Introduction
Maturity-onset diabetes of the young (MODY) is a
monogenic form of diabetes caused by autosomal dominant mutations
in a single gene that impair pancreatic β-cell function.
Mechanistically, MODY differs from type 2 diabetes mellitus (T2DM)
in that it involves the impaired synthesis and secretion of insulin
rather than insulin resistance. MODY accounts for 1-2% of all
diabetes cases in Europe (1).
However, in China, improvements in genetic screening and clinical
diagnostics have increased the diagnostic rate of MODY to ~9%
(2). MODY is currently classified
into 14 genetic subtypes due to mutations in the following genes:
Hepatocyte nuclear factor 4a (HNF4A), HNF1A,
HNF1B, glucokinase (GCK), pancreatic and duodenal
homeobox 1, neuronal differentiation 1, KLF transcription factor
11, carboxyl ester lipase, paired box 4, insulin, B lymphocyte
kinase, ATP-binding cassette subfamily C member 8 (ABCC8),
potassium inwardly rectifying channel subfamily J member 11 and
adaptor protein phosphotyrosine interacting with PH domain and
leucine zipper 1. Among the various subtypes, HNF1A-MODY, GCK-MODY,
HNF4A-MODY and HNF1B-MODY are the most commonly diagnosed (3). In 2011, Fajans and Bell (4) formally described the onset of MODY as
occurring during childhood or early adolescence, typically in
patients aged 9-14 years, although it may present even earlier.
Previous studies have estimated that MODY accounts for ~1-5% of all
diabetes cases in Western countries, such as Europe, North America
and Australia. However, in Asian populations, including China,
owing to differences in screening intensity and genetic
architecture, the true prevalence of MODY may fall below or exceed
this interval (5,6). Over time, MODY can lead to chronic
complications affecting the eyes, kidneys, nerves and blood vessels
(6).
The diagnostic criteria for MODY, proposed by
Tattersall and Fajans (7) in 1975,
comprise: i) Onset of diabetes before age 25 years; ii) diabetes in
at least three family members across successive generations,
indicating autosomal dominant inheritance; iii) presentation as
non-insulin-dependent diabetes mellitus (NIDDM); iv) absence of
obesity; and v) no history of diabetic ketoacidosis (DKA). Despite
these diagnostic definitions, MODY remains rare and is frequently
misdiagnosed as T2DM (8).
The present study describes a MODY-12 pedigree
carrying a heterozygous pathogenic variant in the ABCC8
gene, segregating in an autosomal dominant pattern. The index
patient is a 22-year-old woman with a body mass index within the
normal range (18.5-24.9 kg/m²). Contrary to the classical
diagnostic criteria for MODY, the patient experienced three
episodes DKA within 10 months of diagnosis, along with a transient
but rapid decline in renal function. While most patients with
MODY-12 are currently managed with sulfonylureas, emerging evidence
suggests that sodium-glucose transporter 2 inhibitors (SGLT2is) can
be used in combination with sulfonylureas as an alternative
treatment approach for MODY (9).
Materials and methods
Research subjects
A pedigree was recruited from the Provincial
Hospital Affiliated to Fuzhou University (Fuzhou, China) between
January and December 2024, which comprises five males and seven
females, including five affected individuals (I-2, II-2, II-5,
III-2 and III-4) with onset ages ranging from 19 to 22 years,
consistent with typical MODY phenotypes. A pedigree exhibiting the
MODY-12 phenotype was investigated (Fig. 1A). Next-generation sequencing (NGS)
and Sanger sequencing methods revealed heterozygous pathogenic
ABCC8 variants in five affected individuals (I-2, II-2,
II-5, III-2 and III-4), while seven unaffected relatives (I-1,
II-1, II-3, II-4, II-6, III-1 and III-3) lacked these variants.
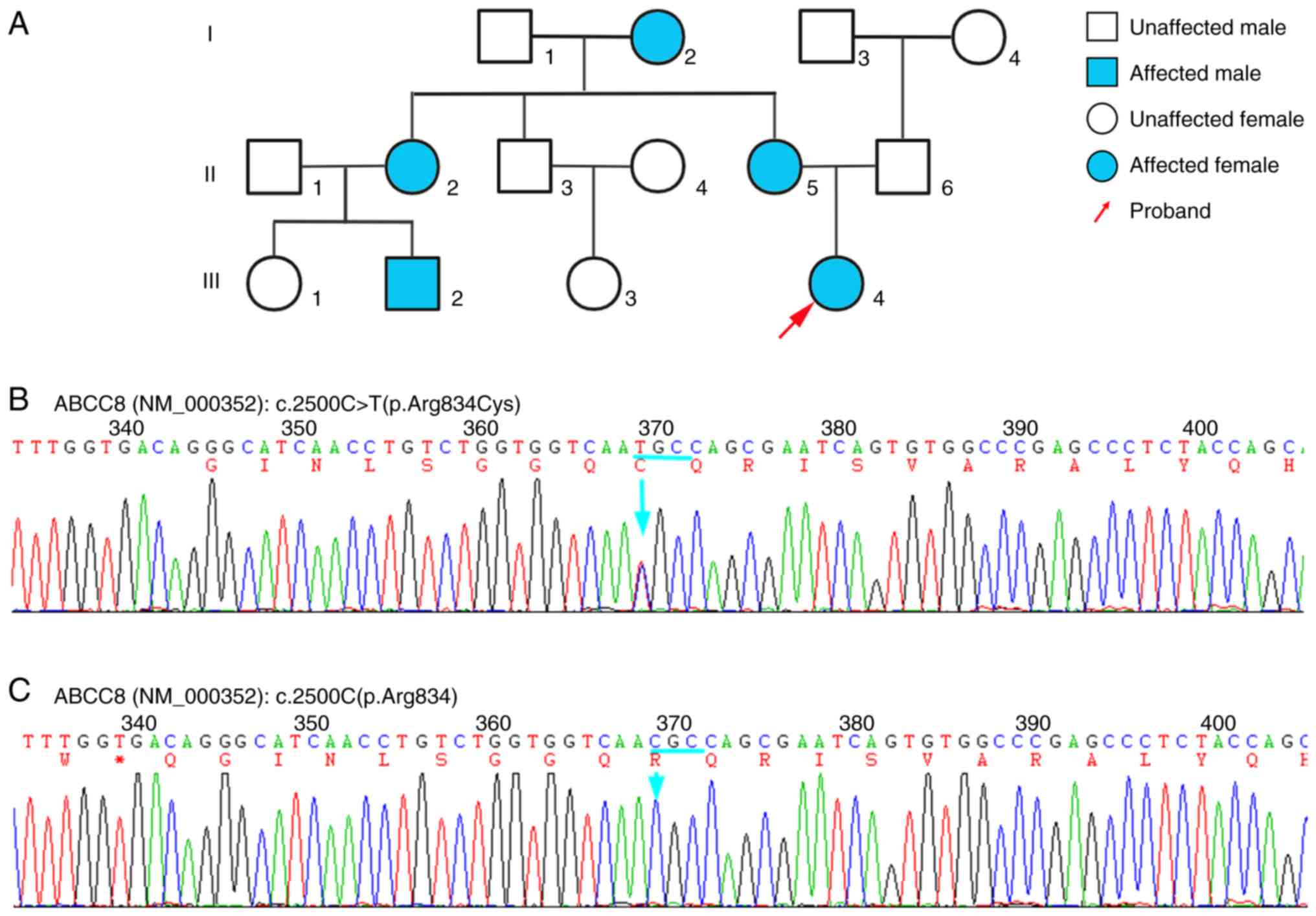 | Figure 1Pedigree and genetic analysis of a
family affected by maturity-onset diabetes of the young-12. (A)
Pedigree of the family. Squares denote males, circles denote
females and blue shading indicates heterozygous carriers of the
ABCC8 c.2500C>T (p.Arg834Cys) variant. (B) Sanger
sequencing chromatogram of the ABCC8 gene, showing the
location of the c.2500C>T (p.Arg834Cys) mutation in exon 21. (C)
Sanger sequencing chromatogram of wild-type ABCC8 exon 21,
with the c.2500C nucleotide peak indicated by an arrow. ABCC8,
ATP-binding cassette, subfamily C, member 8; C, cytosine; T,
thymine; Arg, arginine; Cys, cysteine. |
Clinical phenotype investigation
Detailed clinical information and medical records
were collected for all pedigree members. Subsequently, NGS was
conducted on each participant to identify pathogenic variants. The
study protocol was approved by the Ethics Committee of Fujian
Provincial Hospital (Fuzhou, China). Written informed consent was
obtained from all participants or their legally authorized
representatives.
