Introduction
Diffuse gliomas are a type of neuroepithelial tumor
characterized by astrocytic or oligodendrocytic morphology and
diffuse infiltrative growth patterns (1). Although rare in children compared
with adults (6 per 100,000 vs. 29 per 100,000, respectively), they
represent the most common solid tumors in pediatric patients
globally (1). The World Health
Organization (WHO) guidelines for the ‘Classification of Tumors of
the Central Nervous System (CNS)’ introduced molecular and
histological/phenotypic information for the first time in
2016(2). However, with
advancements in molecular research, notable differences between
adult and pediatric diffuse glioma have been identified in their
pathogenesis, development, molecular variations and epigenetics
(3). Consequently, the 2016 WHO
CNS classification has limitations.
In response, the 2021 fifth edition of the WHO CNS
tumor classification guidelines (4) incorporated molecular and
histopathological criteria with distinct guidelines that could
categorize and diagnose diffuse glioma into adult and pediatric
types. Despite histopathological similarities, these types exhibit
distinct genetic and molecular profiles, treatments and prognoses
(4). The fifth edition further
subdivided pediatric diffuse low-grade glioma (pDLGG) into four
categories: i) Diffuse astrocytoma, MYB- or MYBL1-altered; ii)
angiocentric glioma (AG); iii) polymorphous low-grade
neuroepithelial tumor of the young (PLNTY) and iv) DLGG, MAPK
pathway-altered. Accurate tumor classification is crucial for
guiding precise treatment, assessing clinical prognosis and
developing new therapies.
LGG is the most common pediatric brain tumor;
however, DLGG with infiltrative margins is relatively rare,
accounting for only 8% of cases (5). Genetic alterations such as BRAF
p.V600E mutations, fibroblast growth factor receptor (FGFR)
modifications, and MYB or MYBL1 rearrangements are common in pDLGG
(6,7). Diagnostic classification becomes
challenging when there is an overlap of histopathological and
genetic features, especially in tumors without characteristic
histopathological or radiological findings. Differential diagnosis
is particularly difficult with pilocytic astrocytoma (PA), the
glial component of ganglioglioma, pleomorphic xanthoastrocytoma
(PXA) and dysembryoplastic neuroepithelial tumor (DNT).
The current study presents a case of pDLGG with
oligodendrocyte-like components that was challenging to diagnose.
This case emphasizes the importance of integrating clinical,
histopathological and molecular features for guiding the management
of pediatric diffuse gliomas.
Case report
Patient details
The patient was an 11-year-old girl who was
diagnosed with generalized epilepsy at 1-year-old. The patient
presented to the Shanghai Sixth People's Hospital (Jinjiang, China)
in July 2024 for evaluation and treatment due to symptoms of
intracranial hypertension, including headache and vomiting. The
patient presented to Jinjiang Municipal Hospital for further
treatment in August 2024. Despite long-term use of antiepileptic
drugs, the patient experienced 4-5 seizures per month. The patient
had no history of brain trauma, central nervous system infection or
cerebrovascular disease, and no family history, and the
neurological examination did not reveal any abnormalities.
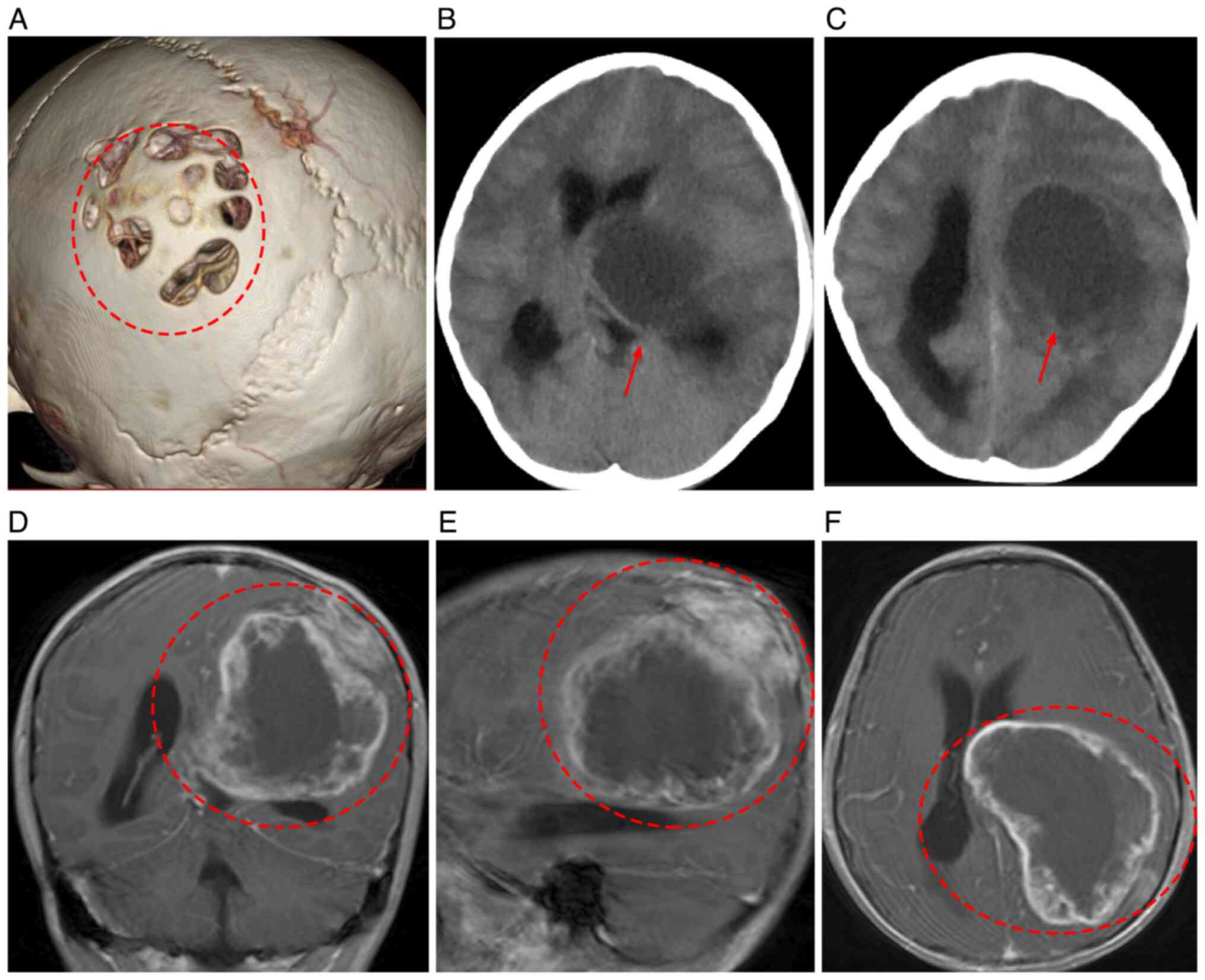
Cranial MRI (Fig.
