Introduction
Febrile seizures (FS) are the most common cause of
convulsive events during childhood. In addition, they also account
for the majority of seizures in children, occurring in 4-10% of all
cases (1,2). A number of children will continue to
experience frequent bouts of FS after the age of 6 years, which is
referred to as FS+. By contrast, generalized epilepsy with FS+
(GEFS+) is an epilepsy syndrome that is typically diagnosed in
families as a whole, which was included in the classification of
epilepsy syndromes by the International League Against Epilepsy
(3) in 2001. This condition was
first reported in a study by Berkovic and Scheffer in 1997
(4,5). Previous studies have shown that
genetic factors are closely associated with GEFS+. Additionally,
the incidence rate of FS is different among different ethnicities,
with an incidence rate of 2-5% in Europe and the Unites States, but
an incidence rate of 5-14% in Asian countries (6-8).
The risk of FS in individuals that have siblings with FS has been
reported to be 9-22%, where the concordance rate is higher in
identical twins compared with in fraternal twins. Furthermore,
25-40% children with FS were found to have a positive family
history of FS. However, to the best of our knowledge, studies
assessing the association between the incidence rate of GEFS+ and
age remain scarce.
γ-aminobutyric acid (GABA) type A (GABAA)
receptors primarily mediate rapid inhibitory neurotransmissions
that occur in the brain. They are mostly formed by the co-assembly
of two α, two β and one γ subunits (9). Amongst these, the β3 subunit is
encoded by the γ-aminobutyric acid type A receptor β3 subunit
(GABRB3) gene, which is located on chromosome 15q11.2-q12
(10-12).
GABAA receptors are mediators of the rapid inhibition of
synaptic ligand-gated chloride channels in the central nervous
system. When GABA binds to GABAA receptors at
β3/α-specific binding sites at the interface between the subunits,
the ion channel opens. This causes an influx of chloride ions into
the cell, decreasing neuronal excitability (13). Therefore, adequate expression of
each subunit is important for the function of GABAA
receptors. GABRB3 is highly expressed during the early
stages of embryonic brain development and is crucial for the
assembly and transportation of GABA receptors upstream of stem cell
differentiation (14). Gene
mutations in GABRB3 can result in changes to the function of
the channel, leading to a decrease in GABAergic postsynaptic
inhibition. This pathological consequence has been reported to
associate with the heterogeneity of epilepsy (15). Recently, GABRB3 mutations
have been identified in patients with infantile spasms and
Lennox-Gastaut syndrome. However, because of the rarity of
GABRB3-associated GEFS+, the phenotype-genotype association
between GABRB3 mutations and GEFS+ remain to be fully
elucidated. Since relevant cases remain limited and further studies
are required (16).
Case report
At the age of 1 year and 8 months (January 2019), a
female patient (child A; the younger individual of twins) was
admitted to the Affiliated Hospital of Jining Medical University
(Jining, China) with a 3-day fever and one occurrence of
convulsions. The temperature of child A was 38.4˚C, who did not
have a cough, sputum, abdominal pain, diarrhea, nausea or vomiting.
The patient had one convulsion at the beginning of the illness,
which manifested as a loss of consciousness, inability to respond
to calls, an upturning of the eyes, clenching of the hands and
rigidity of the limbs. It lasted for several tens of seconds before
stopping without treatment. During the convulsion, the patient did
not foam at the mouth, with no urinary or fecal incontinence. The
past growth and development of child A were in line with normal
ranges. The parents of the patient were healthy and did not have a
consanguineous marriage. Furthermore, the parents of the patient
and family members denied any history of major diseases. However,
the father and twin sister of the patient had a history of similar
febrile convulsions. On the basis of the medical histories and
relevant auxiliary examination results from the twins, the twins
were diagnosed with a clinical phenotype of FS syndrome (FS+), with
seizures that were mainly in the form of cataplexy and focal
seizures.
Results of a physical examination were as follows:
Temperature, 36.8˚C; pulse, 108 beats/min; respiration, 28
breaths/min; weight, 15.5 kg; and height, 97 cm. The patient was
alert, in no acute distress, had achieved age-appropriate
developmental milestones, was well-nourished, and exhibited a ruddy
complexion. There were no observable rashes on the hands, feet or
buttocks. Child A had pharyngeal congestion, with thick respiratory
sounds in both lungs, but dry or wet rales could not be heard. The
heart sound were normal and crisp, where the abdomen was soft,
without abdominal distension. The liver and spleen were not
enlarged, whereas Brudzinski's, Kernig's and Babinski's signs were
all negative. The results of relevant laboratory tests were within
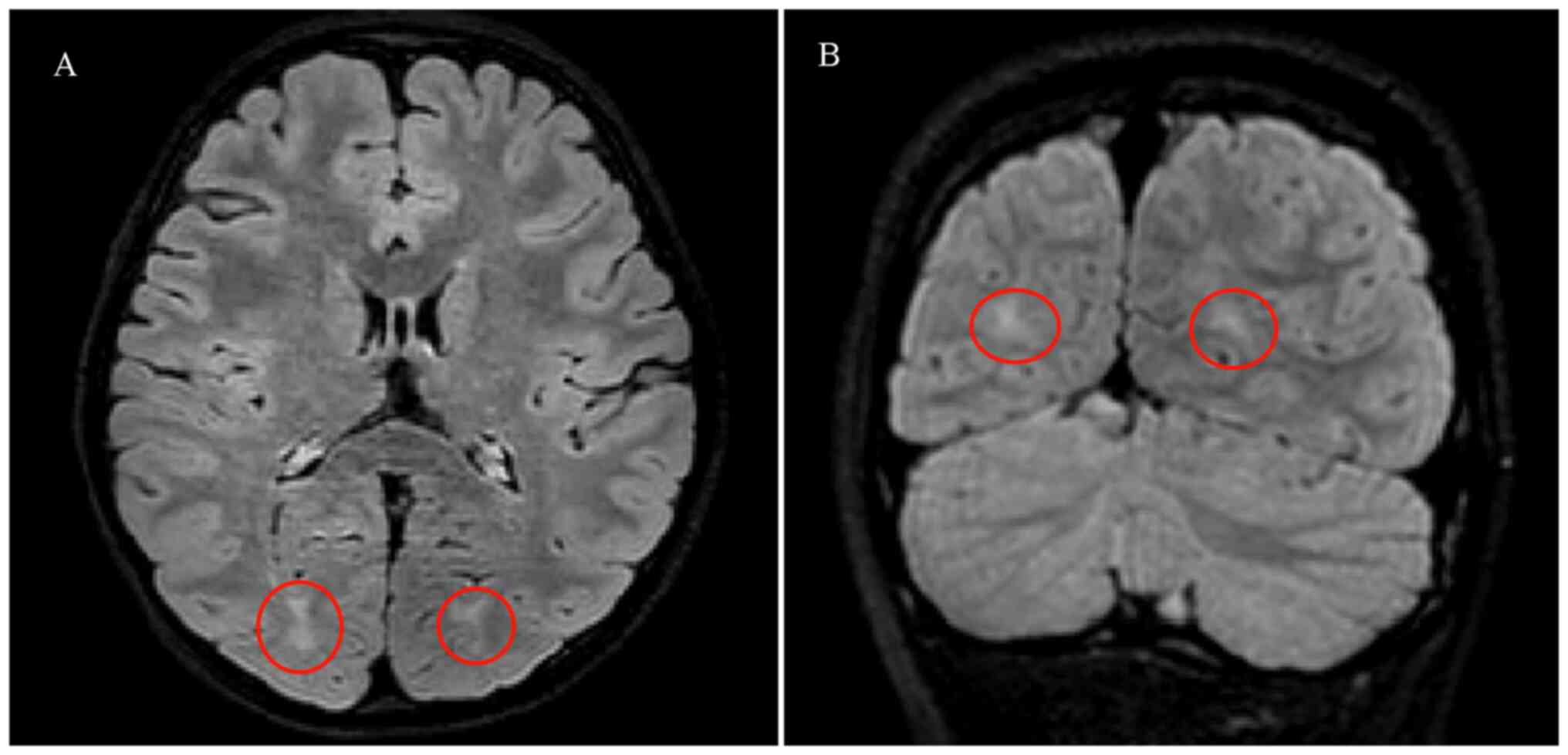
normal limits. From cranial MRI, the bilateral cerebral hemispheric
structure was symmetrical and the brain white matter contrast was
normal. However, a patchy foci of slightly high abnormal
T2-fluid-attenuated inversion recovery signals were observed in the
bilateral occipital lobes (Fig.
