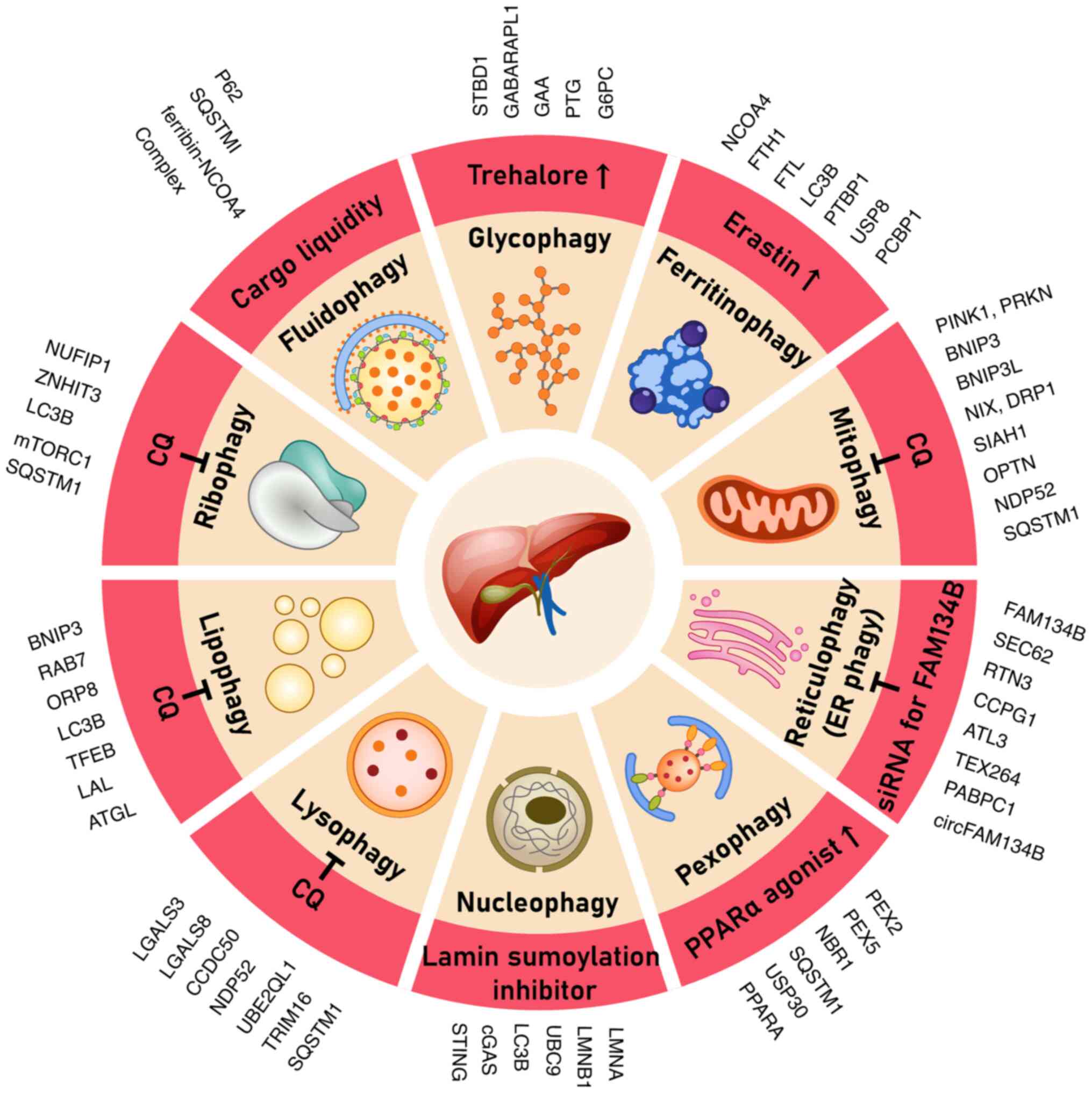
1. Introduction
Hepatocellular carcinoma (HCC) was the third leading
cause of cancer-related mortality in 2020, with ~905,000 new cases
and 830,000 deaths worldwide (1).