1) revealed a large lesion (6.9x9.6x6.4 cm) in the left
temporo-parieto-occipital region, predominantly in the parietal
lobe. The lesion exhibited low T1-weighted imaging and high
T2-weighted imaging signals, with fluid-attenuated inversion
recovery showing hyperintensity and diffusion-weighted imaging
showing predominantly hypointense signals. Peripheral enhancement
was observed, along with marked compression of sulci and
ventricles. Cranial CT showed resorptive bone loss in the left
parietal bone without calcification (Fig. 1). Due to the mass effect of the
lesion, ventricular compression and midline shift to the right, the
patient developed consciousness disturbances and coma (Glasgow coma
scale, E1V1M4=6) 2 days after admission (8). Symptoms of brain herniation,
including anisocoria and absent light reflex in the left pupil,
were noted.
Emergency surgery and intraoperative
findings
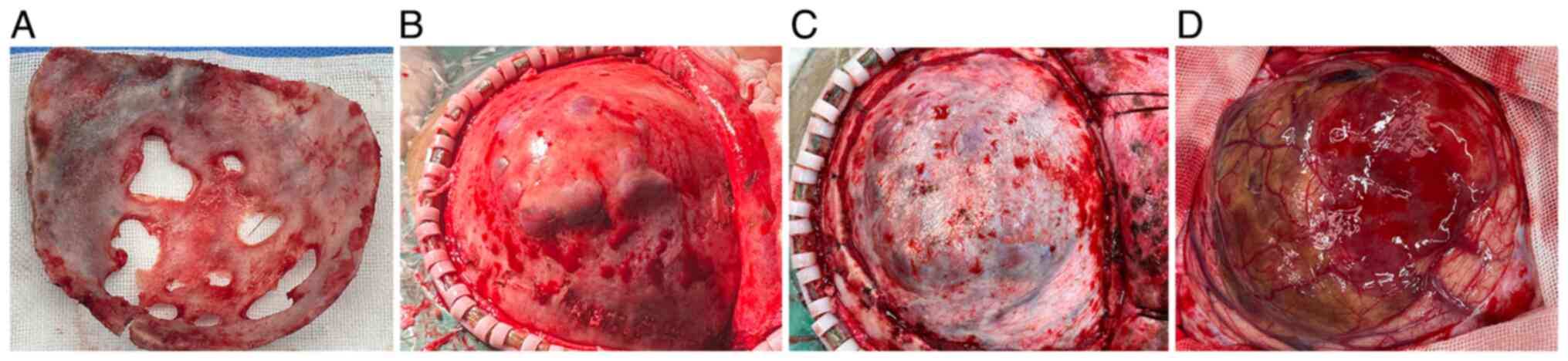
Intraoperatively, the central portion of the
craniotomy window appeared outwardly protruded and thinned due to
tumor compression, with multiple areas of bone destruction
(Fig. 2A). The tumor protruded
onto the cortical surface with the dura mater intact (Fig. 2B and C), the surrounding cortical gyri were
swollen and the sulci were shallow. The tumor was resected in
segments and appeared grayish yellow and grayish red, with a
moderately soft and fibrous consistency (Fig. 2D). The tumor lacked a well-defined
capsule and exhibited unclear boundaries with the surrounding brain
tissue. The tumor was highly vascularized, with areas of necrosis
and hemorrhage (Fig. 2D), and
cystic degeneration was observed in the central portion. The tumor
extended medially to the ventricular trigone, the body of the
lateral ventricle, the internal capsule and the thalamus, and its
dimensions were ~6.5x7.0x10.0 cm. Intraoperative pathological
evaluation suggested features consistent with a LGG.