Extraction of genomic DNA
Peripheral blood (2 ml) was collected from each
participant in an EDTA-coated tube. Genomic DNA was subsequently
extracted from the blood using a MolPure® Magnetic Blood
DNA Kit (Shanghai Yeasen Biotechnology Co., Ltd.).
Clinical and biochemical
assessments
During three hospitalizations of the proband
(III-4), blood and urine samples were obtained to perform routine
biochemistry, urinalysis, and assessment of insulin autoantibodies.
Other affected family members (I-2, II-2, II-5 and III-2) underwent
follow-up visits, during which peripheral blood was collected for
measurement of fasting plasma glucose, serum insulin, insulin
autoantibodies, and standard biochemical analyses.
Comprehensive second-generation
sequencing: Whole-exome, copy number variation (CNV) and
full-length mitochondrial genome profiling
For whole-exome sequencing, the purity and
concentration of the extracted genomic DNA was assessed using a
NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.) and a
Qubit 3.0 fluorometer (Thermo Fisher Scientific, Inc.). Sequencing
libraries were then constructed using the NadPrep DNA Rapid
Digestion Library Preparation Module (cat. no. 1002601; MGI Tech
Co., Ltd.) according to the manufacturer's instructions. This
process included restriction enzyme digestion, end repair and
A-tailing, adapter ligation and amplification by PCR. The ligated
fragments were size-selected for an insert of ~320 bp using 50 µl
NadPrep® SP Beads (cat. no. 1002211; MGI Tech Co.,
Ltd.). The libraries were then pooled and whole-exome regions were
enriched using the NEXome Core Panel [cat. no. 1001852; Nanodigmbio
(Nanjing) Biotechnology Co., Ltd.] and the Hybrid Capture Reagent
[cat. no. 1005102; Nanodigmbio (Nanjing) Biotechnology Co., Ltd.].
Finally, high-throughput sequencing (150 bp; PE150) was performed
on a DNBSEQ-T7 sequencer (MGI Tech Co., Ltd.). Raw sequencing data
were generated and assessed with FastQC v 0.12.0 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/),
Burrows-Wheeler Aligner v 0.7.17 (https://bio-bwa.sourceforge.net/), SAMtools/SAMtools
markdup v 1.17 (https://samtools.sourceforge.net/), Picard
MarkDuplicates v 3.1.0 (https://broadinstitute.github.io/picard/), GATK v 3.8
(https://gatk.broadinstitute.org/hc/en-us) and Exomiser
v 12.1.0 (https://www.sanger.ac.uk/tool/exomiser/) for quality
control. Following the removal of low-quality reads and adapter
sequences, the clean reads were aligned with the reference genome
using Burrows-Wheeler Aligner, and the resulting alignments were
sorted using SAMtools. PCR duplicates were marked and removed using
Picard MarkDuplicates and SAMtools markdup. Finally, variants were
called with the Genome Analysis Toolkit (GATK) following the GATK
Best Practices workflow. The clinical features of the proband were
encoded using standardized Human Phenotype Ontology (HPO)
(https://hpo.jax.org/browse/search?q=MODY&navFilter=all).
Variant prioritization and phenotype-driven ranking were performed
using Exomiser by combining HPO annotations with the detected
variants. Candidate variants were classified according to American
College of Medical Genetics and Genomics (ACMG) guidelines, and
pathogenicity was assigned based on established criteria (10). Allele frequencies were retrieved
from the Genome Aggregation Database (gnomAD v 3.1.2; https://gnomad.broadinstitute.org/).
Pathogenicity of missense variants was predicted using the Rare
Exome Variant Ensemble Learner (REVEL); the functional impact of
amino acid substitutions was assessed with Sorting Intolerant From
Tolerant (SIFT); and the effects of missense variants or small
insertions and deletions (indels) on protein structure and function
were evaluated by the Polymorphism Phenotyping tool (PolyPhen).
A total of 56,705 candidate genes were initially
considered. The following filters were then applied to exclude
likely nonpathogenic variants. Variants with an allele frequency
≥1% in the ExAC (v0.3) (http://exac.broadinstitute.org/) or gnomAD (v 3.1.2)
browsers were excluded. Variants with an allele frequency ≥5% in an
in-house database (iGeneTechDB) were excluded. Synonymous variants
outside of splice-site regions were removed. Variants classified as
‘benign’ or ‘likely benign’ in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar; updated March
2025) were excluded. After these steps, the remaining gene list was
retained for downstream disease-association analysis. The
c.2500C>T (p.Arg834Cys) variant in ABCC8 (NM_000352) was
classified as likely pathogenic. To validate this finding, all
available family members, with the exception of the proband, who
underwent NGS, were screened by Sanger sequencing. Primers flanking
the ABCC8 c.2500C>T site were designed using Primer
Premier 5.0 (Premier Biosoft International). Genomic DNA from the
proband (III-4) and affected relatives (I-2, II-2, II-5 and III-2)
was amplified by polymerase chain reaction using Taq DNA Polymerase
(cat. no. D1806-250UN; Sigma-Aldrich Co., Ltd.) in 25-µl reactions.
Thermal cycling was performed on a VeritiPro™ Thermal Cycler (cat.
no. A48141; Thermo Fisher Scientific Co., Ltd.) with an initial
denaturation at 95˚C for 5 min, followed by 35 cycles of 95˚C for
30 sec, annealing at 58˚C for 30 sec and extension at 72˚C for 45
sec, and a final extension at 72˚C for 7 min. The PCR products were
purified and subjected to bidirectional Sanger sequencing on an ABI
3730XL Genetic Analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Sequencing chromatograms were analyzed with
SeqMan Pro v14.1 (DNASTAR, Inc.), and genotype-phenotype
cosegregation was evaluated across the pedigree. The primers used
were as follows: Forward, 5'-GGAAGTGGCAGCACACATTC-3' and reverse,
5'-GCTCAGCCTGGGGTTGTATT-3'.