The incidence remains the highest in East Asia and Africa, but is
increasing in Western nations. In total, ~54% cases are caused by
chronic hepatitis B infection, whereas 15% are caused by hepatitis
C. Alcohol abuse, aflatoxin, non-alcoholic steatohepatitis and
metabolic syndrome-related diabetes are also contributing factors
(2,3). The male predominance (2-3:1) in
hepatocellular carcinoma (HCC) suggests gender-specific differences
in exposure to risk factors like chronic viral infections and
alcohol consumption, which may contribute to the higher incidence
in men (2).
Autophagy is a cellular self-digestion process that
serves to maintain homeostasis by degrading damaged organelles and
macromolecules through the lysosomes. In cancer, autophagy serves
complex, context-dependent roles, suppressing tumor initiation by
removing harmful cellular components, but can also promote the
survival of established tumors under stress (4,5).
Starvation-induced non-selective macroautophagy randomly engulfs
the cytoplasm and generates nutrients through indiscriminate
lysosomal breakdown (5). Selective
autophagy uses cargo receptors containing LC3-interacting regions,
such as sequestosome 1 (p62), nuclear dot protein 52 kDa or nuclear
receptor coactivator 4 (NCOA4), to bind ubiquitin-tagged substrates
(such as damaged mitochondria, ferritin and aggregates) and recruit
phagophores exclusively to them. This receptor-guided precision
allows for organelle-specific quality control whilst minimizing
biomass loss and can be adjusted independently of bulk autophagy
(6,7). HCC frequently exhibits dysregulated
autophagy (4,5). Selective autophagy involves the
autophagic targeting of specific intracellular organelles or
materials. Key forms of autophagy (Table I) include mitophagy (mitochondria)
(8-11),
lysophagy (lysosomes) (10,12),
reticulophagy [endoplasmic reticulum (ER)] (9,13),
pexophagy (peroxisomes) (14),
nucleophagy (nucleus) (15),
ribophagy (ribosomes) (16,17),
lipophagy (lipid droplets) (11,17-19),
glycophagy (glycogen) (19,20),
ferritinophagy (ferritin/iron) (13,21)
and fluidophagy (autophagy of phase-separated liquid condensates
(18-22).
Each of the aforementioned processes has been implicated in the
pathogenesis and therapeutic response in HCC. Target regulators and
therapeutic agents unique to each type of selective autophagy are
shown in Fig. 1.
 | Table IExperimental findings in selective
autophagy in HCC. |
Table I
Experimental findings in selective
autophagy in HCC.
| Autophagy type | Key experimental
findings | (Refs.) |
|---|
| Mitophagy | • PINK1/Parkin
inhibition leads to dysfunctional mitochondria and increased
apoptosis | (8) |
| | • High mitophagy
gene expression correlates with worse survival | (8) |
| | • Parkin-mediated
mitophagy supports cancer stem cells and neutralizes p53 | (8) |
| Lysophagy | •
Sorafenib-resistant HCC cells rely on lysophagy; polyphyllin D
disrupts this process, causing cell death | (12) |
| | • UBE2QL1 knockdown
leads to damaged lysosome accumulation, sensitizing cells to
lysosomal death | (12) |
| Reticulophagy | • Sorafenib
activates FAM134B-mediated ER-phagy, protecting against
ferroptosis | (34) |
| | • FAM134B knockdown
enhances ferroptotic cell death | (9) |
| | • circFAM134B
silencing enhances lenvatinib-induced ferroptosis | (34) |
| Pexophagy | • Oxidative stress
triggers peroxisomal protein ubiquitination and p62/NBR1-mediated
clearance | (14) |
| | • Blocking
autophagy leads to dysfunctional peroxisome accumulation and DNA
oxidation | (14) |
| | • HCC cells may use
pexophagy to shift metabolism toward mitochondria | (14) |
| Nucleophagy | • Autophagy
degrades micronuclear DNA via cGAS-LC3 interaction, reducing STING