Postoperative course and
complications
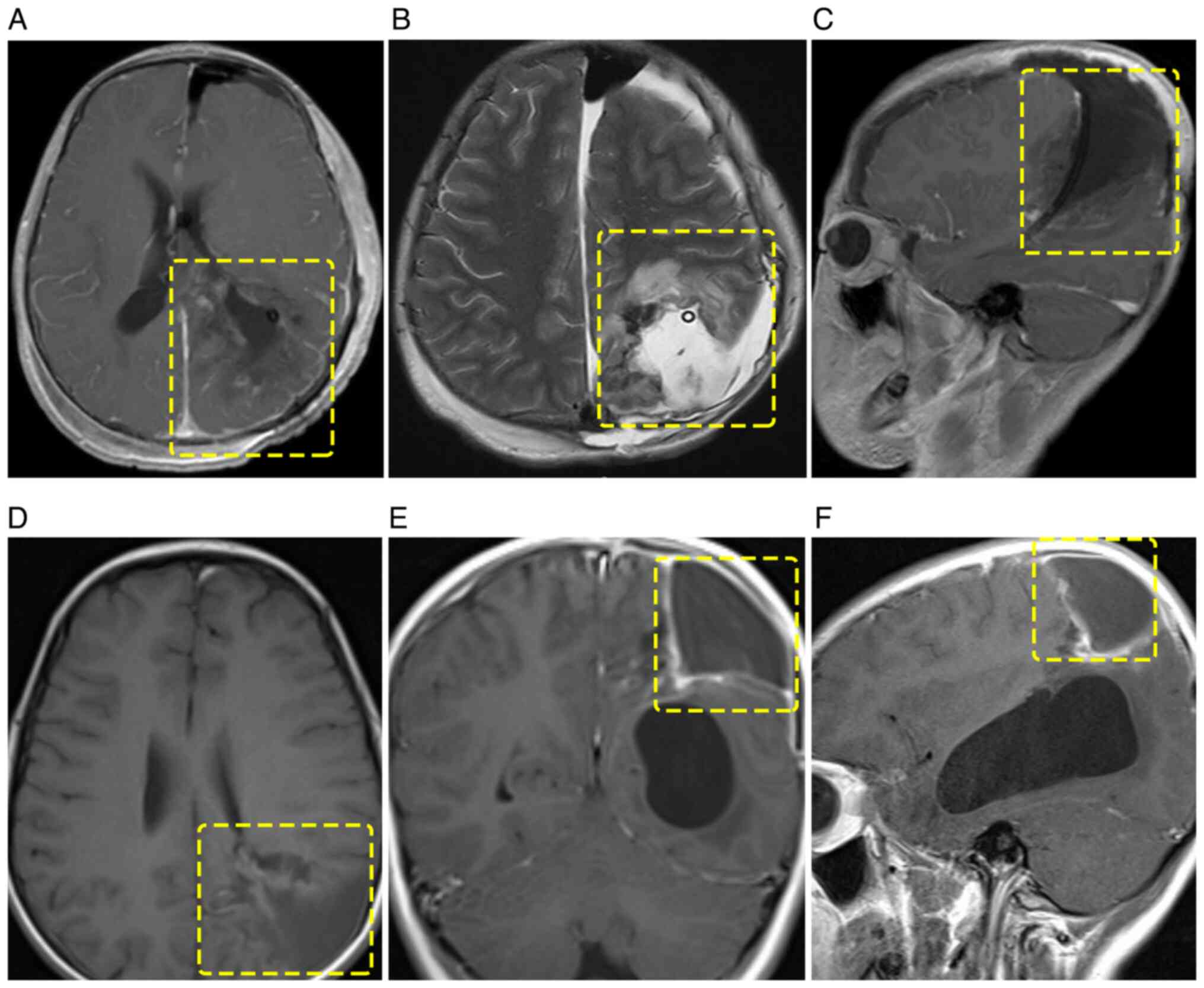
Postoperative cranial MRI confirmed complete tumor
resection (Fig. 3A-C). The patient
was transferred to the intensive care unit (ICU) for observation.
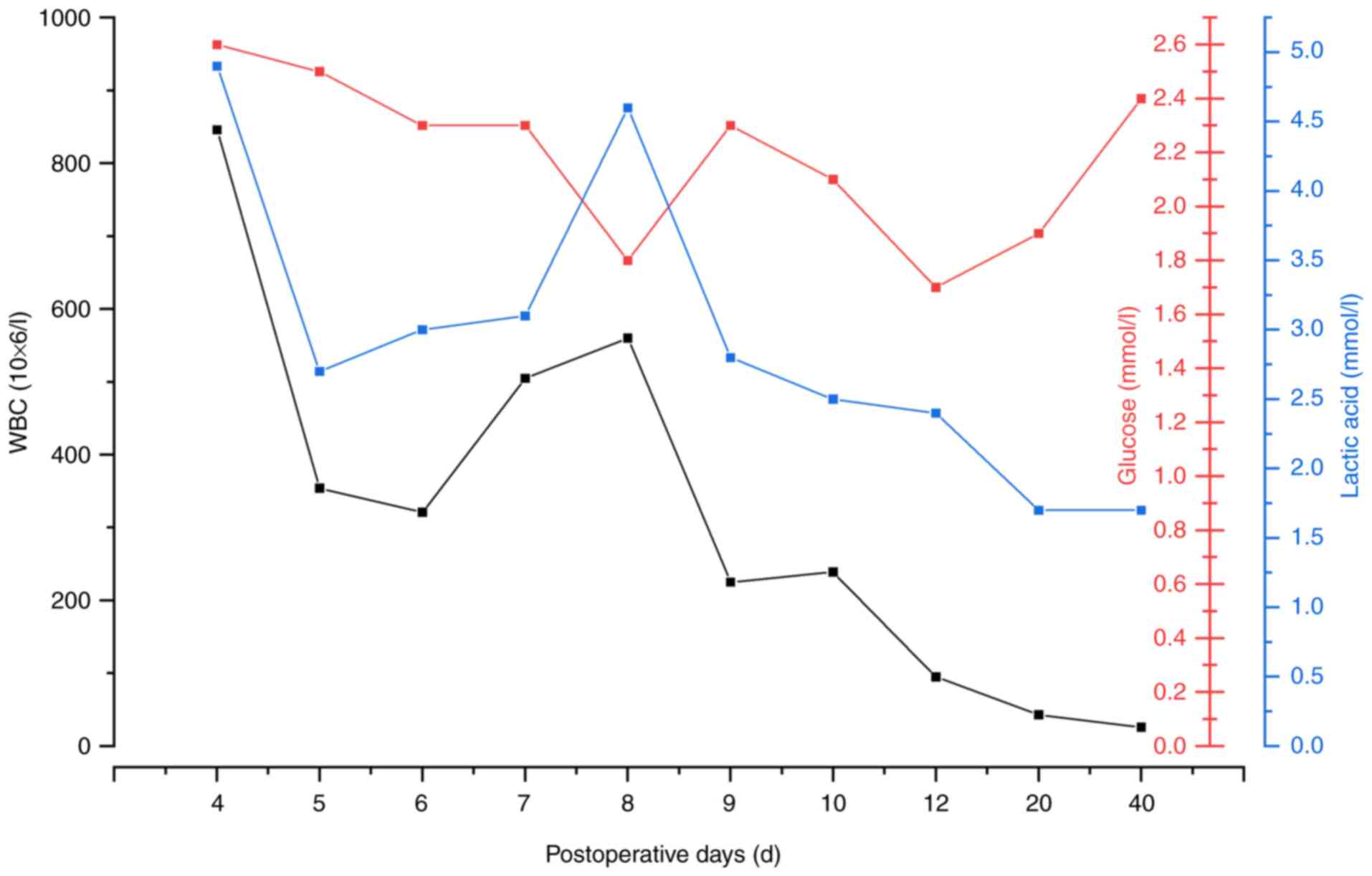
On day 2 post-operation, the patient developed a fever, with a peak
temperature of 40.3˚C. Cerebrospinal fluid (CSF) appeared yellow
and slightly turbid, with a notable elevated white blood cell
count, glucose and lactate levels (Fig. 4). To identify the causative
pathogen, cerebrospinal fluid bacterial culture was performed. The
culture results confirmed Staphylococcus aureus as the
etiological agent of the intracranial infection. The patient was
treated with a regimen of antibiotics, including
piperacillin-tazobactam (120 mg/kg, every 8 h), vancomycin (20
mg/kg, every 12 h) and meropenem (40 mg/kg, every 8 h). By
postoperative day 11, the condition of the patient had stabilized
and they were consequently transferred out of the ICU.