The NGS data were analyzed for CNVkit v 0.9.5
(https://cnvkit.readthedocs.io/en/stable/) using
standard bioinformatics pipelines and mitochondrial DNA variants
were screened against the MITOMAP (https://www.mitomap.org/MITOMAP) and ClinVar
databases.
Bioinformatics analysis
The tertiary structure of the ABCC8 protein, also
known as sulfonylurea receptor 1 (SUR1; UniProt ID: Q09428;
https://www.uniprot.org/uniprotkb/Q09428) was
predicted by homology modeling using SWISS-MODEL (https://swissmodel.expasy.org/repository/uniprot/Q09428)
(11), with the Rattus
norvegicus SUR1 cryo-EM structure (https://www.rcsb.org/structure/6BAA) as a template.
Structural visualization and comparative analyses were conducted
using University of California San Francisco (UCSF) ChimeraX v 1.7
software. Analyses included electrostatic surface potential mapping
and the structural superimposition of wild-type and mutant
(c.2500C>T) models. Deviations between the two structures in key
regions, namely nucleotide-binding domain 2 (NBD2) and
transmembrane helix 14, were quantified by root-mean-square
deviation measurements. UCSF ChimeraX was implemented in Python
3.12.2 (http://www.python.org/). ChimeraX
provides a modular framework comprising a core library providing
fundamental functions, including atomic coordinate manipulation,
molecular surface rendering and basic graphics operations, and
plugin modules for performing advanced functionalities.
Results
Clinical phenotypes
The proband (III-4) was a 22-year-old woman
initially diagnosed with T2DM. Subsequently, NGS led to the
reclassification of her diagnosis as MODY-12 due to the detection
of the heterozygous variant c.2500C>T (p.Arg834Cys) of
ABCC8. Within 10 months of the MODY diagnosis, the patient
experienced three episodes of DKA accompanied by acute kidney
injury (AKI), occurring at 3, 7 and 10 months. The detailed
clinical examination results of the proband are summarized in
Table I. Each episode of DKA was
characterized by fasting blood glucose levels >30 mmol/l, along
with symptoms including nausea, vomiting, fatigue and loss of
appetite. Following insulin therapy and fluid-electrolyte
replacement, the patient's fasting blood glucose is maintained at
<7.0 mmol/l and 2-h postprandial glucose remains <10.0 mmol/l
(12). The patient's renal
function normalized rapidly after the first two AKI episodes, with
serum creatinine returning to baseline within days. During the
third DKA episode, the patient developed bilateral pedal edema,
hyperlipidemia and hypertension, which were suggestive of nephrotic
syndrome. However, renal ultrasound revealed no notable structural
abnormalities.
 | Table IExamination indices of the proband
during three hospital admissions. |
Table I
Examination indices of the proband
during three hospital admissions.
| | Admission | |
|---|
| Test item | First | Second | Third | Normal value |
|---|
| Biochemical
indices | | | | |
|
Cr,
µmol/l | 1,187 | 1,220 | 2,236 | 44-133 |
|
UA,
µmol/l | 490 | 355 | 360 | 142-416 |
|
BUN,
mmol/l | 9.2 | 8.7 | 8.8 | 1.8-7.1 |
|
FBG,
mmol/l | 32.0 | 30.6 | 34.4 | 3.9-7.0 |
| Urine tests | | | | |
|
Glucose | ++++ | ++++ | ++++ | - |
|
Ketone | +++ | ++ | ++ | - |
|
Protein | + | +++ | +++ | - |
In this MODY-12 pedigree, four other affected
individuals (I-2, II-2, II-5 and III-2) were found to harbor
heterozygous pathogenic ABCC8 variants. Notably, all
carriers developed hyperglycemia before the age of 23 years and
exhibited significantly reduced 2-h postprandial insulin secretion.
These observations indicate that the ABCC8 pathogenic
variants impair pancreatic β-cell function, resulting in
early-onset diabetes. Notably, the other affected family members
also experienced DKA episodes accompanied by transient AKI
(Table II). A review of medical
histories revealed that all affected individuals had been treated
with SGLT2is.
| Table IIClinical data of the family members
with maturity-onset diabetes of the young. |
Table II
Clinical data of the family members
with maturity-onset diabetes of the young.
| | Subject | |
|---|
| Variable | III-4 | III-2 | II-2 | II-5 | I-2 | Normal value |
|---|
| Onset age,
years | 22 | 19 | 20 | 20 | 21 | - |
| BMI,
kg/m2 | 20.5 | 20.3 | 19.7 | 20.5 | 19.3 | 18.5-23.9 |
| ABCC8
p.Arg834Cys | Yes | Yes | Yes | Yes | Yes | - |
| Polydipsia | Yes | Yes | Yes | Yes | Yes | - |
| Diuresis | Yes | Yes | Yes | Yes | Yes | - |
| Polyphagia | Yes | Yes | Yes | Yes | Yes | - |
| Obesity | No | No | No | No | No | - |
| AKI | Yes | Yes | Yes | Yes | Yes | - |
| DKA | Yes | Yes | Yes | Yes | Yes | - |
| Hyperlipidemia | Yes | Yes | Yes | No | Yes | - |
| Blurred vision | Yes | Yes | No | No | No | - |
| FPG, mmol/l | 3.0 | 3.7 | 3.3 | 2.9 | 3.5 | 3.9-6.1 |
| FINS, µIU/ml | 24.6 | 23.2 | 25.1 | 24.8 | 26.2 | 5-20 |
| HOMA-IR | 3.3 | 3.8 | 3.7 | 3.2 | 4.1 | ≤1 |
Comprehensive second-generation
sequencing: Whole-exome, copy number variation and full-length
mitochondrial genome profiling
The second-generation gene sequencing results
revealed a heterozygous missense mutation in the ABCC8 gene
of the proband. This mutation involved the substitution of cytosine
(C) with thymine (T) at base 2,500 in exon 21, which resulted in a
change of the encoded amino acid at position 834 from arginine to
cysteine, resulting in an altered protein tertiary structure
(Fig. 1B and C). The ABCC8 c.2500C>T mutation
identified in the proband is registered in the ClinVar database
(https://www.ncbi.nlm.nih.gov/clinvar/variation/303772/).
The mutation site has been reported by Japanese scholars (13). Although annotated as
ambiguous/possibly benign by the Human Gene Mutation Database
(HGMD), this mutation has been reported in patients diagnosed with
NIDDM (10). The allele frequency
of this gene mutation site in the East Asian population is 0.0003.