signaling | (15) |
| | • Autophagy
inhibition increases cytosolic DNA and interferon signaling | (15) |
| | • cGAS-STING can
trigger autophagy-dependent cell death under severe telomere
damage | (15) |
| Ribophagy | • Nutrient
starvation/mTORC1 inhibition relocates NUFIP1 to ribosomes, driving
degradation | (17) |
| | • Blocking
autophagy during nutrient deprivation reduces HCC cell
survival | (17) |
| | • Ribophagy may
help cells remove stalled ribosomes and adapt to stress | (17) |
| Lipophagy | • Autophagy
dysfunction leads to lipid accumulation and HCC development | (18) |
| | • Blocking
autophagy causes lipid droplet buildup and energy depletion | (19) |
| | • BNIP3-driven
lipophagy restrains tumor growth; low BNIP3 yields lipid-rich
tumors | (30) |
| Glycophagy | • STBD1/PTG loss
sensitizes HCC cells to glucose withdrawal | (19) |
| | • Autophagy
inhibition reduces glucose output during fasting | (20) |
| | • G6PC deficiency
impairs glycophagy, leading to adenomas/HCC | (64) |
| Ferritinophagy | • NCOA4 releases
iron from ferritin, enabling ferroptosis | (13) |
| | • NCOA4
overexpression increases labile iron and slows tumor growth | (49) |
| | • PTBP1 silencing
upregulates NCOA4 and sensitizes tumors to sorafenib | (13) |
| Fluidophagy | • Optineurin/NDP52
form droplets on damaged mitochondria to facilitate clearance | (22) |
| | • p62 forms liquid
droplets that sequester ubiquitinated proteins | (22) |
| | • In
autophagy-deficient tumors, p62 aggregates activate NRF2, promoting
tumorigenesis | (18) |
In this review work, PubMed and Google Scholar were
searched for entries added between January 2015 and March 2025 with
Boolean strings combining ‘selective autophagy’, ‘hepatocellular
carcinoma’ and each pathway name (such as ‘mitophagy’ and
‘ferritinophagy’) for a narrative-review approach. In addition, the
reference lists of the retrieved articles were hand-screened. The
inclusion criteria were as follows: English-language primary data
or systematic reviews detailing mechanisms, biomarkers or
therapeutic manipulation of selective autophagy in HCC or liver
models. The exclusion criteria were non-HCC cancers, conference
abstracts without the full text, studies lacking mechanistic
details and commentaries. Two authors independently screened
titles/abstracts, and any disagreements were resolved by a third
reviewer. Data on the pathway effectors, experimental design and
translational relevance were extracted and tabulated to ensure
consistency.
2. Mitophagy in HCC
Mitophagy is the selective autophagic removal of
mitochondria that is critical for mitochondrial quality control.
This is primarily mediated by the phosphatase and tensin
homolog-induced kinase 1 (PINK1)/Parkin pathway, wherein PINK1
accumulates in depolarized mitochondria and recruits Parkin to tag
the mitochondria for autophagy (23,24).
In HCC, Parkin expression is frequently lost or reduced
(downregulated in ~50% HCC tumors) (25). Previous studies have identified
siah E3 ubiquitin protein ligase 1 (SIAH1) as a key Parkin
substitute in HCC. Specifically, SIAH1 can cooperate with PINK1 to
ubiquitinate mitochondrial proteins and initiate mitophagy
(26,27).
Sorafenib-induced mitophagy serves as a
cytoprotective mechanism, clearing drug-damaged mitochondria and
preventing the excess accumulation of reactive oxygen species (ROS)
(26). Sorafenib actively induces
mitophagy as a defense mechanism against HCC, by inducing PINK1
accumulation and Parkin recruitment to damaged mitochondria, in
turn activating mitophagy in HCC cells. Inhibition or silencing of
PINK1 or SIAH1 has been found to suppress mitophagy and increase
sorafenib-induced cell death, confirming their role as a
cytoprotective component (28,29).