Histopathological examination
The excised tumor tissue was fixed using 10% neutral
buffered formalin, with fixation performed at room temperature for
24-48 h. After fixation and paraffin embedding, the tissue blocks
were sectioned into slices with a thickness of 4 µm. Hematoxylin
and eosin staining was conducted at room temperature with a
staining duration of 5-10 min for hematoxylin and 1-3 min for
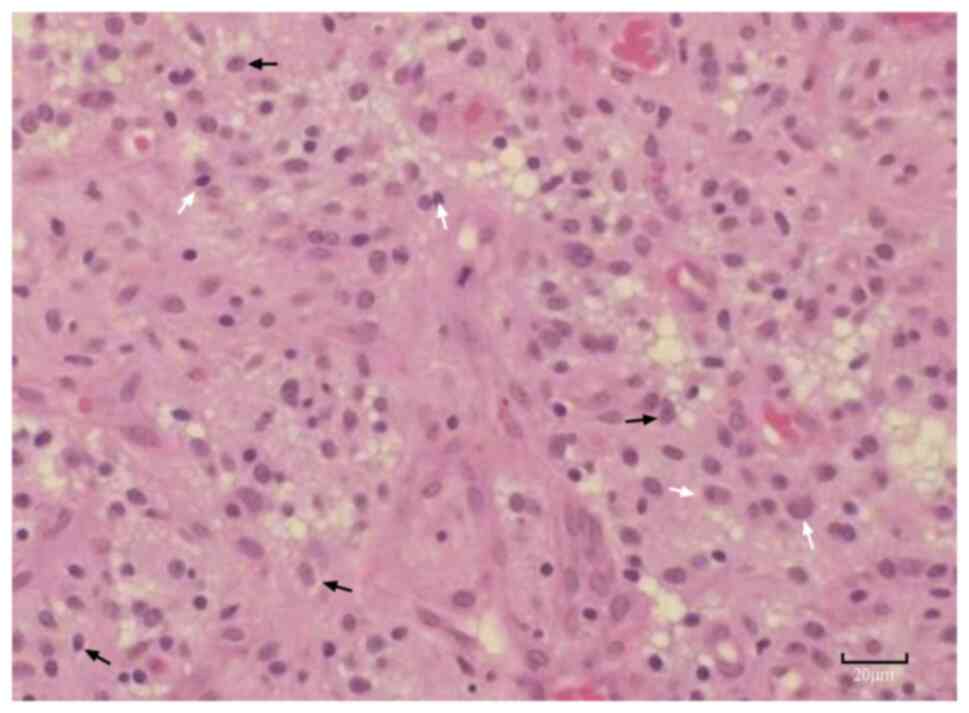
eosin. The staining revealed that the tumor cells exhibited diffuse
infiltrative growth with low to moderate cellular density (Fig. 5). The tumor showed polymorphic
cellular morphology and architecture, forming microcystic and
oligodendroglioma-like astrocytic proliferations. Some areas
displayed spindle-shaped cells, mucinous degeneration,
epithelial-like features and perivascular arrangements. Notable
findings included prominent microvascular proliferation, focal
areas of clear cytoplasm, necrosis and calcification. The tumor
demonstrated moderate cellular proliferation, with mitotic figures
observed at a rate of 2-3 per 10 high-power fields. Focal regions
resembling oligodendroglioma were observed; however, hallmark
features such as Rosenthal fibers, perivascular pseudorosettes and
eosinophilic granular bodies were absent. The morphological
characteristics were consistent with diffuse astrocytoma (9).
Immunohistochemical (IHC) staining and
molecular analysis
For the IHC analyses of the tumor tissue involved in
the present study, the following experimental details were
conducted in accordance with standard pathological procedures and
the information documented in the study. For detecting specific
molecular markers (including glial fibrillary acidic protein,
microtubule-associated protein 2, Olig-2, S-100, vimentin, ATRX,
CD34, and Ki-67), IHC staining was performed at room temperature
for 60 min. All stained sections were observed and analyzed under a
light microscope. Initial IHC staining, including analysis for
GFAP, Ki-67, S-100, Vimentin and CD34, was performed by the
Department of Pathology Service at Jinjiang Municipal Hospital. Due
to the diagnostically challenging nature of the case, an
independent expert consultation was sought from the Pathology
Department of The First Affiliated Hospital of Fujian Medical
University (Fuzhou, China). This secondary consultation encompassed
additional IHC staining for MAP2, Olig-2, and ATRX, as well as
high-throughput tumor sequencing. IHC staining was performed on
formalin-fixed (10% neutral buffered formalin), paraffin-embedded
tissue sections (4-µm thick) as previously described (10). The staining process was conducted
at room temperature for a duration of 60 min. Primary antibodies
included glial fibrillary acidic protein (GFAP),
microtubule-associated protein 2 (MAP2), Olig-2, S-100, vimentin,
ATRX, CD34 and Ki-67 (Dako, Abcam, and Millipore; dilutions
1:100-1:500) as previously described (10). Appropriate antigen retrieval and
detection procedures were applied. Negative and positive controls
were included in each run.