1). Abnormalities were not observed in the patient during
electroencephalography (Fig. 2).
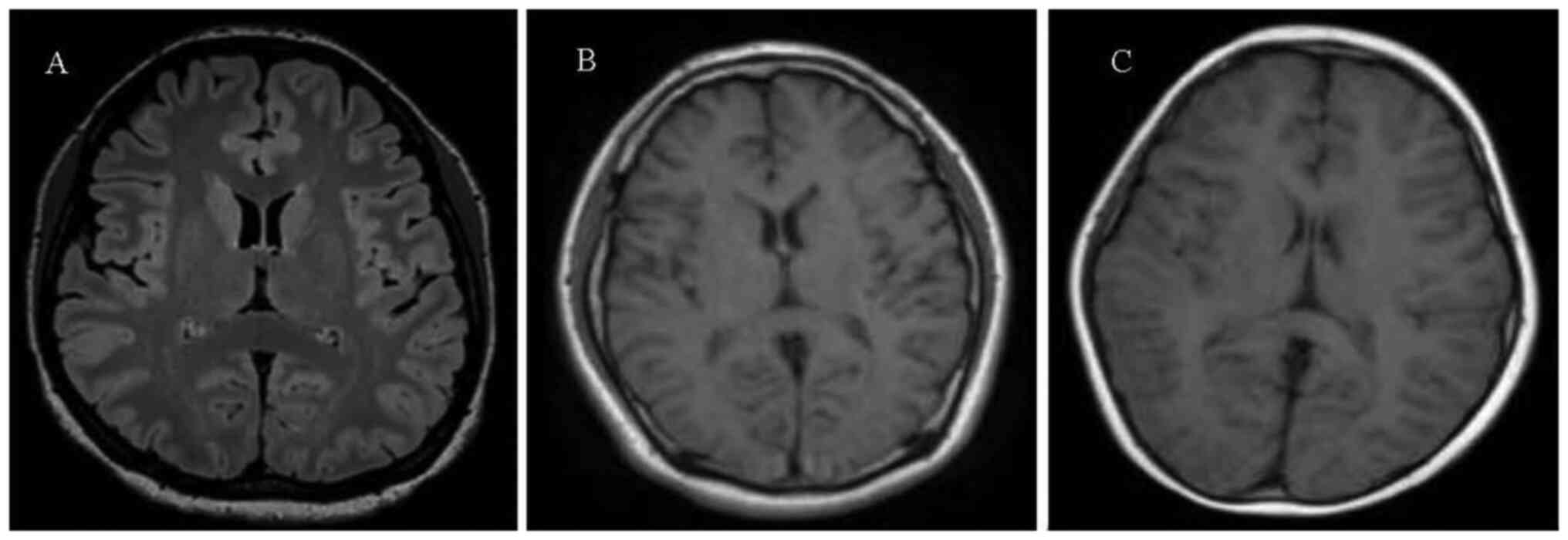
In addition, abnormalities were not observed in the cranial MRI
examinations of the mother (Fig.
3A), father (Fig. 3B) and the
twin sister (child B; Fig. 3C) of
child A.