In orthotopic Huh7 xenografts, combining sorafenib with the
dynamin-related protein 1 (DRP1) inhibitor mdivi-1 (50 mg/kg) was
observed to cut tumor volume by 63 vs. 41% for sorafenib
monotherapy, indicating that blocking mitophagy can magnify
sorafenib lethality (30).
Consequently, HCC cells can survive more readily after sorafenib
treatment when mitophagy remains intact. Although basal mitophagy
can prevent liver tumor initiation by removing dysfunctional
ROS-producing mitochondria in established HCC, it frequently
promotes tumor cell metabolism and therapy resistance (5). Chemotherapy (such as cisplatin) has
been reported to induce mitophagy in surviving HCC cells through
DRP1-mediated mitochondrial fission (31). Inhibiting DRP1 with mdivi-1 can
block this mitophagy and increase the apoptosis of
cisplatin-treated HCC cells (31).
Excessive mitophagy activation has been explored as
a therapeutic strategy for inducing HCC cell death (Table II). The iron chelator deferiprone
triggers PINK1/Parkin-independent mitophagy in liver cells and
slows HCC tumor growth in mice, presumably by forcing mitochondrial
turnover and compromising tumor bioenergetics (8). Chronic hepatitis B virus infection, a
major etiological factor of HCC, can influence mitophagy (32,33).
Hepatitis B virus X protein has been shown to induce NIP3-like
protein X-dependent mitophagy in liver cells, which reprograms
metabolism towards glycolysis to enhance cancer stem cell
properties (34).
| Table IITherapeutic strategies for selective
autophagy in HCC. |
Table II
Therapeutic strategies for selective
autophagy in HCC.
| Autophagy type | Therapeutic
targets/strategies |
Mechanism/rationale | (Refs.) |
|---|
| Mitophagy | • Parkin/PINK1
pathway inhibition | • Increases
dysfunctional mitochondria, ROS and apoptosis | (8) |
| | •
Chloroquine/Hydroxychloroquine | • Prevents
autolysosome formation, increases mitochondrial ROS | (10) |
| | • AMPK activators
(Metformin) | • Removes damaged
mitochondria, reduces steatosis | (11) |
| | • BNIP3
upregulation | • Triggers
excessive mitophagy and cell death | (9) |
| Lysophagy | • ASM inhibitors
(Desipramine) | • Destabilizes
lysosomes, mimicking polyphyllin D effects | (10,12) |
| | • Lysosomal BH3
mimetics (BMH-21) | • Activates
autophagy to overwhelm lysosomes | (10) |
| | • TFEB activators
(Trehalose) | • Increases
lysosome biogenesis, generates autophagy trap | (10) |
| | • Lysosomal pH
alkalinizers | • Impairs
degradation, causing substrate accumulation | (10) |
| Reticulophagy | • FAM134B
inhibition | • Enhances
sorafenib-induced ferroptosis | (9) |
| | • PABPC1
inhibitors | • Reduces FAM134B
translation and ER-phagy | (9) |
| | • Ferroptosis
inducers + autophagy inhibitors | • Prevents
ferroptosis mitigation via ER-phagy | (9) |
| Pexophagy | • USP30
inhibitors | • Increases
peroxisome ubiquitination and clearance | (14) |
| | • ROS inducers +
autophagy inhibitors | • Forces peroxisome
damage accumulation | (14) |
| | • PPARα agonists +
autophagy inhibitors | • Increases
peroxisome load then blocks turnover | (14) |
| Nucleophagy | • STING agonists +
autophagy inhibitors | • Amplifies innate
immune activation by sustaining STING signals | (15) |
| | • Immune checkpoint
inhibitors + autophagy inhibitors | • Promotes
immunogenic cell death via DNA release | (15) |
| | • p53 reactivators
+ mTOR inhibitors | • Induces death in
genomically unstable cells | (15) |
| Ribophagy | • mTORC1
inhibitors | • Induces autophagy
including ribophagy | (16) |
| | • Dual PI3K/mTOR
inhibitors + autophagy blockers | • Traps
non-functional ribosomes in autophagosomes | (16) |
| | • NUFIP1/ZNHITE3
disruptors | • Prevents ribosome