Tumor cells showed positive staining for GFAP, MAP2,
Olig-2, S-100, vimentin and ATRX, CD34 exhibited focal positivity,
and the Ki-67 proliferation index was ~10%. Analysis by tumor cell
genetic sequencing demonstrated the absence of 1p/19q co-deletion
and chromosome +7/-10 alterations but confirmed the presence of
mutations in BRAF p.V600E, FGFR1 and FGFR4.
Molecular testing
Genomic DNA was extracted from FFPE tumor tissue
using the MagMAX™ FFPE DNA/RNA Ultra Kit (cat. no. A31881; Thermo
Fisher Scientific, Inc.). DNA purity and integrity were assessed by
spectrophotometry (NanoDrop 2000; A260/A280 and A260/230 ratios;
NanoDrop Technologies; Thermo Fisher Scientific, Inc.) and
microcapillary electrophoresis (DNA Integrity Number; Agilent 2100
Bioanalyzer; Agilent Technologies, Inc.). Sequencing libraries were
prepared using the ThruPLEX DNA-seq Kit (cat. no. R400406; Takara
Biotechnology Co., Ltd.) and quantified by qPCR (KAPA Library
Quantification Kit; cat. no. KK4824; Kapa Biosystems; Roche
Diagnostics) to a final loading concentration of 1.2-1.8 nM.
High-throughput sequencing was performed on an Illumina platform
with paired-end 150 bp reads (2x150 bp).
High-throughput sequencing of brain tumor-related
genes revealed mutations in the following genes: ALK, BRCA2, DAXX,
FAT1, FGFR1, FGFR4, MSH2, NRAS, SMO and TSC2 (Table I). Although ATRX IHC staining was
positive, no ATRX mutation was detected via genetic analysis. Given
the higher specificity and reliability of genetic testing, the IHC
finding was considered less definitive. Based on the morphological
and IHC features, the pathology department initially diagnosed the
tumor as a PLNTY. However, considering the genetic analysis
results, the diagnosis of the tumor was revised to a pDLGG with
MAPK pathway alterations.
 | Table IHigh-throughput sequencing of mutated
brain tumor-related genes. |
Table I
High-throughput sequencing of mutated
brain tumor-related genes.
| Mutated gene | Mutation type and
location | Mutation abundance,
% |
|---|
| ALK | Missense mutation
in exon 9 | 45.52 |
| BRCA2 | Missense mutation
in exon 11 | 3.92 |
| DAXX | Missense mutation
in exon 5 | 46.7 |
| FAT1 | Missense mutation
in exon 19 | 40.36 |
| FGFR1 | Insertion mutation
in exons 9-18 | 20.72 |
| FGFR4 | Missense mutation
in exon 16 | 52.82 |
| MSH2 | 5' untranslated
region mutation in exon 1 | 63.24 |
| NRAS | Mutation at splice
site in intron 3 | 49.87 |
| SMO | Missense mutation
in exon 12 | 51.0 |
| TSC2 | Missense mutation
in exon 34 | 45.46 |
Follow-up 3 months post-surgery
During the 3-month postoperative follow-up period,
the frequency of seizures in the patient decreased to 1-2 per
month, with periods of no seizures. Therefore, the dosage of
antiepileptic medications was gradually reduced. Repeat
contrast-enhanced MRI showed no residual or recurrent lesions
(Fig. 3D-F).
Discussion
DLGG is the most common brain tumor in children and
adolescents (11,12). When these tumors are located in the
temporal lobe, patients often experience difficult-to-control
seizures, which are resistant to anti-epileptic drugs (13). Since Hughlings Jackson's classic
study in the late 19th century (14), the association between brain tumors
and epilepsy has been well recognized. In 2003, Luyken et al
(15) referred to these tumors as
'long-term epilepsy-associated tumors' (LEATs). LEATs differ from
traditional brain tumors in that they tend to present at a younger
age (seizures are typically the primary and often the only
neurological symptom), grow slowly, are localized to the neocortex
and are often predominantly found in the temporal lobe (16); seizure control is closely
associated with the tumor subtype and the timing of intervention.
In most cases, surgical resection of LEATs yields favorable
outcomes in terms of both seizure reduction and tumor management
(17). Differential diagnoses
include ganglioglioma, DNT, desmoplastic infantile ganglioglioma,
papillary glioneuronal tumor, PA, PXA, AG and PLNTY (18). The patient in the current case
study presented in a similar manner, with seizures as the initial
symptom. The condition was not clearly diagnosed and the patient
suffered from drug-resistant epilepsy for >10 years.
Preoperative imaging did not provide a clear diagnosis, and the
postoperative tumor histopathological features were not
characteristic enough to definitively identify the type of glioma.
Thus, imaging and histopathological features alone could not
establish the definitive glioma type. The importance of
immunohistochemistry and molecular genetic testing is increasingly
apparent in characterizing and refining tumor classification. In
particular, whole-genome DNA methylation analysis and
identification of recurrent genomic alterations (such as mutations,
rearrangements and copy number abnormalities) are essential for
describing biologically distinct disease entities and fine-tuning
tumor classification.
CD34 is a single-chain transmembrane glycoprotein
that is primarily expressed on immature hematopoietic stem cells,
myeloid cells and endothelial cells. It is commonly used as a
marker for certain tumor cells and is widely used to assess LEATs,
glioneuronal lesions and dysplastic cells (19). CD34 is also associated with the
diagnosis of epilepsy-associated tumors (19). CD34 expression is observed in 80%
of ganglioglioma and 84% of PXA cases (18,19).
BRAF p.V600E is an important member of the MAPK pathway that
influences cell proliferation, and BRAF p.V600E mutations,
especially BRAF p.V600E, are commonly seen in glioma subgroups,
including ganglioglioma, PXA, DNT and a subgroup of PA (20,21).
The FGFR family consists of transmembrane tyrosine kinase receptors
(FGFR1-4). Among these, fusions involving FGFR2 and FGFR3 are the
most frequently observed in PLNTY, with FGFR3::TACC3, FGFR2::SHTN1
(KIAA198) and FGFR2::INA fusions being specifically identified
(22).