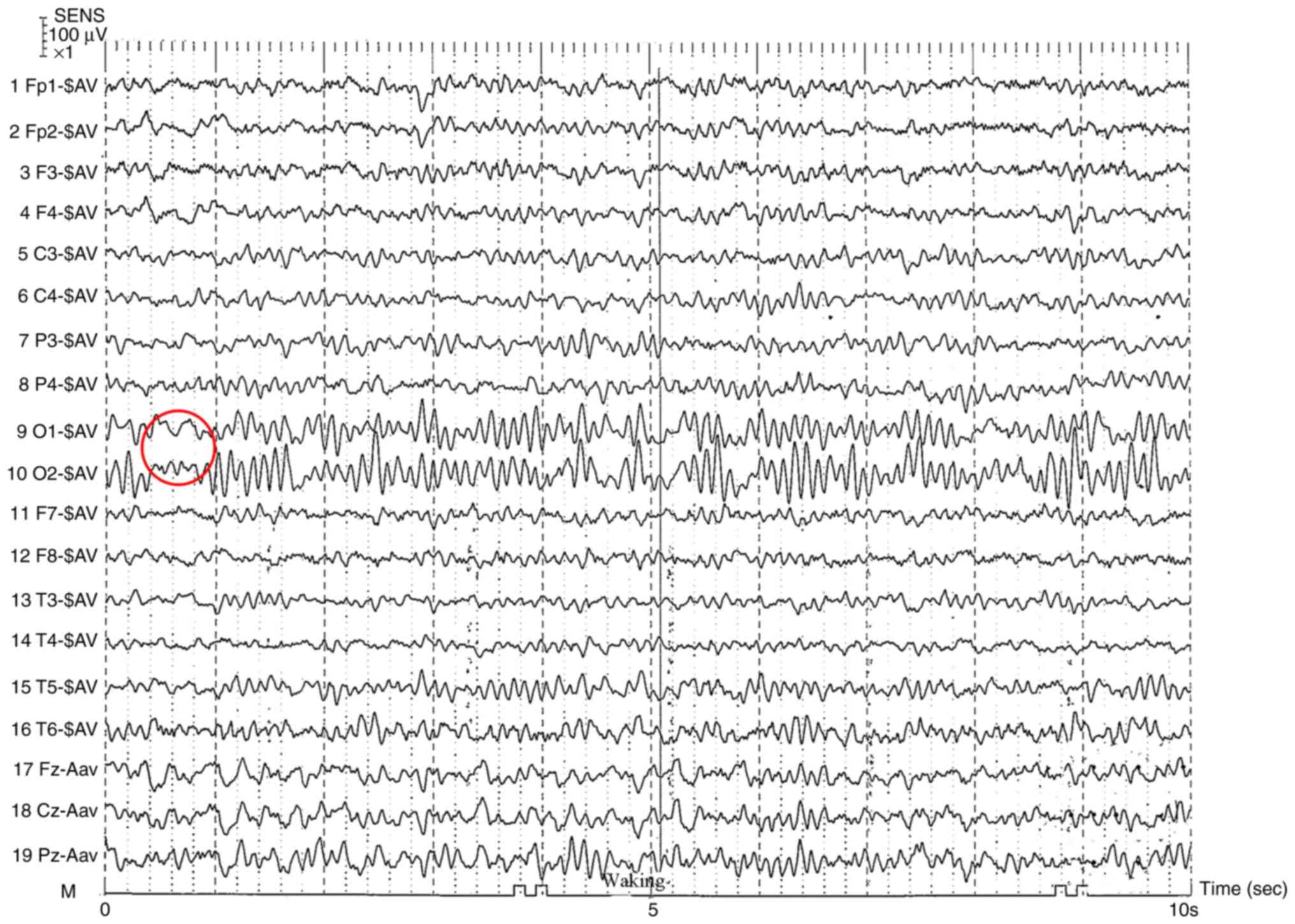 | Figure 2Electroencephalography of the
patient. During the awake, quiet, and eye-closed state, bilateral
occipital regions exhibited 10.0-11.0 Hz alpha rhythm at 40-60 µV,
intermixed with sparse irregular waves. Activity was approximately
symmetrical on the left and right sides, indicating that the
regulation of amplitude modulation was acceptable. With the eyes
closed α inhibition and appearance was observed. Peak, spindle and
slow waves were observed during the sleep period, and the left and
right sides were generally symmetrical, indicating the absence of a
sleep cycle disorder. SENS, sensitivity; Fp1, frontopolar left;
Fp2, frontopolar right; F3, frontal left; F4, frontal right; C3,
central left; C4, central right; P3, parietal left; P4, parietal
right; O1, occipital left; O2, occipital right; F7, anterior
temporal left; F8, anterior temporal right; T3, mid-temporal left;
T4, mid-temporal right; T5, posterior temporal left; T6, posterior
temporal right; Fz, frontal midline; Cz, central midline; Pz,
parietal midline; -SAV, scalp average reference; -Aav, auricular
average reference; M, ground electrode. |
Child A was diagnosed with ‘epilepsy observation’
from a referral hospital (specific details unknown) in December
2019, and immediately started to take 2 ml levetiracetam oral
solution (100 mg/ml) twice daily. No seizures occurred during the
treatment period. However, after stopping the medication for 2 days
in June 2020, the patient had one seizure. Therefore, the patient
continued to take 2 ml levetiracetam (100 mg/ml) twice daily. In
July 2020, child A experienced a seizure after another fever, which
was characterized by a trance-like consciousness, dazed eyes,
staggering when walking and an inability to respond to calls. The
seizure was also accompanied by tachypnea and bilateral manual
automatisms, more prominent on the right side, followed by gradual
assumption of a prone position. No clonic movements were observed
during the ictal phase. The seizure lasted for ~5 min before it was
relieved. Subsequently, in the outpatient department of the
hospital, the medication was adjusted to 2.5 ml levetiracetam (100
mg/ml) twice daily, following which no further seizures were
reported. In September 2022, the levetiracetam dosage was reduced
by 0.5 ml every ~2 months. In April 2023, at which time the
levetiracetam dosage was 0.5 ml in the morning and 1.0 ml in the
evening), the patient had another fever (body temperature of
38.0˚C) and again experienced a seizure. After a consultation in
our epilepsy clinic, it was recommended to increase the dosage of
levetiracetam (100 mg/ml) back to 2.5 ml twice daily. Child A was
followed up to the present day and was aged 6 years and 5 months at
the last follow-up appointment (November 2023). Despite regular
medication, the patient still experiences seizures, which manifest
as trance-like consciousness, dazed eyes and an inability to answer
calls. The seizures typically lasted 5-6 min before resolving.
Child B, the elder twin sister, had symptoms similar
to FS. Fevers >38˚C occasionally induced convulsions, which
manifested as a loss of consciousness, inability to respond to
calls, staring eyes, cyanosis, limb convulsions and foaming at the
mouth. These seizures lasted for 1-2 min before relieving. In
January 2020, child B began oral treatment of 2 ml levetiracetam
(100 mg/ml) twice daily. The patient discontinued the medication
for 3 days during treatment and then continued oral administration,
during which time there were no seizures. In May 2022, the patient
began to reduce the dosage of levetiracetam (100 mg/ml) by 0.5 ml
every ~2 months, before discontinuing the medication in December
2022. However, in the middle of December 2022, the patient
experienced a fever that was induced by coronavirus-19 disease.
Convulsions occurred twice when the body temperature of the patient
reached 38˚C, with each seizure lasting for ~2 min at an interval
of 8 h. In March 2023, convulsions again occurred as the result of
a fever that was caused by influenza A infection. The patient was
in a standing position and suddenly fell, which was followed by
convulsions that lasted for 2-3 min before they were relieved. The
patient then slept and woke up with no abnormalities. In September
2023, the patient again developed a fever, which reached 39.4˚C and
convulsions occurred (the specific details are unknown). From
December 2022 until October 2023, no medication was used. However,
in October 2023, the patient came to our outpatient service and was
prescribed 2.5 ml levetiracetam (100 mg/ml). At present, although
the patient is maintained on 2.5 ml levetiracetam twice daily, the
patient still experiences FS.
Neither child A nor child B received long-term
systematic hospitalization treatment, meaning that they did not
receive any specialist care. When the twins do not experience
fever-associated convulsions, their quality of life was not
affected and they exhibit regular growth and development.