It has a REVEL score of 0.852 (indicates a potential impact on
protein function) and is predicted to be damaging or deleterious by
SIFT and PROVEAN, respectively. According to ACMG guidelines, the
combination of PS4_Supporting (indicates that the variant is
observed at a significantly higher frequency in the patient cohort
than in the control cohort), PP3 (indicates that multiple
statistical methods predict the variant will exert a deleterious
effect on the gene or its gene product) and PP1 criteria (denotes
that the mutation co-segregates with the disease within the
pedigree) indicates a likely pathogenicity. In summary, these
findings indicate that the mutation at this site is pathogenic and
deleterious. In the investigated family, the heterozygous carriers
of this variant (III-4, III-2, II-5, II-2 and I-2) all presented
with diabetes, whereas the remaining family members did not carry
this variant or have a history of diabetes.
NGS data were analyzed for CNVs and mitochondrial
DNA variants, but no phenotype-associated CNVs or pathogenic/likely
pathogenic variants were detected.
Bioinformatics analysis
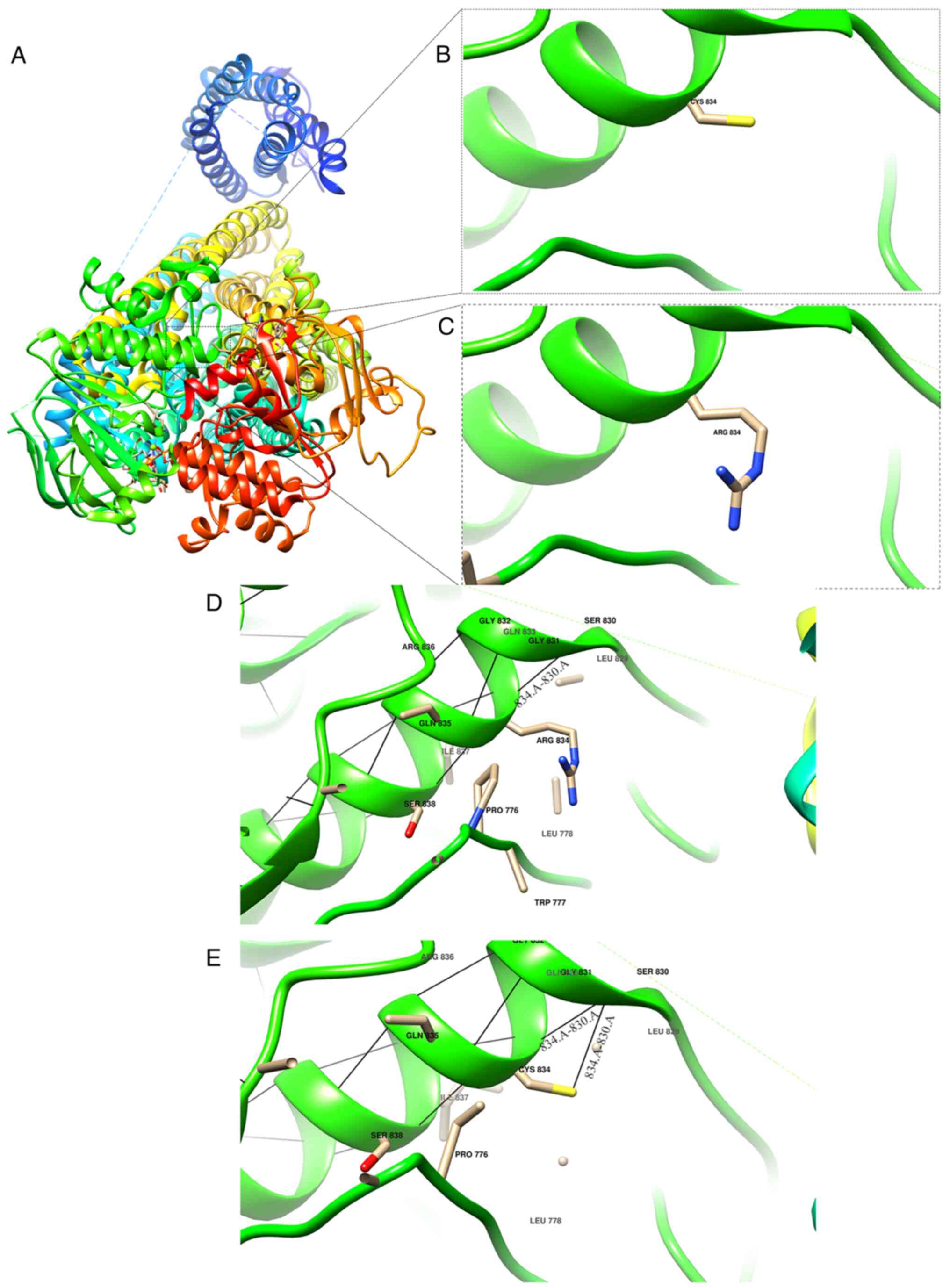
Homology models of the wild-type and p.Arg834Cys
mutant proteins were generated using SWISS-MODEL (Fig. 2A). Structural superimposition
revealed that the p.Arg834Cys substitution, where the positively
charged arginine at residue 834 in the NBD2 region of SUR1 is
replaced with neutral cysteine, induces local conformational
alterations (Fig. 2B and C). In the wild-type structure, Arg834
forms a single hydrogen bond with Ser830, whereas in the
p.Arg834Cys mutant, Cys834 forms two hydrogen bonds with Ser830
(Fig. 2D and E). These alterations induce
conformational alterations that disrupt the native tertiary fold of
the protein. Since protein function is associated with its tertiary
structure, these perturbations are expected to impair SUR1
activity.
Discussion
Among all monogenic forms of diabetes, MODY is the
most prevalent. However, despite the diversity of its subtypes, the
overall occurrence of MODY in clinical practice remains relatively
low, particularly for the rarer subtypes, including MODY-4 and
MODY-6 to MODY-14(1). The
ABCC8 gene is associated with MODY-12. As of April 2023, the
HGMD had cataloged 737 distinct mutations in ABCC8,
highlighting high genetic heterogeneity. Of these, missense and
nonsense mutations are the most common, accounting for 502 cases.
Other reported mutation types include splicing mutations (100
cases), small deletions (81 cases), small insertions (25 cases),
gross deletions (16 cases), small indels (5 cases), gross
insertions/duplications (5 cases), complex rearrangements (2 cases)
and regulatory mutations (1 case).
In the present study, NGS and Sanger sequencing
revealed that all affected individuals in the pedigree with MODY-12
harbored the ABCC8 c.2500C>T (p.Arg834Cys) mutation. To
assess the potential structural impact of this variant, the
SWISS-MODEL server was employed to construct the three-dimensional
structure of the mutant protein. This demonstrated that the genetic
mutation results in the substitution of arginine at residue 834
with cysteine, which induces spatial conformational changes,
primarily due to changes in local electrostatic interactions and
steric effects. The most fundamental difference between the mutant
and wild-type protein is in the properties of the amino acid side
chains; the substitution of the positively charged, elongated side
chain of Arg834 by the neutral, shorter side chain of Cys834
disrupts the local electrostatic potential and markedly diminishes
steric hindrance. In addition, the hydrogen bonding network is
critical for the structural integrity of the protein. However,
following the mutation, an additional hydrogen bond is predicted to
form between the residue at position 834 and Ser830, altering both
the number and orientation of hydrogen bonds. This restricts the
conformational flexibility of these residues and ultimately
distorts the secondary structure of the 830-834 region. In
addition, the reduction in steric hindrance may lead to an adaptive
displacement of adjacent amino acid side chains, thereby
compromising the stability of local hydrophobic residues.