The case described in the present study was
histologically diagnosed as a diffuse astrocytoma, although despite
improving molecular phenotyping, it was still difficult to
definitively categorize the tumor as a specific LGG. The molecular
markers included Olig-2 positivity, BRAF p.V600E mutation, FGFR1
mutation, FGFR4 mutation and focal CD34 positivity. Notably, there
were no IDH mutations or 1p/19q co-deletions, which led to
differential considerations including oligodendroglioma, PA, PXA,
clear cell ependymoma, ganglioglioma, DNT and PLNTY.
Oligodendrogliomas are typically CD34-negative and exhibit IDH
mutations and 1p/19q co-deletions, which were absent in the tumor.
Oligodendroglioma was initially considered in the differential
diagnosis due to the tumor's low-grade neuroglial morphology;
however, it was ruled out as the tumor lacked the defining IDH
mutation and 1p/19q co-deletion, and was positive for CD34. PA was
excluded due to the lack of Rosenthal fibers and the presence of
focal CD34 expression. PXA and clear cell ependymoma were excluded
based on the positive Olig-2 expression. Diffuse astrocytoma
usually expresses CDKN2A/B, which was absent in the present case.
Ganglioglioma is characterized by MAPK pathway activation gene
alterations, and in high-grade forms, TERT mutations, TP53
mutations, ATRX loss and H3K27M mutations may be detected.
DNT and PLNTY were the most difficult to
differentiate from the tumor in the present case. CD34 may be
positive in the FGFR1 tyrosine kinase fusion or mutation subtype,
but this positivity is limited to a few cells, not as diffuse as in
PLNTY and PLNTY typically shows a Ki-67 expression of <5%
(23). DNT often shows
calcifications on CT, no mass effect or edema, and histologically,
it exhibits oligodendroglioma-like cells arranged in columns with a
microcystic structure, mucinous matrices and scattered dysmorphic
neurons that appear to ‘float’ within the mucinous microcysts. DNT
commonly exhibits FGFR1 gene fusions and BRAF p.V600E mutations
(24). DLGG with MAPK pathway
alterations share similarities with PLNTY, as both are classified
under childhood DLGG. However, in MAPK pathway-altered glioma, CD34
does not show strong diffuse positivity, which differentiates DLGG
from PLNTY (25). Yang et
al (26) categorized DLGG with
MAPK pathway alterations and BRAF p.V600E mutations as intermediate
risk tumors with a high risk of recurrence and progression.
Therefore, based on this information, the current case was
diagnosed as a pDLGG with MAPK pathway alterations. This diagnosis
encourages more frequent follow-up and heightened vigilance in
monitoring for potential changes or complications. Therefore, in
future follow-ups, potential biological evolution should be
assessed in combination with clinical manifestations, imaging
changes and molecular retesting when necessary.
In recent years, with the rapid advancement of
molecular biology techniques, research on pDLGG has shifted its
focus from conventional histopathological classification toward
more refined molecular subtyping (23). The majority of pediatric LGG cases
harbor distinct driver alterations that commonly lead to activation
of the MAPK pathway, along with downstream activation of the
mammalian target of rapamycin (mTOR) signaling cascade (27-29).
The MAPK pathway, mediated through receptor tyrosine kinases (RTK)
and downstream metabolic and transcriptional effectors, plays a
central role in cellular signal transduction (30). In addition to canonical
alterations, such as the BRAF p.V600E mutation (6,30)
and FGFR fusions (19,31), studies have identified other
genetic events. These events, include TERT promoter mutations,
CDKN2A/B deletions, MYB/MYBL1 rearrangements, and alterations in
NTRK, KRAS and IDH1, also play a notable role in a subset of
pediatric patients with LGG (7,27-28,32).
The role of IDH1 mutations in the formation of pDLGG remains
unclear; however, a recent case series demonstrated that while
patients with IDH1-mutant pDLGG exhibited good short-term survival,
the 5-year progression-free survival rate was 42.9%, and
glioma-related mortality emerged at the 10-year mark (33).
These molecular alterations not only facilitate
precise tumor classification but are also closely associated with
patient prognosis (23,34). Molecular targeted therapies for
patients with pDLGG represent a promising frontier in pediatric
neuro-oncology. Targeting hyperactivation of the Ras-MAPK pathway
has been a major focus of recent research, with Raf and MEK
inhibitors either already approved by the US Food and Drug
Administration or currently under investigation in clinical trials
for patients with pLGG (35,36).
Current research is also investigating the role of mTOR inhibitors
and RTK inhibitors as monotherapies or in combination with other
treatment modalities (37).
In addition, increasing attention has been directed
toward the study of the tumor immune microenvironment (38-41).
Targeted therapies against BRAF-altered tumors have proven to be
highly effective; Sievert et al (42) demonstrated that second-generation
BRAF p.V600E inhibitors, such as PLX PB-3, can successfully target
the KIAA1549-BRAF p.V600E fusion, a result of the 7q34 tandem
duplication, which is particularly dominant in pediatric PA. This
provides a novel therapeutic opportunity for tumors with BRAF
p.V600E mutations, with efficacy dependent on the level of Ras
activation in tumor cells (43).
In the future, multi-omics approaches integrating techniques such
as genetic testing, single-cell RNA sequencing and genomic
structural variations may become crucial for precise tumor
classification and personalized treatment strategies.
Meanwhile, the application of BRAF p.V600E or FGFR
inhibitors (such as vemurafenib, dabrafenib and trametinib) in
pDLGG is being explored in clinical trials and has shown promising
efficacy (35-37,43,44).
Immunotherapy and small-molecule targeted therapies are expected to
offer new treatment options for these tumors, warranting further
attention and investigation.
In conclusion, the present case highlights the
importance of enhancing the recognition of potential low-grade
tumors in pediatric and adolescent patients with temporal lobe
epilepsy in clinical practice, and emphasizes the need for timely
molecular testing to achieve precise diagnosis since
histopathological classification can be challenging. The diagnosis
of DLGG with MAPK pathway alterations, in cases such as that
presented in the present study, remains controversial and
additional evidence-based research is needed to establish risk
stratification and intervention guidelines for these tumors. With
the advancement of targeted therapies, such as those targeting BRAF
p.V600E and FGFR, the treatment of these tumors will become more
precise and individualized, and recognizing molecular abnormalities
will ensure targeted treatment options for future rare cases of
malignant transformation or epilepsy control.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The sequencing data generated in the present study
may be found in the SRA database at the following URL: https://www.ncbi.nlm.nih.gov/sra/PRJNA1330527. The
other data generated in the present study may be requested from the
corresponding author.