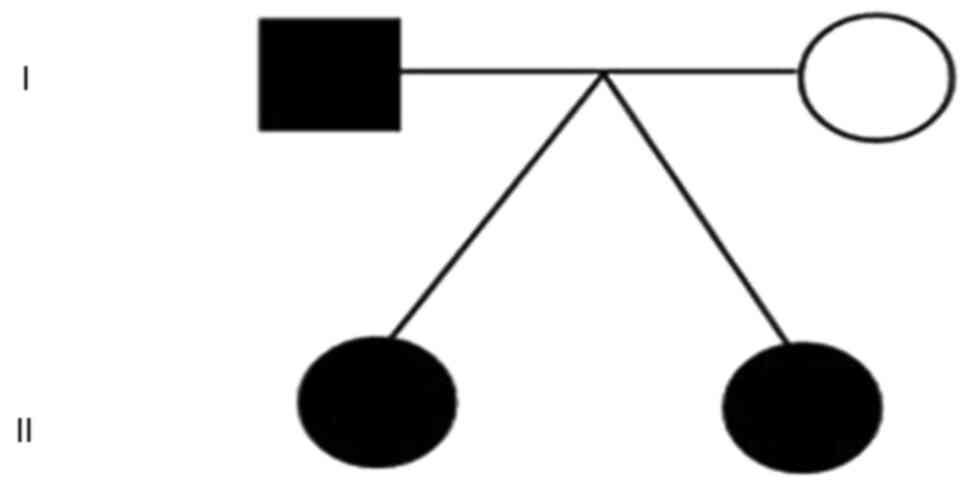
The father of the patients also experienced FS
during childhood, although the exact manifestations of the seizures
were not recalled. However, the father did not take any
antiepileptic drugs and does not currently experience seizures (as
of November 2023, the individual was 38 years old.). The disease
status of each family member is presented in Fig. 4.
In total, 4 ml venous blood of the proband and 2 ml
blood samples of Child A's parents and sister were collected and
sent to Beijing Kangxu Medical Laboratory for whole-exon detection.
Genomic DNA was extracted from blood samples using the Qiagen
FlexiGene DNA Kit (cat. no. 51206; Qiagen GmbH) and the genomic DNA
quality control was performed. Agarose gel electrophoresis was used
to analyze the degree of DNA degradation and whether there was RNA
and protein contamination. Qubit 2.0 fluorometer was used to
quantify DNA concentrations. The Covaris Ultrasonic DNA
Fragmentation Instrument (Covaris LLC) was used to randomly
interrupt fragments of genomic DNA with a growth of 180-280 bp
using the SureSelect XT Library Prep Kit ILM (cat. no. 5190-8863;
Agilent Technologies, Inc.). After repairing the end of DNA
fragment and adding A-tail, DNA library was prepared by connecting
splices at both ends of the fragment. The DNA library was amplified
using the TransNGS Index Primers (384) Kit (cat. no. 3KI241;
TransGen Biotech Co., Ltd.) according to the manufacturer's
protocol: Initial denaturation was at 95˚C for 10 min; followed by
35 cycles of 30 sec at 95˚C, 30 sec at 60˚C and 45 sec at 72˚C,
with a final extension at 72˚C for 5 min. The subsequent
adaptor-specific primers were employed for the amplification of the
DNA library: Forward, 5'-GGGGAGTCAGGTGCAAGAG-s-T-3' and reverse
5'-GAAGCGACAGTCACAACTTCC-s-T-3' (-s-represents a phosphorothioate
bond).
A library with a specific index tag was hybridized
in liquid phase with up to 500,000 biotin-labeled Agilent
SureSelect Human ALL Exon V6 probes (cat. no. 5190-8863; Agilent
Technologies, Inc.), before being captured using streptomycin
beads. After PCR linear amplification, the library was inspected
and sequenced. After the library construction is completed, use
Qubit 2.0 fluorometer (Thermo Fisher Scientific, Inc.) was used for
preliminary quantification, before Agilent 2200 (Agilent
Technologies, Inc.) was used to detect the insert size of the
library. The final concentration of the library was 95.1 ng/µl for
Child A, 71.3 ng/µl for Child A's father, and 96.6 ng/µl for Child
A's mother. Subsequently, NovaSeq6000 S4 Reagent Kit v1.5 (300
cycles; cat. no. 20012866; Illumina, Inc.) was used to perform
double-ended sequencing on the NovaSeq 6000 (Illumina, Inc.)
platform, with each end measuring 150 bp and an average sequencing
depth of 100X and the data in fastq format was obtained.
The sequencing read was compared to the Human
reference Genome (hg19 version) using the BWA tool (v0.7.15). The
comparison results were converted into bam format for variation
analysis. GATK (v3.6; https://www.broadinstitute.org) was used to detect
single nucleotide mutations and small insertion loss variations.
CODEX (v1.14.1; https://www.bioconductor.org/packages/release/bioc/html/CODEX.html),
XHMM (v1.0; (https://zzz.bwh.harvard.edu/xhmm/index.shtml) and Kang
Xu Capture sequencing copy number variation detection software
V1.0) were used to analyze possible copy number variations.
Gene-related annotation analysis was performed using the reference
genome version GRCH37/Hg19 (https://www.ncbi.nlm.nih.gov/refseq), Ensembl
(http://grch37.ensembl.org/Homo_sapiens/Info/Index) and
UCSC (version GRCH37/Hg19 reference genome; https://genome.ucsc.edu) databases. The 1000G (2015
update; http://www.1000genomes.org), dbSNP
(v150; https://www.ncbi.nlm.nih.gov/SNP) and ExAC (v0.3; ExAC
is now in gnomAD; www.gnomad-sg.org) tools were used to annotate the
frequency of variation in a population. PolyPhen2 (version 2;
http://genetics.bwh.harvard.edu/pph2), SIFT (version
2; https://sift.bii.a-star.edu.sg) and
MutationTaster (NCBI 37/Ensembl 69; http://www.mutationtaster.org) were used for protein
gene locus mutation function damage prediction. OMIM (https://www.omim.org/), HGMD (http://www.hgmd.org) and ClinVar annotations
(https://submit.ncbi.nlm.nih.gov/clinvar/) were used to
make disease-related annotations associated with disease. Sites
were classified using the American Society for Medical Genetics and
Genomics (ACMG) variation scale with the Society for Molecular
Pathology system (pathogenic, possibly pathogenic, of uncertain
significance, possibly benign and benign). The annotation results
are filtered according to the characteristics of the mutation sites
in the database and the variation that may be associated with the
disease is retained.
Subsequently, candidate mutation sites were
investigated using Sanger sequencing. The human genome GenBank
database-gene sequences, the Primer design website design primers
and Primer Z (http://genepipe.ncgm.sinica.edu.tw/primerz/primerz4.do).
PCR amplification was then performed, before the ABI 3730
generation sequencer (Thermo Fisher Scientific, Inc.) was used for
the sequencing of the amplification products. The obtained data
were visualized using the Chromas software (V2.6.6) and compared
with the results of whole exome sequencing.