clearance during stress | (17) |
| Lipophagy | • Autophagy
inhibitors in NAFLD-HCC | • Blocks fat
mobilization, causing energy deficit | (17-19) |
| | • AMPK activators +
autophagy inhibitors | • Creates autophagy
trap during peak lipid mobilization | (11) |
| | • DGAT1/2
inhibitors + autophagy blockers | • Prevents
triglyceride formation, increasing toxic free fatty acids | (11) |
| Glycophagy | • Glycogen
phosphorylase inhibitors + autophagy blockers | • Redirects to
lysosomal route then blocks it, causing energy crisis | (19) |
| | • STBD1
stabilizers | • Increases
glycogen autophagy beyond handling capacity | (19) |
| | • Glucose
restriction/SGLT2 inhibitors | • Forces tumors to
consume internal glycogen | (19,20) |
|
Ferritinophagy/ | • NCOA4
upregulation | • Increases iron
release from ferritin | (13) |
| Ferroptosis | • Ferroptosis
inducers | • Promotes
iron-dependent lipid peroxidation | (13) |
| | • Iron
mobilizers | • Primes cells for
ferroptosis | (13,21) |
In summary, mitophagy in HCC appears to have a dual
role. It can either protect cells by clearing damaged mitochondria
(limiting oxidative stress and cell death) or enable tumor survival
under treatments, such as kinase inhibitors or chemotherapy
(5,31). Therapeutically, mitophagy can be
manipulated by inhibition (to prevent tumor cells from escaping
apoptosis during therapy) or overactivation (to induce metabolic
crisis in tumors) (8,31).
3. Lysophagy in HCC
Lysophagy is the selective removal of lysosomes
damaged by autophagy. When lysosomal membranes are compromised,
cells can target these leaky lysosomes for autophagic degradation
to prevent the release of hydrolytic enzymes into the cytosol
(9). The process involves damage
recognition by cytosolic galectins (whereby galectin-3/8 binds to
exposed luminal glycans on ruptured lysosomes) and the
ubiquitination of lysosomal membrane proteins, followed by the
recruitment of autophagy receptors (35,36).
Recently, coiled-coil domain containing 50 (CCDC50)
was identified as a dedicated lysophagy receptor in human melanoma
cells (9). CCDC50 can bind to
galectin-3-decorated K63-ubiquitinated lysosomes and facilitate
their clearance through autophagy (9). In HCC, CCDC50 serves a
tumor-promoting role by sustaining lysosomal homeostasis. It was
previously shown that CCDC50 expression is upregulated in HCC
tumors and cell lines, where its high expression associates with
more aggressive tumor features and worse patient prognosis
(9).
Functionally, CCDC50 loss led to the accumulation of
broken lysosomes in HCC cells, impaired autophagic flux, elevated
ROS levels and cell death (9). In
mouse models, CCDC50 deficiency was found to significantly suppress
HCC tumor growth, presumably due to the cytotoxic buildup of
dysfunctional lysosomes and oxidative damage (9). Therapeutically, inhibiting lysophagy
could harm cancer cells by allowing lysosomal damage to accumulate.
Polyphyllin D has been observed to cause lysosomal membrane
permeabilization in HCC cells, resulting in the loss of lysophagy
and spillage of enzymes to trigger cell death (37). Lysosome-targeting agents, such as
chloroquine or the palmitoyl-protein thioesterase 1 (PPT1)
inhibitor GNS561, can effectively kill HCC cells by overwhelming
the lysosomal system (38). GNS561
(ezurpimtrostat) showed potent anti-tumor activity in preclinical
HCC models and has since entered clinical trials (39).
Overall, these aforementioned findings suggest that
lysophagy allows HCC cells to maintain the functionality of their
digestive organelles. Disruption of lysophagy (either genetically
or with lysosomotropic drugs) will likely cause catastrophic
lysosomal failure and tumor suppression (9,37).