Furthermore, Cys834 has the potential to form disulfide bonds under
oxidative conditions, which could promote aberrant dimerization
(14). Disulfide bond formation
may also interfere with the normal folding process of the SUR1
protein, as observed by 200-nsec molecular dynamics simulations and
documented in KATP channelopathies (15). Collectively, these structural
alterations are predicted to lead to a pronounced conformational
rearrangement of the ATP-binding domain, disrupting key residue
interactions essential for ATPase activity and compromising the
regulatory function of the protein.
A comprehensive review of the LitVar database
indicates that ABCC8 mutations are associated with numerous
disorders, including congenital hyperinsulinemia, neonatal diabetes
and MODY-12 (16,17). For patients harboring these
mutations, particularly those leading to SUR1 subunit dysfunction,
clinical management with oral sulfonylureas has demonstrated
short-term efficacy and safety, as evidenced by significant
reductions in glycated hemoglobin levels within 3 months. However,
careful monitoring and the individualized adjustment of
sulfonylurea dosage is essential to optimize treatment outcomes
(18). In addition, a review of
the literature on ABCC8 gene mutations reveals several
molecular mechanisms underlying MODY, as summarized in Fig. 3A (19,20).
Mutations in the ABCC8 gene primarily impair the expression
and function of the KATP channel, which is essential for
the release of insulin by pancreatic β cells. The KATP
channel is an octamer composed of four Kir6.x pore-forming units
and four regulatory SUR1 subunits (Fig. 3B). ABCC8 mutations markedly
reduce the surface expression of SUR1, thereby decreasing the
overall abundance of functional KATP channels and
disrupting glucose-stimulated insulin secretion (21). Furthermore, ABCC8 modulates
multiple facets of KATP channel activity, including ATP
and Mg²+ sensitivity, β-cell membrane excitability and
the function of voltage-dependent Ca²+ channels
(22). Notably, certain
ABCC8 mutations have been shown to reduce the ATP
sensitivity of KATP channels by ~4-fold. Consequently,
even under hyperglycemic conditions with compensatory changes in β
cells that maintain normal intracellular ATP levels and ATP/ADP
ratios, the closure of KATP channels is delayed,
ultimately delaying the triggering of insulin secretion. The
electrical activity of pancreatic β cells is essential for insulin
exocytosis (19). However, when
KATP channel function is compromised, β cells fail to
depolarize normally, likely due to diminished K+ efflux.
This prevents the proper opening of voltage-dependent
Ca²+ channels and reduces intracellular Ca²+
concentrations, thereby impairing Ca²+-dependent insulin
exocytosis (23). These findings
have been substantiated by quantitative experiments using
patch-clamp and calcium imaging techniques. Furthermore, the
KATP channel contains two nucleotide-binding sites, NBF1
and NBF2, on the SUR1 subunit. NBF1 lacks ATPase activity and
functions independently of Mg²+, whereas NBF2 exhibits
ATPase activity and is highly dependent on Mg²+,
particularly in pancreatic β cells (22). Certain ABCC8 gene mutations
increase the Mg²+ dependence of NBF2, thereby enhancing
overall potassium currents (20),
impairing KATP channel inhibition and ultimately
suppressing insulin secretion.
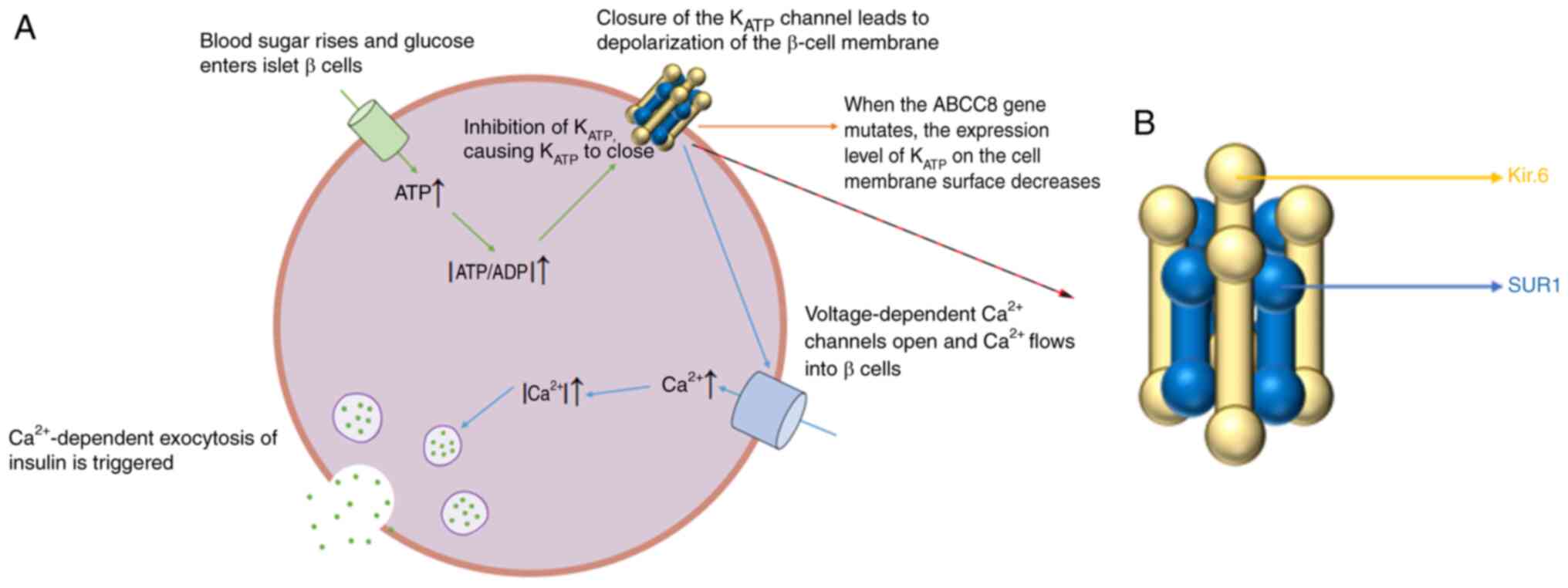 | Figure 3Function and structural organization
of the KATP channel. (A) Role of the KATP
channel in the secretion of insulin by pancreatic β cells.
ABCC8 c.2500C>T (p.Arg834Cys) mutation impairs
KATP channel expression and activity, thereby disrupting
membrane electrophysiology and reducing insulin secretion. (B)
Schematic of the KATP channel octameric assembly,
comprising four Kir6.2 pore-forming subunits and four SUR1
regulatory subunits. KATP, ATP-sensitive potassium;
ABCC8, ATP-binding cassette, subfamily C, member 8; SUR1,
sulfonylurea receptor 1; C, cytosine; T, thymine; Arg, arginine;
Cys, cysteine. |
SGLT-2is are widely used to manage hyperglycemia in
patients with diabetes. They primarily function by inhibiting
glucose reabsorption in the proximal renal tubules, thereby
increasing urinary glucose excretion. This induces osmotic
diuresis, resulting in a notable loss of body fluids (24). In its 2018 consensus report, the
American Diabetes Association and the European Association for the
Study of Diabetes emphasized the clinical value of SGLT-2is,
recommending their use, supported by high-level evidence, in
patients with chronic kidney disease (CKD) and heart failure
(25). A 2022 meta-analysis of
cardiovascular and renal outcomes further demonstrated that
SGLT-2is not only confer cardio-renal benefits but also effectively
reduce mortality in patients with T2DM and diabetic kidney disease.