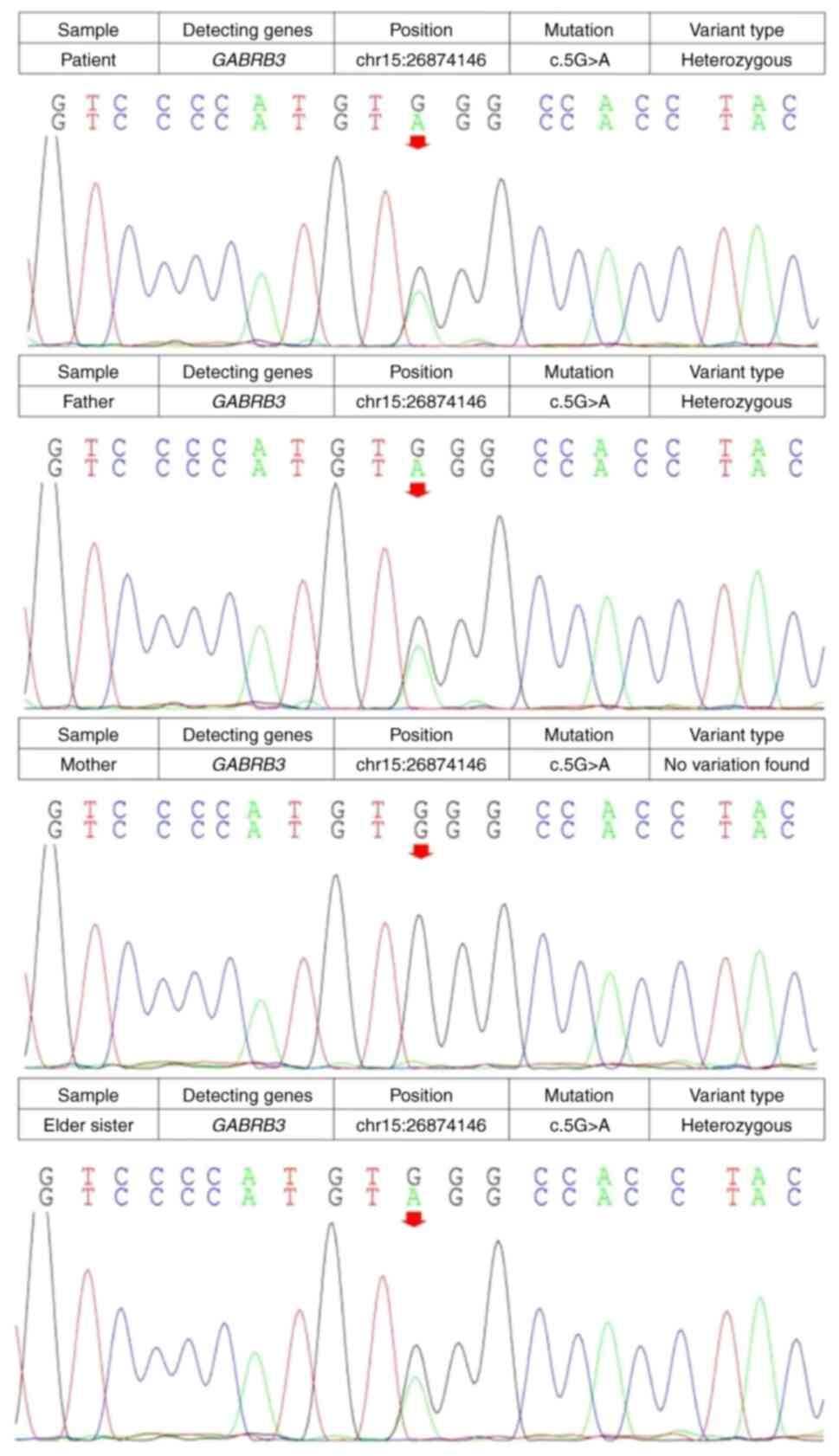
A nonsense mutation in GABRB3 (c.5G>A,
p.Trp2*) was detected in both of the patients and their father.
This mutation was present in the father of the patients but not in
their mother, suggesting that it was of paternal origin (Fig. 5). c.5G>A, p.Trp2* is Exon 1, the
second amino acid encoded by this gene changes from tryptophan
(TRP) to a stop codon, causing protein translation to terminate
early.
The American College of Medical Genetics and
Genomics criteria indicated very strong pathogenic evidence
(PVS1_VeryStrong; PS4). ‘PVS1_VeryStrong’ signify the pathogenic
mechanism of a gene is through the loss of function, non-sense
mutations, frameshift mutations, ±1 or 2 position splicing
mutations, start codon variations, or single or multiple exon
deletion mutations on the gene. Strong pathogenic evidence ‘PS4’
means that the frequency of mutations occurring in the relevant
patient population is notably higher compared with that in the
control group.
Discussion
GEFS+ is an ion channel disease for which the
pathogenesis has not been fully elucidated, although previous
studies have revealed a genetic susceptibility. The mechanism of
GEFS+ inheritance is complex, although the majority of the cases
autosomal dominant. This condition is characterized by phenotypic
and genetic heterogeneity, with diverse clinical phenotypes
(17). Numerous candidate genes
have been reported to be associated with FS or FS+, including GABA
receptor-associated genes (such as GABRA1, GABRB3,
GABRD and GABRG2), voltage-gated sodium ion
channel-associated genes (SCN1A, SCN1B, SCN2A
and SCN9A) and transport-associated genes, such as
SLC12A5 and SLC32A1 (18-21).
This type of genetically associated FS+ is known as GEFS+, which is
a common familial epilepsy syndrome. Therefore, a history of FS and
FS+ in family members is important for the diagnosis of GEFS+ in an
individual.
GABA is one of the most important inhibitory
neurotransmitter in the brain. It serves an important role in
epilepsy onset and development (22). Abnormalities in various aspects of
GABA metabolism can lead to seizures. Under normal circumstances,
GABA maintains a balance of neurotransmitter content in the brain
through the glutamate/GABA/glutamate cycle (23), to ensure its inhibitory effects are
exerted. When genetic alterations result in the inability of
mutated subunits to pair with normal subunits, GABA receptor
assembly becomes impaired to obstruct receptor function (14). This frequently results in epileptic
syndromes with different clinical phenotypes, either due to
downregulated surface receptor expression or decreased magnitudes
of GABA-induced inhibitory potentials (22,24).
All GABA-related receptors have similar structures, with four
transmembrane (TM) domains (TM1 to TM4) and a long extracellular
N-terminal domain. Ion channel pores are formed by the TM2 of each
subunit. The pathogenesis of GABRB3 mutations is due to the
function of the protein domain (11,12).
Regardless of the receptor, mutations located in genes encoding the
TM regions of a receptor subunit are associated with more severe
phenotypes, whereas mutations in the N-terminal appear to be
associated with milder phenotypes. Rather than being associated
with the mutated gene, the clinical phenotypes of
GABRB3-related mutations are more likely associated with the
location of the mutation within the structure of the protein
(25-27).