4. Reticulophagy (ER-phagy) in HCC
Reticulophagy is the selective autophagic
degradation of portions of the ER. Several ER-resident receptor
proteins [family with sequence similarity 134 member B (FAM134B),
SEC62 homolog, preprotein translocation factor (SEC62), reticulon 3
(RTN3) and cell cycle progression 1 (CCPG1)] can mediate
reticulophagy by binding LC3/γ-aminobutyric acid
receptor-associated protein (GABARAP), a type B autophagy-related
gene 8 family protein receptor, to form autophagosomes and capture
ER fragments (40). FAM134B is a
critical ER-phagy receptor that binds to LC3 through a conserved
LIR motif and selectively fragments the ER membrane for autophagic
engulfment (40).
Sorafenib induces ER stress and activates
FAM134B-dependent reticulophagy (41). Liu et al (41) reported that sorafenib can stimulate
ER-phagy through FAM134B, potentially mitigating the cytotoxicity
of sorafenib. FAM134B knockdown blocked ER-phagy and enhanced
sorafenib-induced ferroptosis in HCC cells (40). Intact FAM134B-mediated
reticulophagy was protective because it contains ER components,
including misfolded proteins, calcium-overloaded ER fragments, and
damaged ER membranes, along with oxidized lipids and proteins
generating ROS, that contribute to oxidative stress and ferroptosis
(40).
A recent study on the circular (circ)RNA of the
FAM134B gene (circFAM134B) showed that this circRNA increases
FAM134B expression and promotes ER-phagy, leading to the
suppression of ferroptosis in HCC cells treated with lenvatinib.
Silencing circFAM134B increased the sensitivity of HCC cells to
Lenvatinib (40). From a
therapeutic perspective, inhibiting FAM134B-mediated ER-phagy may
be beneficial by eliminating ferroptosis in tyrosine kinase
inhibitor-treated tumors (40).
This hypothesis is based on the observation that FAM134B knockdown
potentiated sorafenib and lenvatinib lethality in preclinical
models (40).
5. Pexophagy in HCC
Pexophagy is the selective autophagic degradation of
peroxisomes. Peroxisomes are vital organelles involved in lipid
metabolism, ROS detoxification, biosynthesis of bile acids and
plasmalogens (42). Quality
control of peroxisomes is essential because dysfunctional
peroxisomes can produce excessive ROS and toxic byproducts.
Pexophagy removes surplus or damaged peroxisomes to maintain the
cellular redox balance (5).
Pexophagy malfunction leads to peroxisomal
accumulation, which is associated with oxidative stress and
inflammation in the liver (43).
In catalase-knockout mice, extended fasting triggered excessive
pexophagy and hepatocyte cell death (44). This suggests that under conditions
of high oxidative stress burden, pexophagy may be activated to the
point of cell death, a mechanism potentially relevant in chronic
liver disease and HCC development (5).
To the best of our knowledge, direct studies of
pexophagy in HCC are sparse. However, analyses of HCC tissues have
revealed the dysregulation of peroxisomal proteins and redox
imbalance, which could be partly due to altered pexophagic activity
(5). Pexophagy in HCC remains
underexplored therapeutically. Peroxisome proliferator-activated
receptor-α agonists (such as clofibrate) can induce peroxisome
proliferation and subsequent pexophagy in the liver, such that they
trigger pexophagy in fatty liver contexts (45). Another approach is to target
autophagy adapters, such as neighbor of BRCA1 gene 1 protein which,
along with p62, can act as receptors for ubiquitinated peroxisomes
to enhance the efficiency of pexophagy (46).