However, the meta-analysis excluded patients with an estimated
glomerular filtration rate (eGFR) <30 ml/min/1.73 m², a subgroup
that may respond differently to treatment, thus limiting the
generalizability of its findings (26).
The long-term renal protection provided by SGLT-2is
is attributed not only to their beneficial effects on
cardiovascular risk factors, such as the lowering of blood pressure
and improvement of glycemic control, but also to direct renal
mechanisms, including the reduction of intraglomerular pressure and
anti-fibrotic effects. These combined actions have been associated
with a slower decline in eGFR and reduced proteinuria in clinical
studies (27). Investigation of
the potential cardiorenal protective benefits of SGLT-2is in
patients with heart disease has revealed that the osmotic diuresis
induced by these agents may exacerbate a hyperosmolar state and
increase the risk of urinary retention (28). Several mechanisms may account for
the occurrence of AKI in patients treated with SGLT-2is. First,
these agents induce osmotic diuresis, leading to dehydration and a
subsequent reduction in blood volume (29). Second, increased delivery of sodium
to the distal nephron activates tubuloglomerular feedback,
resulting in afferent arteriolar constriction. Third, SGLT-2is may
promote renal artery vasoconstriction, further compromising renal
blood flow. These hemodynamic alterations can collectively lead to
a gradual decline in eGFR (30).
Furthermore, SGLT2is may exacerbate hypoxia in the renal medulla by
reducing local oxygen availability and increasing metabolic demand.
This hypoxic state stabilizes hypoxia-inducible factor (31), resulting in its upregulation, which
in turn elevates plasma erythropoietin levels and promotes
reticulocytosis (32).
Pseudo-AKI is a clinical phenomenon characterized by
transient increases in serum creatinine or reductions in eGFR
caused by extrarenal factors, rather than intrinsic damage to the
renal parenchyma (33). Pseudo-AKI
in patients with MODY-12 is particularly complex. In addition to
the transient eGFR reductions induced by SGLT2i therapy, recurrent
DKA episodes and various comorbidities act as critical triggers.
Firstly, recurrent DKA can impair renal function through two main
mechanisms: i) Severe dehydration and a reduction in effective
circulating blood volume may lead to prerenal AKI (29) and ii) metabolic acidosis may
trigger renal vasoconstriction, further decreasing renal perfusion
and the eGFR (34). Additionally,
CKD reduces the intrinsic resilience of the kidney to acute insults
by inducing structural damage and impairing adaptive repair
mechanisms, with reported odds ratios indicating a significantly
increased risk. Furthermore, cardiovascular diseases, such as heart
failure, reduce cardiac output below critical thresholds, thereby
diminishing renal perfusion and imposing additional hemodynamic
stress on the kidneys. The use of nephrotoxic agents further
exacerbates renal impairment (35), for example, non-steroidal
anti-inflammatory drugs inhibit cyclooxygenase-2, leading to a
reduction in the production of prostaglandin E2, which
is essential for tubular repair (36). Similarly, contrast agents can
trigger oxidative stress, as evidenced by increased malondialdehyde
levels, thereby inducing cellular injury (37). Genetic factors also contribute to
renal vulnerability. Specific mutations in the ABCC8 gene,
such as p.Arg834Cys, disrupt the function of KATP
channels in β-cells, resulting in abnormal lipolysis and elevated
free fatty acid levels. These metabolic disturbances can overwhelm
the capacity of the kidney to adapt, further destabilizing renal
function. In the present study, it was hypothesized that the
pseudo-AKI observed in this pedigree may result from a synergistic
interaction between pharmacological and genetic factors.
Specifically, treatment with SGLT2is increases sodium excretion,
potentially reducing the effective circulating volume and altering
renal tubular dynamics, thereby mimicking AKI. Concurrently, the
ABCC8 c.2500C>T (p.Arg834Cys) mutation may impair
KATP channel function, leading to metabolic disturbances
that increase susceptibility to renal dysfunction. Together, these
factors may contribute synergistically to the development of
pseudo-AKI. A schematic summary of the study is presented in
Fig. 4.
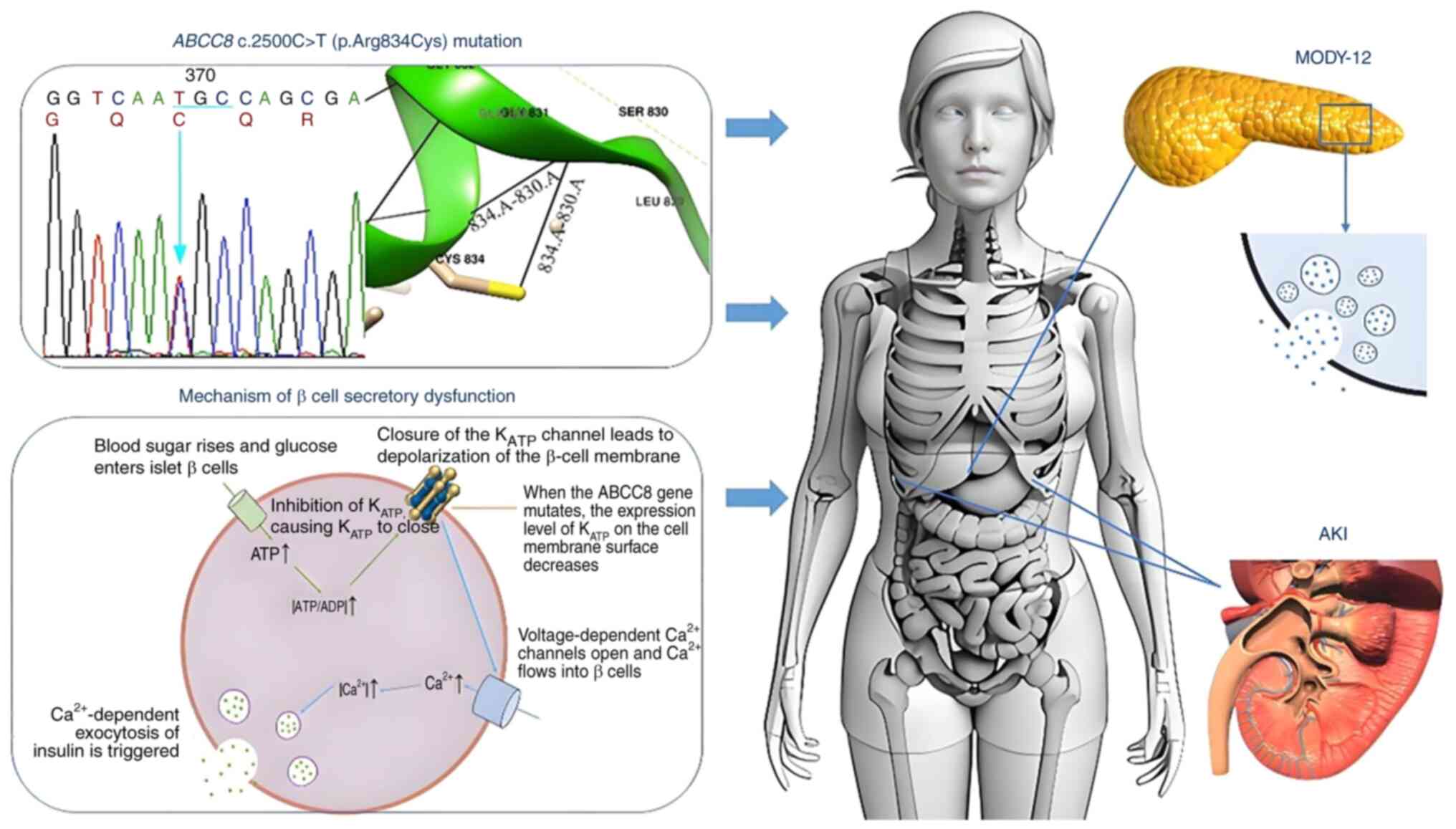 | Figure 4Graphical abstract illustrating the
effects of the pathogenic ABCC8 variant. The ABCC8
c.2500C>T (p.Arg834Cys) pathogenic variant disrupts the tertiary
structure of the protein and compromises KATP channel
activity and glucose-stimulated insulin secretion in pancreatic β
cells, thereby culminating in the clinical manifestations of
MODY-12 and AKI. KATP, ATP-sensitive potassium; ABCC8,
ATP-binding cassette, subfamily C, member 8; C, cytosine; T,
thymine; Arg, arginine; Cys, cysteine; MODY-12, maturity-onset
diabetes of the young-12; AKI, acute kidney injury. |
There are several limitations in the present study.