GABRB3 has nine exons in total, is ~230 kb in
length and is highly expressed in various regions of the brain. The
β3 subunit encoded by this gene is the earliest β subunit to appear
in the embryonic brain, where its expression level in the perinatal
brain is typically 150% higher compared with that in the adult
brain (24). Data from previous
studies using rodent models suggest that β3 subunits are expressed
during development, but that the expression of β3 declines
postnatally (28,29). As a GABAA receptor
subunit, β3 serves a central role in GABAA receptor
assembly and trafficking to the cell surface (30). Unlike other GABAA
receptor subunits, which require assembly with accompanying
subunits for surface localization, the β3 subunit can be
transported to the cell surface and form homologous pentamers when
expressed alone because it contains four amino acids (arginine 180,
glutamate 179, lysine 173 and glycine 171). This is specific to the
β3 subunit, suggesting its unique ability for homologous
oligomerization and membrane targeting (14). Considering the etiology of
neurodevelopmental disorders, GABRB3 is abundant in the
early brain and has been reported to induce stem cell
differentiation (31).
Furthermore, β3 subunit-dependent phosphorylation mediates
GABAA receptor accumulation and the immobilization of
the inhibitory synaptic scaffold protein gephyrin at synapses.
These events are crucial for the long-term potentiation of
inhibition and may modulate network excitability (32). In addition, β3 subunit-adaptor
protein 2 interactions stabilize GABAA receptors at
endocytic zones and may serve a role in regulating the number of
synaptic receptors during inhibitory synaptic plasticity (33). These aforementioned findings
indicate that β3 subunits serve a role in regulating synaptic
strength and brain development. In the present case, a
GABRB3 mutation was identified, which may cause changes to
the structure and function of the β3 subunit, resulting in it being
unable to pair and bind with other subunits normally, thereby
reducing the expression levels of the GABAA receptor.
This may lead to the decreased postsynaptic inhibitory effect of
GABA, preventing it from exerting its inhibitory neurotransmitter
effects and affecting the excitability of the neural network,
increasing the risk of seizures.
Exons 1 and 1A of GABRB3, along with exons
2-9, produce selective mRNA transcripts that two specific signal
peptide sequences are derived from (34). These sequences encode two mature
polypeptides that have slight variations in the N-terminal residues
and produce different signaling peptide sequences. Therefore, a
mutation may lead to structural modifications of the β3 subunit of
GABAA receptors (35).
Previous studies of GABA gene-associated childhood catatonic
epilepsy have identified various missense mutations, such as
(c.31C>T; p.Pro11Ser), (c.44C>T; p.Ser15Phe) and (c.94G>A;
p.Gly32Arg), all of which are located in exon 1A of GABRB3.
These mutations presumably cause the hyperglycosylation of
GABAA receptor β3 subunit proteins, causing a decrease
in GABA-evoked currents, leading to seizures (16). In the present case, it was
hypothesized that the identified mutation (c.5G>A, p.Trp2*) may
alter the structure of the GABAA receptors and lead to
its dysfunction, resulting in an impaired GABA-mediated inhibition
in the brain and the development of GEFS+ in children.
The first case of epilepsy associated with a
GABRB3 mutation was reported in 2008(35). An increasing number of reports have
shown a strong association between GABRB3 mutations and
epilepsy (36,37). The majority of known cases of
GABRB3 mutations are in children with early onset epileptic
encephalopathy (34). Furthermore,
Tanaka et al (35)
previously reported that GABRB3 mutations are associated
with childhood absence epilepsy. GABRB3 mutation-associated
epileptic seizures can take a variety of forms, including
generalized tonic-clonic, tonic, infantile spasms, myoclonic and
atonic (16,35,38).
The majority of such seizures tended to have a febrile trigger in
affected patients. Patients tend to exhibit different degrees of
delays in language, motor and intellectual development. In
addition, a number of patients have demonstrated various
mental/behavioral disorders, such as restlessness, autism spectrum
disorder, attention deficit hyperactivity disorder or aggressive
behavior. In terms of clinical phenotypes, GABRB3
variant-associated epilepsy can manifest as multiple forms of
epileptic syndromes (16,35,38).
The prognosis is generally favorable for catatonic epilepsy, FS and
additional febrile convulsions. However, prognosis is poor for
patients with Lennox-Gastaut syndrome, West syndrome and infantile
spasms, which are frequently associated with parenchymal damage).
In addition, a number of children with epileptic phenotypes of FS
with additional symptoms and catatonic epilepsy have normal
intellectual, motor and language development (16,39).
Upon examination using imaging, the majority of patients show a
normal MRI of the cranium, although a small number of patients will
show abnormalities, such as reduced myelin sheaths, multiple gyrus
malformations, cerebellar hypoplasia, abnormalities of the corpus
callosum or severe diffuse cerebral atrophy (16). In the present case, the twins and
their father had a GABRB3 mutation that was not identified
in the mother. Brain MRI scans of this family revealed no notable
abnormalities, further indicating that patients with GABRB3
mutation-associated FS have a mild phenotype and favorable
prognosis.
In the present case report, the twins presented with
seizures in early childhood, where the majority of seizures
occurred after a fever of >38˚C. The seizures were characterized
by a loss of consciousness, staring, cyanosis, staggering and
convulsions of the limbs with or without foaming at the mouth,
which lasted for a period of a few sec to lasting seconds to
minutes. This was confirmed from the medical histories of the twins
and the results of auxiliary examinations. Upon follow-up (November
2023), it was revealed that the twins still experienced seizures
despite being aged >6 years. A nonsense mutation in
GABRB3 (c.5G>A, p.Trp2*) was detected in the twins and
their father, which revealed a familial predisposition, with a
history of FS in the father. After performing a literature review,
it was considered that this gene may be associated with the
seizures in the twins (they may have GABRB3
mutation-associated GEFS+). However, in the present case, the twins
and their father had different seizure patterns. Since
GABRB3 expression is high in the embryonic brain and
gradually decreases in adulthood, although the mutated genes were
the same, the associated clinical phenotypes may differ because of
the differences in expression levels among different individuals.
However, notable genotype-phenotype associations have previously
been reported in terms of the localization of variants within the
protein domain of GABAA receptor subunits. Additionally,
an in-depth historical and literature review has corroborated that
the prevalence of GEFS+ may be age-independent. In the present
case, the seizures of the twins may have decreased compared with
before. The reasons for this may be as follows: i) The twins now
regularly use medication and the drug-controlled effect is
satisfactory; ii) the frequency of the fevers that the twins
experience has decreased compared with before, thereby reducing the
likelihood of seizures; and iii) compared with infancy, the
expression of GABRB3 is reduced in adulthood, meaning that
the human body may be less affected by its level of expression.
Therapeutically, generally acceptable seizure
control has been reported in cases of GABRB3
mutation-associated epilepsy after treatment with sodium valproate,
perampanel and clonazepam (40).