6. Nucleophagy in HCC
Nucleophagy is the selective autophagic degradation
of nuclear material. In mammalian cells, nucleophagy is less common
but has been observed during senescence and after DNA damage
(21). One key mechanism described
in cancer cells is DNA damage-induced nucleophagy through lamin
tagging (21). Nucleophagy excises
and degrades damaged nuclear constituents when chromatin breaks or
lamin scaffolds become irreparable. During this process,
SUMO-modified lamin A/C binds to LC3, allowing autophagosomes to
sequester micronuclei or ruptured envelope fragments for lysosomal
digestion (47). In hepatocytes,
inhibiting lamin A/C SUMOylation increases γ-H2AX foci and
micronuclei, reflecting elevated DNA damage. Activation of
nucleophagy mitigates this damage by removing damaged nuclear
components, thereby preserving genome stability. However, this
protective process may allow tumor cells to survive genotoxic
stress. Cells do not necessarily die immediately, because
nucleophagy selectively engulfs and digests damaged nuclear
material, maintaining nuclear integrity and cellular viability
despite persistent DNA lesions. Consequently, nucleophagy functions
as a double-edged sword, limiting DNA-damage signaling while
potentially promoting tumor cell survival (21).
Li et al (21) demonstrated that when DNA damage
causes small fragments of DNA to leak from the nucleus, the cell
responds by SUMOylating nuclear lamin A/C through the E2 enzyme
ubiquitin-conjugating enzyme E2 I. SUMOylation creates a signal
that recruits LC3, effectively bridging the nuclear lamina to the
autophagosome (21). In the
context of HCC, direct evidence of nucleophagy is limited. However,
the liver tumor environment such as DNA-damaging chemotherapeutic
agents, metabolic stress or lamina deformations, trigger
nucleophagy in HCC. HCC cells are frequently exposed to genotoxic
stress, which can result in double-stranded DNA breaks and
micronucleus formation (21,47).
From a therapeutic standpoint, targeting nucleophagy
is challenging because it is not well defined and can harm normal
cells. One potential angle is that inhibiting SUMOylation can block
nucleophagy and perhaps make HCC cells more prone to accumulating
nuclear damage, leading to cell death.
7. Ribophagy in HCC
Ribophagy involves the selective autophagic
degradation of ribosomes. During nutrient starvation, cells degrade
their ribosomes to recycle amino acids and nucleotides (48). Autophagy-dependent degradation of
ribosomal RNA has been shown to be crucial for maintaining
nucleotide pools during development (13). During metabolic stress, ribophagy
dismantles superfluous or stalled ribosomes to recycle nucleotides
and amino acids. Starvation or mTOR1 inhibition drives nuclear
FMR1-interacting protein 1 (NUFIP1) from the nucleoplasm to the 60S
subunits, where its LC3-interacting region, aided by zinc finger
HIT domain-containing protein 3, targets ribosomes to
autophagosomes, whereas concurrent p62 recruitment clears
ubiquitinated ribosomal proteins (49). Suppressing NUFIP1 in
nutrient-deprived HCC cells heightens proteotoxic stress, ATP
depletion and apoptosis, underscoring the role of ribophagy in
balancing translation load and energy supply (49). Direct studies on the role of
ribophagy in HCC are lacking. However, given that solid tumors
frequently experience nutrient-poor and hypoxic microenvironments,
it is conceivable that HCC cells can invoke ribophagy as an
adaptive mechanism. A study has previously shown that blocking
late-stage autophagy with chloroquine similarly impaired ribophagic
recycling in HCC cells (22). From
a functional standpoint, ribophagy in HCC could contribute to
therapy resistance, where when exposed to metabolic stress (such as
glycolysis inhibitors or anti-angiogenic therapy), HCC cells may
digest ribosomes and survive longer.
8. Lipophagy in HCC
Lipophagy is the autophagic degradation of lipid
droplets. In liver cells, lipophagy is crucial for mobilizing
stored triglycerides and cholesterol esters, delivering them to
lysosomes where lipases break them down to free fatty acids
(50). This process is central to
energy homeostasis. In the context of liver disease and HCC,
lipophagy has significant implications in counteracting lipid
accumulation (51).
In HCC tumors, lipophagy appears to have a
context-dependent role. Cancer cells can use lipophagy to tap into
lipid droplet reserves to meet energy demands (4). During starvation or therapy-induced
nutrient stress, HCC cells can upregulate factors, such as
CCAAT/enhancer binding protein α, enhancing lipophagy to release
fatty acids from lipid droplets to sustain ATP production (4).
