review and editing. ACN, HM and MB
provided visualization (transforming the raw medical data, such as
medical reports, imaging results or patient information, into
visual representations such as charts, graphs or images to
facilitate understanding and interpretation). DCG and HM were in
charge of supervision. HM, MB and MED were responsible for project
administration. ACN and HM confirm the authenticity of all the raw
data. All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The study was conducted in accordance with The
Declaration of Helsinki, and was approved by the Ethics Committee
of the ‘Titu Maiorescu’ University (approval no. 16/06.12.2024;
Bucharest, Romania). All legal guardians (parents) provided
informed written consent.
Patient consent for publication
The patient's legal guardians (parents) provided
written informed consent for the publication of data from the
proband, proband's brother and themselves.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mocanu H, Mocanu AI, Dragoi AM and
Radulescu M: Long-term histological results of ossicular chain
reconstruction using bioceramic implants. Exp Ther Med.
21(260)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mocanu H: The role of perinatal hearing
screening in the normal development of the infant's language. In:
Debating Globalization. Identity, Nation and Dialogue. 4th edition.
Boldea I and Sigmirean C (eds). Arhipeleag XXI Press, Tirgu Mures,
pp562-569, 2017.
|
|
3
|
Mocanu H: The economic impact of early
diagnosis of congenital hearing loss. In: Debating Globalization.
Identity, Nation and Dialogue. 4th edition. Boldea I and Sigmirean
C (eds). Arhipeleag XXI Press, Tirgu Mures, pp556-561, 2017.
|
|
4
|
Palmer CG, Lueddeke JT and Zhou J: Factors
influencing parental decision about genetics evaluation for their
deaf or hard-of-hearing child. Genet Med. 11:248–255.
2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Smith R, Hildebrand M and Van Camp G:
Deafness and Hereditary Deafness Overview. 2010. Available from:
http://www.ncbi.nlm.nih.gov/pubmed/20301607. Accessed
at February 16, 2022.
|
|
6
|
Neagu AC, Budișteanu M, Gheorghe DC,
Mocanu AI and Mocanu H: rare gene mutations in romanian hypoacusis
patients: Case series and a review of the literature. Medicina
(Kaunas). 58(1252)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Németh AH, Kwasniewska AC, Lise S, Parolin
Schnekenberg R, Becker EB, Bera KD, Shanks ME, Gregory L, Buck D,
Zameel Cader M, et al: Next generation sequencing for molecular
diagnosis of neurological disorders using ataxias as a model.
Brain. 136:3106–3118. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pichardo MXA, Bernate F, Trujillo-Ángel
JF, Alba MCS, Lubo MP, Perdigon NA, Ramirez LB, Jimenez D, Escobar
SA, Gonzalez IF and Regalado LGC: Radio-Tartaglia Syndrome: A rare
cause of delay in neurodevelopment - A case report. Iran J
Neonatol. 15:60–64. 2024.
|
|
9
|
National Center for Biotechnology
Information. Radio-Tartaglia Syndrome (Concept ID: C5543339) -
MedGen - NCBI. Available from: https://www.ncbi.nlm.nih.gov/medgen/1778557.
|
|
10
|
Jordan VK, Zaveri HP and Scott DA: 1p36
deletion syndrome: An update. Appl Clin Genet. 8:189–200.
2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Online Mendelian Inheritance in Man,
OMIM®. Johns Hopkins University, Baltimore, MD, 2021. Available
from: https://omim.org/.
|
|
12
|
Radio FC, Pang K, Ciolfi A, Levy MA,
Hernández-García A, Pedace L, Pantaleoni F, Liu Z, de Boer E,
Jackson A, et al: SPEN haploinsufficiency causes a
neurodevelopmental disorder overlapping proximal 1p36 deletion
syndrome with an episignature of X chromosomes in females. Am J Hum
Genet. 108:502–516. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Mocanu H and Oncioiu I: The influence of
clinical and environmental risk factors in the etiology of
congenital sensorineural hearing loss in the Romanian population.
Iran J Public Health. 48:2301–2303. 2019.PubMed/NCBI
|
|
14
|
Ma M, Moulton MJ, Lu S and Bellen HJ:
‘Fly-ing’ from rare to common neurodegenerative disease mechanisms.
Trends Genet. 38:972–984. 2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shimada S, Shimojima K, Okamoto N, Sangu
N, Hirasawa K, Matsuo M, Ikeuchi M, Shimakawa S, Shimizu K, Mizuno
S, et al: Microarray analysis of 50 patients reveals the critical
chromosomal regions responsible for 1p36 deletion syndrome-related
complications. Brain Dev. 37:515–526. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Battaglia A, Hoyme HE, Dallapiccola B,
Zackai E, Hudgins L, McDonald-McGinn D, Bahi-Buisson N, Romano C,
Williams CA, Brailey LL, et al: Further delineation of deletion
1p36 syndrome in 60 patients: A recognizable phenotype and common
cause of developmental delay and mental retardation. Pediatrics.
121:404–410. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Radio-Tartaglia Syndrome genetic and rare
diseases information center. Department of Health and Human
Services. Available from: https://www.ncbi.nlm.nih.gov/medgen/1778557. Accessed
at: February 20, 2024.
|
|
18
|
Sobreira N, Schiettecatte F, Valle D and
Hamosh A: GeneMatcher: A matching tool for connecting investigators
with an interest in the same gene. Hum Mutat. 36:928–930.
2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Firth HV, Richards SM, Bevan AP, Clayton
S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM and Carter
NP: DECIPHER: Database of chromosomal imbalance and phenotype in
humans using ensembl resources. Am J Hum Genet. 84:524–533.
2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Pober BR: Williams-Beuren syndrome. N Engl
J Med. 362:239–252. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Williams Syndrome Genetic and Rare
Diseases Information Center. Department of Health and Human
Services. Available from: https://www.ncbi.nlm.nih.gov/medgen/?term=Williams+syndrome.
Accessed at: July 15, 2024.
|
|
22
|
Stanley TL, Leong A and Pober BR: Growth,
body composition, and endocrine issues in Williams syndrome. Curr
Opin Endocrinol Diabetes Obes. 28:64–74. 2021.PubMed/NCBI View Article : Google Scholar
|