The method of drug selection for GEFS+ is mainly based on the
clinical phenotype and pathogenesis. Valproic acid and
carbamazepine are typically used as first-line drugs, though newer
antiepileptic drugs, such as lamotrigine and levetiracetam, are
becoming more widely used because of their higher safety profiles
and milder side effects. Amongst these aforementioned new drugs,
levetiracetam selectively inhibits high-voltage-activated calcium
channels and reduces calcium release from intraneural stores.
Furthermore, it has no inhibitory effects on simple seizures
induced by convulsant stimulation and shows only weak activity in
maximal stimulation and threshold tests (41). In vitro, levetiracetam has
been reported to exert no effects on neuronal voltage-gated sodium
channels or T-type calcium currents, nor did it directly predispose
cells to GABAergic neurotransmission. However, it did counteract
the activity of negative regulators of GABA-activated currents and
glycine-gated currents, whilst partially inhibiting N-type calcium
currents in neuronal cells (40).
Furthermore, levetiracetam has been documented to inhibit
hippocampal epileptiform bursts of discharge without affecting
normal neuronal excitability, suggesting that it may selectively
inhibit epileptiform bursts of discharge, super-synchronization and
seizure propagation (42). In the
present case, levetiracetam was administered to the twins for
seizure control with favorable outcomes. However, they both had
difficulties in discontinuing the drug. At the latest follow-up,
they remain dependent on levetiracetam regularly for seizure
control. Therefore, the present case may provide assistance to
clinicians when treating children with GEFS+.
Acknowledgements
Not applicable.
Funding
Funding: The present case was supported by the Key Research and
Development Plan of Jining City (grant nos. 2023YXNS061,
2023YXNS028 and 2023YXNS111), the Shandong Provincial Natural
Science Foundation (grant no. ZR2023QH076), and Lin He's
Academician Workstation of New Medicine and Clinical Translation in
Jining Medical University (grant no. JYHL2021FMS18).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The novel c.5G>A
(p.Trp2*) missense mutation in GABRB3 was deposited in SRA
(https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1272306/;
accession no. PRJNA1272306).
Authors' contributions
SL, RL and QK designed the study. YW, KF, SR, LW, JG
and XC collected the data. SL, JL, RL and QL contributed to data
analysis and interpretation. SL and RL drafted the manuscript. QK
and RL contributed to the revision. SL and JL confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The present case was approved by The Ethics
Committee of The Affiliated Hospital of Jining Medical University
(approval no. 2023-09-C031; Jining, China).
Patient consent for publication
The patients' parents provided written informed
consent for the publication of any associated data, as well as
accompanying images and videos.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Smith DK, Sadler KP and Benedum M: Febrile
seizures: Risks, evaluation, and prognosis. Am Fam Physician.
99:445–450. 2019.PubMed/NCBI
|
|
2
|
Gupta A: Febrile seizures. Continuum
(Minneap Minn). 22:51–59. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Specchio N, Wirrell EC, Scheffer IE,
Nabbout R, Riney K, Samia P, Guerreiro M, Gwer S, Zuberi SM,
Wilmshurst JM, et al: International league against epilepsy
classification and definition of epilepsy syndromes with onset in
childhood: Position paper by the ILAE task force on nosology and
definitions. Epilepsia. 63:1398–1442. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Liu XW, Li W, Han T, Wei K, Qiao S, Su L
and Chi Z: The finding of a new heterozygous mutation site of the
SCN2A gene in a monozygotic twin family carrying and
exhibiting genetic epilepsy with febrile seizures plus (GEFS+)
using targeted next-generation sequencing. Clin Neurol Neurosurg.
169:86–91. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Berkovic SF and Scheffer IE: Epilepsies
with single gene inheritance. Brain Dev. 19:13–18. 1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Canpolat M, Per H, Gumus H, Elmali F and
Kumandas S: Investigating the prevalence of febrile convulsion in
Kayseri, Turkey: An assessment of the risk factors for recurrence
of febrile convulsion and for development of epilepsy. Seizure.
55:36–47. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Subcommittee on Febrile Seizures; American
Academy of Pediatrics. Neurodiagnostic evaluation of the child with
a simple febrile seizure. Pediatrics. 127:389–394. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Paul SP, Seymour M, Flower D and Rogers E:
Febrile convulsions in children. Nurs Child Young People. 27:14–15.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hörtnagl H, Tasan RO, Wieselthaler A,
Kirchmair E, Sieghart W and Sperk G: Patterns of mRNA and protein
expression for 12 GABAA receptor subunits in the mouse
brain. Neuroscience. 236:345–372. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kim JJ, Gharpure A, Teng J, Zhuang Y,
Howard RJ, Zhu S, Noviello CM, Walsh RM Jr, Lindahl E and Hibbs RE:
Shared structural mechanisms of general anaesthetics and
benzodiazepines. Nature. 585:303–308. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Laverty D, Desai R, Uchański T, Masiulis
S, Stec WJ, Malinauskas T, Zivanov J, Pardon E, Steyaert J, Miller
KW and Aricescu AR: Cryo-EM structure of the human α1β3γ2
GABAA receptor in a lipid bilayer. Nature. 565:516–520.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Masiulis S, Desai R, Uchański T, Serna
Martin I, Laverty D, Karia D, Malinauskas T, Zivanov J, Pardon E,
Kotecha A, et al: GABAA receptor signalling mechanisms
revealed by structural pharmacology. Nature. 565:454–459.
2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hannan S, Minere M, Harris J, Izquierdo P,
Thomas P, Tench B and Smart TG: GABAAR isoform and
subunit structural motifs determine synaptic and extrasynaptic
receptor localisation. Neuropharmacology.
169(107540)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shi YW, Zhang Q, Cai K, Poliquin S, Shen
W, Winters N, Yi YH, Wang J, Hu N, Macdonald RL, et al: Synaptic
clustering differences due to different GABRB3 mutations
cause variable epilepsy syndromes. Brain. 142:3028–3044.
2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hernandez CC, Zhang Y, Hu N, Shen D, Shen
W, Liu X, Kong W, Jiang Y and Macdonald RL: GABAA
receptor coupling junction and pore GABRB3 mutations are
linked to early-onset epileptic encephalopathy. Sci Rep.
7(15903)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Møller RS, Wuttke TV, Helbig I, Marini C,
Johannesen KM, Brilstra EH, Vaher U, Borggraefe I, Talvik I, Talvik
T, et al: Mutations in GABRB3: From febrile seizures to
epileptic encephalopathies. Neurology. 88:483–492. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang YH, Burgess R, Malone JP, Glubb GC,
Helbig KL, Vadlamudi L, Kivity S, Afawi Z, Bleasel A, Grattan-Smith
P, et al: Genetic epilepsy with febrile seizures plus: Refining the
spectrum. Neurology. 89:1210–1219. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ye XG, Liu ZG, Wang J, Dai JM, Qiao PX,
Gao PM and Liao WP: YWHAG mutations cause childhood
myoclonic epilepsy and febrile seizures: Molecular Sub-regional
effect and mechanism. Front Genet. 12(632466)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Heron SE, Regan BM, Harris RV, Gardner AE,
Coleman MJ, Bennett MF, Grinton BE, Helbig KL, Sperling MR, Haut S,
et al: Association of SLC32A1 missense variants with genetic
epilepsy with febrile seizures plus. Neurology. 96:e2251–e2260.