Authors' contributions
PWH conceived the study, and analyzed and
interpreted imaging data. CZC and MYL collected and analyzed
clinical data, and drafted the manuscript. XLG and MFC contributed
to the analysis and interpretation of data, and revised the
manuscript for important intellectual content. All authors
reviewed, discussed, read and approved the final manuscript. CZC
and MYL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient's parents for the publication of this case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferris SP, Hofmann JW, Solomon DA and
Perry A: Characterization of gliomas: From morphology to molecules.
Virchows Arch. 471:257–269. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Purkait S, Mahajan S, Sharma MC, Sarkar C
and Suri V: Pediatric-type diffuse low grade gliomas:
Histomolecular profile and practical approach to their integrated
diagnosis according to the WHO CNS5 classification. Indian J Pathol
Microbiol. 65 (Suppl):S42–S49. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Patel T, Singh G and Goswami P: Recent
updates in pediatric diffuse glioma classification: Insights and
conclusions from the WHO 5th edition. J Med Life. 17:665–670.
2024.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lassaletta A, Zapotocky M, Bouffet E,
Hawkins C and Tabori U: An integrative molecular and genomic
analysis of pediatric hemispheric low-grade gliomas: An update.
Childs Nerv Syst. 32:1789–1797. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ellison DW, Hawkins C, Jones DTW,
Onar-Thomas A, Pfister SM, Reifenberger G and Louis DN: cIMPACT-NOW
update 4: Diffuse gliomas characterized by MYB, MYBL1, or FGFR1
alterations or BRAFV600E mutation. Acta Neuropathol.
137:683–687. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Suh YY, Lee K, Shim YM, Phi JH, Park CK,
Kim SK, Choi SH, Yun H and Park SH: MYB/MYBL1::QKI fusion-positive
diffuse glioma. J Neuropathol Exp Neurol. 82:250–260.
2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Teasdale G and Jennett B: Assessment of
coma and impaired consciousness. A practical scale. Lancet.
2:81–84. 1974.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wirsching HG, Galanis E and Weller M:
Glioblastoma. Handb Clin Neurol. 134:381–397. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hu Y, Zhang S, Ye H, Wang G, Chen X and
Zhang Y: Lateral ventricle chordoid meningioma presenting with
inflammatory syndrome in an adult male: A case report. Exp Ther
Med. 25(236)2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pallud J and McKhann GM: Diffuse low-grade
glioma-related epilepsy. Neurosurg Clin N Am. 30:43–54.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
PDQ Pediatric Treatment Editorial Board.
Childhood astrocytomas, other gliomas, and glioneuronal/neuronal
tumors treatment (PDQ®): Health professional version.
In: PDQ Cancer Information Summaries [Internet]. Bethesda (MD):
National Cancer Institute (US), 2002.
|
|
13
|
Lombardi D, Marsh R and de Tribolet N: Low
grade glioma in intractable epilepsy: Lesionectomy versus epilepsy
surgery. Acta Neurochir Suppl. 68:70–74. 1997.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hughlings-Jackson J: Observations on the
physiology and pathology of hemi-chorea. Edinb Med J. 14:294–303.
1869.PubMed/NCBI
|
|
15
|
Luyken C, Blümcke I, Fimmers R, Urbach H,
Elger CE, Wiestler OD and Schramm J: The spectrum of long-term
epilepsy-associated tumors: Long-term seizure and tumor outcome and
neurosurgical aspects. Epilepsia. 44:822–830. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Thom M, Blümcke I and Aronica E: Long-term
epilepsy-associated tumors. Brain Pathol. 22:350–379.
2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Baticulon RE, Wittayanakorn N and Maixner
W: Low-grade glioma of the temporal lobe and tumor-related epilepsy
in children. Childs Nerv Syst. 40:3085–3098. 2024.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Giulioni M, Marucci G, Cossu M, Tassi L,
Bramerio M, Barba C, Buccoliero AM, Vornetti G, Zenesini C,
Consales A, et al: CD34 expression in low-grade epilepsy-associated
tumors: Relationships with clinicopathologic features. World
Neurosurg. 121:e761–e768. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Huse JT, Snuderl M, Jones DTW, Brathwaite
CD, Altman N, Lavi E, Saffery R, Sexton-Oates A, Blumcke I, Capper
D, et al: Polymorphous low-grade neuroepithelial tumor of the young
(PLNTY): An epileptogenic neoplasm with oligodendroglioma-like
components, aberrant CD34 expression, and genetic alterations
involving the MAP kinase pathway. Acta Neuropathol. 133:417–429.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Schindler G, Capper D, Meyer J, Janzarik
W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C,
Hasselblatt M, et al: Analysis of BRAF V600E mutation in 1,320
nervous system tumors reveals high mutation frequencies in
pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar
pilocytic astrocytoma. Acta Neuropathol. 121:397–405.
2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Vuong HG, Altibi AMA, Duong UNP, Ngo HTT,
Pham TQ, Fung KM and Hassell L: BRAF mutation is associated with an
improved survival in glioma-a systematic review and meta-analysis.
Mol Neurobiol. 55:3718–3724. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251.
2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Orhan O, Eray HA, Alpergin BC, Zaimoglu M,
Ozpiskin OM, Aras N, Heper A and Eroglu U: Polymorphous low-grade
neuroepithelial tumor of the young (PLNTY): A case report with
surgical and neuropathological differential diagnosis. Clin
Neuropathol. 43:83–91. 2024.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Surrey LF, Jain P, Zhang B, Straka J, Zhao
X, Harding BN, Resnick AC, Storm PB, Buccoliero AM, Genitori L, et
al: Genomic analysis of dysembryoplastic neuroepithelial tumor
spectrum reveals a diversity of molecular alterations dysregulating
the MAPK and PI3K/mTOR pathways. J Neuropathol Exp Neurol.