First, the small sample size limits the ability to perform a
comprehensive analysis of the heterogeneity of renal complications
in MODY-12. To address this, a dedicated MODY-12 disease database
is under development and multi-center cooperative studies with a
larger cohort are planned, thereby increasing statistical power and
improving the generalizability of the findings. Second, although
the structure of the mutant ABCC8-encoded protein was
preliminarily predicted using Swiss-Model, the impact of the
mutation on the dynamic conformation of the protein was not
thoroughly investigated. In future studies, molecular dynamics
simulation software, such as Gromacs, Sybyl or Discovery Studio,
will be employed to simulate and analyze the wild-type and mutant
proteins over an extended time scale. This approach will help to
reveal mutation-induced conformational changes, alterations in the
free energy landscape, and modifications in the interaction network
of key residues. These future studies will provide deeper insights
into the molecular mechanisms underlying the functional impact of
the ABCC8 mutation and lay a theoretical foundation for the
development of precise treatment strategies.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Fujian Province Natural
Science Fund Project (grant nos. 2022J01996, 2022J01409,
2023J011159, 2024J011017, 2022J01417, 2020Y9027 and 2024Y0033),
Joint Funds for the Innovation of Science and Technology in Fujian
Province (grant no. 2023Y9284), the Fujian Province Medical
Innovation Foundation (grant no. 2022CXA001), Startup Fund for
Scientific Research, Traditional Chinese Medicine Science and
Technology Project of Fujian Provincial Health Commission (grant
no. 2021zyjc06), Fujian Medical University (grant nos. 2022QH2042
and 2023QH2038) and the National famous and old Chinese medicine
experts (Xuemei Zhang, Xiaohua Yan, Shaoguang Lv, Chunjin Yi)
inheritance studio construction project.
Availability of data and materials
The data generated in the present study may be found
in the ClinVAR repository under accession number VCV000303772.9 or
at the following URL: https://www.ncbi.nlm.nih.gov/clinvar/variation/303772/,
and in the NCBI BioProject database under accession number
PRJNA1251783 or at the following URL: https://dataview.ncbi.nlm.nih.gov/object/PRJNA1251783.
Authors' contributions
JZ, XC, HPY, YC, DDR, JZ and KYH acquired the data.
QC, JHZ, LZ, MZG and YMG conducted the data analysis and
interpretation. JXL, JZ, XC, LC and HPY wrote the manuscript. LC,
JXL and TMW made critical revisions to the manuscript. JXL, TMW,
LSL and JWL conceived and designed the study. JWL and JZ confirm
the authenticity of all the raw data. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Fujian Provincial Hospital (Fuzhou, China;
K2024-01-027), and all procedures were performed in accordance with
the tenets of the Declaration of Helsinki. All participants and
legal guardians of the minors involved in the present study
provided written informed consent.
Patient consent for publication
Written informed consent for publication was
obtained from the patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Aarthy R, Aston-Mourney K, Mikocka-Walus
A, Radha V, Amutha A, Anjana RM, Unnikrishnan R and Mohan V:
Clinical features, complications and treatment of rarer forms of
maturity-onset diabetes of the young (MODY) - A review. J Diabetes
Complications. 35(107640)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li J, Wang X, Mao H, Wen L, Deng A, Li Y,
Zhang H and Liu C: Precision therapy for three Chinese families
with maturity-onset diabetes of the young (MODY12). Front
Endocrinol (Lausanne). 13(858096)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yahaya TO and Ufuoma SB: Genetics and
pathophysiology of maturity-onset diabetes of the young (MODY): A
review of current trends. Oman Med J. 35(e126)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fajans SS and Bell GI: MODY: History,
genetics, pathophysiology, and clinical decision making. Diabetes
Care. 34:1878–1884. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Shepherd M, Shields B, Hammersley S,
Hudson M, McDonald TJ, Colclough K, Oram RA, Knight B, Hyde C, Cox
J, et al: Systematic population screening, using biomarkers and
genetic testing, identifies 2.5% of the U.K. pediatric diabetes
population with monogenic diabetes. Diabetes Care. 39:1879–1888.
2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Irgens HU, Molnes J, Johansson BB, Ringdal
M, Skrivarhaug T, Undlien DE, Søvik O, Joner G, Molven A and
Njølstad PR: Prevalence of monogenic diabetes in the
population-based norwegian childhood diabetes registry.
Diabetologia. 56:1512–1519. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tattersall RB and Fajans SS: A difference
between the inheritance of classical juvenile-onset and
maturity-onset type diabetes of young people. Diabetes. 24:44–53.
1975.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kant R, Davis A and Verma V:
Maturity-onset diabetes of the young: Rapid evidence review. Am Fam
Physician. 105:162–167. 2022.PubMed/NCBI
|
|
9
|
Ovsyannikova AK, Rymar OD, Shakhtshneider
EV, Voropaeva EN, Ivanoshchuk DE and Voevoda MI: MODY in
Siberia-molecular genetics and clinical characteristics. Diabetes
Mellitus. 20:5–12. 2017.(In Russ).
|
|
10
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF Chimera-a
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004.PubMed/NCBI View Article : Google Scholar
|
|
12
|
American Diabetes Association. 6. Glycemic
targets: Standards of medical care in diabetes-2019. Diabetes Care.