2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Deng H, Zheng W and Song Z: The genetics
and molecular biology of fever-associated seizures or epilepsy.
Expert Rev Mol Med. 20(e3)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Carvill GL, Weckhuysen S, McMahon JM,
Hartmann C, Møller RS, Hjalgrim H, Cook J, Geraghty E, O'Roak BJ,
Petrou S, et al: GABRA1 and STXBP1: Novel genetic causes of
Dravet syndrome. Neurology. 82:1245–1253. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Feng Y, Wei ZH, Liu C, Li GY, Qiao XZ, Gan
YJ, Zhang CC and Deng YC: Genetic variations in GABA metabolism and
epilepsy. Seizure. 101:22–29. 2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Schevon CA, Weiss SA, McKhann G Jr,
Goodman RR, Yuste R, Emerson RG and Trevelyan AJ: Evidence of an
inhibitory restraint of seizure activity in humans. Nat Commun.
3(1060)2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tanaka M, DeLorey TM, Delgado-Escueta A
and Olsen RW: GABRB3, Epilepsy, and Neurodevelopment. In:
Jasper's Basic Mechanisms of the Epilepsies. Noebels JL, Avoli M,
Rogawski MA, Olsen RW and Delgado-Escueta AV (eds.) National Center
for Biotechnology Information (US) Copyright ©. 2012, Michael A
Rogawski, Antonio V Delgado-Escueta, Jeffrey L Noebels, Massimo
Avoli and Richard W Olsen. Bethesda (MD), 2012.
|
|
25
|
Maillard PY, Baer S, Schaefer É, Desnous
B, Villeneuve N, Lépine A, Fabre A, Lacoste C, El Chehadeh S, Piton
A, et al: Molecular and clinical descriptions of patients with
GABAA receptor gene variants (GABRA1, GABRB2, GABRB3,
GABRG2): A cohort study, review of literature, and
genotype-phenotype correlation. Epilepsia. 63:2519–2533.
2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Maljevic S, Møller RS, Reid CA,
Pérez-Palma E, Lal D, May P and Lerche H: Spectrum of
GABAA receptor variants in epilepsy. Curr Opin Neurol.
32:183–190. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Johannesen KM, Iqbal S, Guazzi M,
Mohammadi NA, Pérez-Palma E, Schaefer E, De Saint Martin A,
Abiwarde MT, McTague A, Pons R, et al: Structural mapping of
GABRB3 variants reveals genotype-phenotype correlations.
Genet Med. 24:681–693. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Grabenstatter HL, Russek SJ and
Brooks-Kayal AR: Molecular pathways controlling inhibitory receptor
expression. Epilepsia. 53 (Suppl 9):S71–S78. 2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
El Achkar CM, Harrer M, Smith L, Kelly M,
Iqbal S, Maljevic S, Niturad CE, Vissers LELM, Poduri A, Yang E, et
al: Characterization of the GABRB2-associated
neurodevelopmental disorders. Ann Neurol. 89:573–586.
2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Jacob TC, Moss SJ and Jurd R:
GABA(A) receptor trafficking and its role in the dynamic
modulation of neuronal inhibition. Nat Rev Neurosci. 9:331–343.
2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Andäng M, Hjerling-Leffler J, Moliner A,
Lundgren TK, Castelo-Branco G, Nanou E, Pozas E, Bryja V, Halliez
S, Nishimaru H, et al: Histone H2AX-dependent GABA(A)
receptor regulation of stem cell proliferation. Nature.
451:460–464. 2008.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Petrini EM, Ravasenga T, Hausrat TJ,
Iurilli G, Olcese U, Racine V, Sibarita JB, Jacob TC, Moss SJ,
Benfenati F, et al: Synaptic recruitment of gephyrin regulates
surface GABAA receptor dynamics for the expression of
inhibitory LTP. Nat Commun. 5(3921)2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Smith KR, Muir J, Rao Y, Browarski M,
Gruenig MC, Sheehan DF, Haucke V and Kittler JT: Stabilization of
GABA(A) receptors at endocytic zones is mediated by an
AP2 binding motif within the GABA(A) receptor β3
subunit. J Neurosci. 32:2485–2498. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang Y, Lian Y and Xie N: Early onset
epileptic encephalopathy with a novel GABRB3 mutation
treated effectively with clonazepam: A case report. Medicine
(Baltimore). 96(e9273)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Tanaka M, Olsen RW, Medina MT, Schwartz E,
Alonso ME, Duron RM, Castro-Ortega R, Martinez-Juarez IE,
Pascual-Castroviejo I, Machado-Salas J, et al: Hyperglycosylation
and reduced GABA currents of mutated GABRB3 polypeptide in
remitting childhood absence epilepsy. Am J Hum Genet. 82:1249–1261.
2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Khair AM and Salvucci AE: Phenotype
expression variability in children with GABRB3 heterozygous
mutations. Oman Med J. 36(e240)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Epi4K Consortium. De novo mutations in
SLC1A2 and CACNA1A are important causes of epileptic
encephalopathies. Am J Hum Genet. 99:287–298. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bamborschke D, Pergande M, Daimagüler HS,
Mangold E, Dötsch J, Herkenrath P, Cirak S and Fazeli W: Cleft
palate as distinguishing feature in a patient with GABRB3
epileptic encephalopathy. Neuropediatrics. 50:378–381.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhang Y, Kong W, Gao Y, Liu X, Gao K, Xie
H, Wu Y, Zhang Y, Wang J, Gao F, et al: Gene mutation analysis in
253 Chinese children with unexplained epilepsy and
intellectual/developmental disabilities. PLoS One.
10(e0141782)2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yang Y, Zeng Q, Cheng M, Niu X, Xiangwei
W, Gong P, Li W, Ma J, Zhang X, Yang X, et al:
GABRB3-related epilepsy: Novel variants, clinical features
and therapeutic implications. J Neurol. 269:2649–2665.
2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhu Y, Yang J and Zhu X: Combined effects
of levetiracetam and sodium valproate on paediatric patients with
epilepsy: A systematic review and meta-analysis. Seizure. 95:17–25.
2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sonmezturk HH and Azar NJ: Levetiracetam
extended release as adjuvant therapy for the control of
partial-onset seizures. J Cent Nerv Syst Dis. 3:17–25.
2011.PubMed/NCBI View Article : Google Scholar
|