78:1100–1111. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ryall S, Zapotocky M, Fukuoka K, Nobre L,
Guerreiro Stucklin A, Bennett J, Siddaway R, Li C, Pajovic S,
Arnoldo A, et al: Integrated molecular and clinical analysis of
1,000 pediatric low-grade gliomas. Cancer Cell. 37:569–583.e5.
2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yang RR, Aibaidula A, Wang WW, Chan AK,
Shi ZF, Zhang ZY, Chan DTM, Poon WS, Liu XZ, Li WC, et al:
Pediatric low-grade gliomas can be molecularly stratified for risk.
Acta Neuropathol. 136:641–655. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhang J, Wu G, Miller CP, Tatevossian RG,
Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, et
al: Whole-genome sequencing identifies genetic alterations in
pediatric low-grade gliomas. Nat Genet. 45:602–612. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Jones DTW, Hutter B, Jäger N, Korshunov A,
Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, et
al: Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic
astrocytoma. Nat Genet. 45:927–932. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Bandopadhayay P, Ramkissoon LA, Jain P,
Bergthold G, Wala J, Zeid R, Schumacher SE, Urbanski L, O'Rourke R,
Gibson WJ, et al: MYB-QKI rearrangements in angiocentric glioma
drive tumorigenicity through a tripartite mechanism. Nat Genet.
48:273–282. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
30
|
Manoharan N, Liu KX, Mueller S, Haas-Kogan
DA and Bandopadhayay P: Pediatric low-grade glioma: Targeted
therapeutics and clinical trials in the molecular era. Neoplasia.
36(100857)2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Rivera B, Gayden T, Carrot-Zhang J, Nadaf
J, Boshari T, Faury D, Zeinieh M, Blanc R, Burk DL, Fahiminiya S,
et al: Germline and somatic FGFR1 abnormalities in dysembryoplastic
neuroepithelial tumors. Acta Neuropathol. 131:847–863.
2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Qaddoumi I, Orisme W, Wen J, Santiago T,
Gupta K, Dalton JD, Tang B, Haupfear K, Punchihewa C, Easton J, et
al: Genetic alterations in uncommon low-grade neuroepithelial
tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and
align with morphology. Acta Neuropathol. 131:833–845.
2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yeo KK, Alexandrescu S, Cotter JA,
Vogelzang J, Bhave V, Li MM, Ji J, Benhamida JK, Rosenblum MK, Bale
TA, et al: Multi-institutional study of the frequency, genomic
landscape, and outcome of IDH-mutant glioma in pediatrics. Neuro
Oncol. 25:199–210. 2023.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bale TA and Rosenblum MK: The 2021 WHO
classification of tumors of the central nervous system: An update
on pediatric low-grade gliomas and glioneuronal tumors. Brain
Pathol. 32(e13060)2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kakadia S, Yarlagadda N, Awad R, Kundranda
M, Niu J, Naraev B, Mina L, Dragovich T, Gimbel M and Mahmoud F:
Mechanisms of resistance to BRAF and MEK inhibitors and clinical
update of US food and drug administration-approved targeted therapy
in advanced melanoma. Onco Targets Ther. 11:7095–7107.
2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Nicolaides T, Nazemi KJ, Crawford J,
Kilburn L, Minturn J, Gajjar A, Gauvain K, Leary S, Dhall G, Aboian
M, et al: Phase I study of vemurafenib in children with recurrent
or progressive BRAFV600E mutant brain tumors: Pacific
pediatric neuro-oncology consortium study (PNOC-002). Oncotarget.
11:1942–1952. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ullrich NJ, Prabhu SP, Reddy AT, Fisher
MJ, Packer R, Goldman S, Robison NJ, Gutmann DH, Viskochil DH,
Allen JC, et al: A phase II study of continuous oral mTOR inhibitor
everolimus for recurrent, radiographic-progressive
neurofibromatosis type 1-associated pediatric low-grade glioma: A
neurofibromatosis clinical trials consortium study. Neuro Oncol.
22:1527–1535. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
de Visser KE and Joyce JA: The evolving
tumor microenvironment: From cancer initiation to metastatic
outgrowth. Cancer Cell. 41:374–403. 2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bejarano L, Jordāo MJC and Joyce JA:
Therapeutic targeting of the tumor microenvironment. Cancer Discov.
11:933–959. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Rajendran S, Hu Y, Canella A, Peterson C,
Gross A, Cam M, Nazzaro M, Haffey A, Serin-Harmanci A, Distefano R,
et al: Single-cell RNA sequencing reveals immunosuppressive myeloid
cell diversity during malignant progression in a murine model of
glioma. Cell Rep. 42(112197)2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Jones DTW, Kocialkowski S, Liu L, Pearson
DM, Bäcklund LM, Ichimura K and Collins VP: Tandem duplication
producing a novel oncogenic BRAF fusion gene defines the majority
of pilocytic astrocytomas. Cancer Res. 68:8673–8677.
2008.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sievert AJ, Lang SS, Boucher KL, Madsen
PJ, Slaunwhite E, Choudhari N, Kellet M, Storm PB and Resnick AC:
Paradoxical activation and RAF inhibitor resistance of BRAF protein
kinase fusions characterizing pediatric astrocytomas. Proc Natl
Acad Sci USA. 110:5957–5962. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wang H, Long-Boyle J, Winger BA,
Nicolaides T, Mueller S, Prados M and Ivaturi V: Population
pharmacokinetics of vemurafenib in children with
recurrent/refractory BRAF gene V600E-mutant astrocytomas. J Clin
Pharmacol. 60:1209–1219. 2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wen PY, Stein A, van den Bent M, De Greve
J, Wick A, de Vos FYFL, von Bubnoff N, van Linde ME, Lai A, Prager
GW, et al: Dabrafenib plus trametinib in patients with
BRAFV600E-mutant low-grade and high-grade glioma (ROAR):
A multicentre, open-label, single-arm, phase 2, basket trial.
Lancet Oncol. 23:53–64. 2022.PubMed/NCBI View Article : Google Scholar
|