42 (Suppl 1):S61–S70. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ohta Y, Tanizawa Y, Inoue H, Hosaka T,
Ueda K, Matsutani A, Repunte VP, Yamada M, Kurachi Y, Bryan J, et
al: Identification and functional analysis of sulfonylurea receptor
1 variants in Japanese patients with NIDDM. Diabetes. 47:476–481.
1998.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Olson TM and Terzic A: Human K(ATP)
channelopathies: Diseases of metabolic homeostasis. Pflugers Arch.
460:295–306. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fukuda Y, Aguilar-Bryan L, Vaxillaire M,
Dechaume A, Wang Y, Dean M, Moitra K, Bryan J and Schuetz JD:
Conserved intramolecular disulfide bond is critical to trafficking
and fate of ATP-binding cassette (ABC) transporters ABCB6 and
sulfonylurea receptor 1 (SUR1)/ABCC8. J Biol Chem. 286:8481–8492.
2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
De Franco E, Saint-Martin C, Brusgaard K,
Knight Johnson AE, Aguilar-Bryan L, Bowman P, Arnoux JB, Larsen AR,
Sanyoura M, Greeley SAW, et al: Update of variants identified in
the pancreatic β-cell K(ATP) channel genes KCNJ11 and ABCC8 in
individuals with congenital hyperinsulinism and diabetes. Hum
Mutat. 41:884–905. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Babenko AP, Polak M, Cave H, Busiah K,
Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M
and Froguel P: Activating mutations in the ABCC8 gene in neonatal
diabetes mellitus. N Engl J Med. 355:456–466. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rafiq M, Flanagan SE, Patch AM, Shields
BM, Ellard S and Hattersley AT: Neonatal Diabetes International
Collaborative Group. Effective treatment with oral sulfonylureas in
patients with diabetes due to sulfonylurea receptor 1 (SUR1)
mutations. Diabetes Care. 31:204–209. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tarasov AI, Nicolson TJ, Riveline JP,
Taneja TK, Baldwin SA, Baldwin JM, Charpentier G, Gautier JF,
Froguel P, Vaxillaire M and Rutter GA: A rare mutation in
ABCC8/SUR1 leading to altered ATP-sensitive K+ channel activity and
beta-cell glucose sensing is associated with type 2 diabetes in
adults. Diabetes. 57:1595–1604. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Proks P, Girard C and Ashcroft FM:
Functional effects of KCNJ11 mutations causing neonatal diabetes:
Enhanced activation by MgATP. Hum Mol Genet. 14:2717–2726.
2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yan FF, Lin YW, MacMullen C, Ganguly A,
Stanley CA and Shyng SL: Congenital hyperinsulinism associated
ABCC8 mutations that cause defective trafficking of ATP-sensitive
K+ channels: Identification and rescue. Diabetes. 56:2339–2348.
2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
de Wet H, Rees MG, Shimomura K, Aittoniemi
J, Patch AM, Flanagan SE, Ellard S, Hattersley AT, Sansom MS and
Ashcroft FM: Increased ATPase activity produced by mutations at
arginine-1380 in nucleotide-binding domain 2 of ABCC8 causes
neonatal diabetes. Proc Natl Acad Sci USA. 104:18988–18992.
2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ashcroft FM: ATP-sensitive potassium
channelopathies: Focus on insulin secretion. J Clin Invest.
115:2047–2058. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zinman B, Wanner C, Lachin JM, Fitchett D,
Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ,
et al: Empagliflozin, cardiovascular outcomes, and mortality in
type 2 diabetes. N Engl J Med. 373:2117–2128. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Davies MJ, D'Alessio DA, Fradkin J, Kernan
WN, Mathieu C, Mingrone G, Rossing P, Tsapas A, Wexler DJ and Buse
JB: Management of hyperglycemia in type 2 diabetes, 2018. A
consensus report by the American diabetes association (ADA) and the
European association for the study of diabetes (EASD). Diabetes
Care. 41:2669–2701. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kaze AD, Zhuo M, Kim SC, Patorno E and
Paik JM: Association of SGLT2 inhibitors with cardiovascular,
kidney, and safety outcomes among patients with diabetic kidney
disease: A meta-analysis. Cardiovasc Diabetol.
21(47)2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
DeFronzo RA, Norton L and Abdul-Ghani M:
Renal, metabolic and cardiovascular considerations of SGLT2
inhibition. Nat Rev Nephrol. 13:11–26. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hahn K, Ejaz AA, Kanbay M, Lanaspa MA and
Johnson RJ: Acute kidney injury from SGLT2 inhibitors: Potential
mechanisms. Nat Rev Nephrol. 12:711–712. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Myers SR, Glaser NS, Trainor JL, Nigrovic
LE, Garro A, Tzimenatos L, Quayle KS, Kwok MY, Rewers A, Stoner MJ,
et al: Frequency and risk factors of acute kidney injury during
diabetic ketoacidosis in children and association with
neurocognitive outcomes. JAMA Netw Open. 3(e2025481)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yang H, Yang L, Jardine MJ, Arnott C,
Neuen BL, Xu K, Zhao X, Qian D, Cui B, Qiu Y, et al: The
association between sodium-glucose cotransporter 2 inhibitors and
contrast-associated acute kidney injury in patients with type 2
diabetes undergoing angiography: A propensity-matched study. Eur J
Med Res. 29(621)2024.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chang YK, Choi H, Jeong JY, Na KR, Lee KW,
Lim BJ and Choi DE: Dapagliflozin, SGLT2 inhibitor, attenuates
renal ischemia-reperfusion injury. PLoS One.
11(e0158810)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Heyman SN, Rosenberger C, Rosen S and
Khamaisi M: Why is diabetes mellitus a risk factor for
contrast-induced nephropathy? Biomed Res Int.
2013(123589)2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Errabelli P, Lathiya M and Singh D: Case
report: A case of pseudo-acute kidney injury due to
cyclin-dependent kinase inhibitor. Front Nephrol.
4(1389562)2024.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Huang SK, Huang CY, Lin CH, Cheng BW,
Chiang YT, Lee YC, Yeh SN, Chan CI, Chua WK, Lee YJ and Ting WH:
Acute kidney injury is a common complication in children and
adolescents hospitalized for diabetic ketoacidosis. PLoS One.
15(e0239160)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Damman K and Testani JM: The kidney in
heart failure: An update. Eur Heart J. 36:1437–1444.
2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Drozdzal S, Lechowicz K, Szostak B, Rosik
J, Kotfis K, Machoy-Mokrzyńska A, Białecka M, Ciechanowski K and
Gawrońska-Szklarz B: Kidney damage from nonsteroidal
anti-inflammatory drugs-Myth or truth? Review of selected
literature. Pharmacol Res Perspect. 9(e00817)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Deng K, Pei M, Li B, Yang N, Wang Z, Wan
X, Zhong Z, Yang Z and Chen Y: Signal pathways involved in
contrast-induced acute kidney injury. Front Physiol 2024; 15:
1490725.
|