Conversely, lipophagy can act as a tumor-suppressive
mechanism by preventing excessive lipid accumulation and
lipotoxicity. Preventing lipotoxicity suppresses tumors because
BCL2-interacting protein 3 (BNIP3)-mediated lipophagy tethers lipid
droplets to autophagosomes, ensuring their degradation. By clearing
excess lipids, this process avoids lipid-driven energy supply and
toxic lipid accumulation, restraining tumor growth. Loss of BNIP3
leads to lipid-rich tumors and poorer survival, underscoring
lipophagy's tumor-suppressive role. Chen et al (34) previously showed that BNIP3 can
tether lipid droplets to autophagosomes through LC3 binding in a
process termed ‘mitolipophagy’, which restrained HCC development.
In a genetic HCC mouse model, loss of BNIP3 led to the earlier
development of larger tumors that contained markedly more lipid
droplets (51).
In human HCC specimens, low BNIP3 expression has
been associated with higher lipid content in tumors and poor
patient survival (51). These
findings indicate that insufficient lipophagy (at least in part due
to BNIP3 inactivation) can promote HCC progression by allowing
lipids to accumulate and fuel tumor growth (51). Therapeutically, there are two
potential strategies: Inhibition of lipophagy to starve HCC cells
or enhancement of lipophagy to induce lipotoxic stress in tumors.
It has been previously demonstrated that combining an autophagy
inhibitor (e.g., chloroquine or its analog hydroxychloroquine) with
a metabolic stressor increased lipid accumulation and cell death in
HCC (22,38).
9. Glycophagy in HCC
Glycophagy is the selective autophagy of glycogen.
The liver is the main storage site for glycogen, where in addition
to canonical cytosolic glycogenolysis, hepatocytes possess a
lysosomal route to degrade glycogen. This pathway is mediated by
starch-binding domain-containing protein 1, which binds glycogen
and recruits it to the autophagosome by interacting with
γ-aminobutyric acid receptor-associated protein-like 1(52). Inside lysosomes, glycogen is broken
down by acid α-glucosidase (52).
In HCC, the role of glycophagy is not well
characterized but is likely relevant, given that numerous HCC cells
exhibit metabolic flexibility and the ability to store glucose as
glycogen (53). HCC cells may
invoke glycophagy when extracellular glucose is scarce. During
transarterial embolization therapy, which induces acute nutrient
starvation in the tumor region, cancer cells can survive by
consuming their glycogen through autophagy (53). Previous studies have shown that
depriving HCC cells of glucose triggers autophagy activation
(26,53). Therapeutically, targeting
glycophagy is challenging. A conceivable approach during
transarterial embolization or fasting-mimicking diet therapy for
HCC is to add an agent that stimulates glycophagy to rapidly
deplete tumor glycogen, thereby rendering the nutrient cut-off more
lethal to cancer cells.
10. Ferritinophagy in HCC
Ferritinophagy is the selective autophagy of
ferritin, a cellular iron storage complex. This process is mainly
mediated by the cargo receptor NCOA4, which binds to ferritin and
delivers it to autophagosomes (54). Through ferritinophagy, iron stored
in ferritin can be released upon lysosomal degradation, increasing
the labile iron pool within the cell (54). Ferritinophagy links autophagy to
iron homeostasis and ferroptosis, which is a form of cell death
caused by iron-dependent lipid peroxidation.
Aberrant iron metabolism is common in HCC. Previous
studies have indicated that ferritinophagy serves a role in HCC
cell death and survival (10,55).
The induction of ferritinophagy can kill HCC cells by ferroptosis.
NCOA4 mediates ferritinophagy, targeting ferritin for lysosomal
degradation to release stored iron. This NCOA4-driven iron
liberation expands the cellular labile iron pool, providing
redox-active Fe²+ that catalyzes ROS generation through
Fenton chemistry (56). The
resulting oxidative stress triggers the peroxidation of
polyunsaturated fatty acids in membranes, which is a hallmark of
ferroptosis (10).
Mechanistically, NCOA4-mediated ferritinophagy sensitizes cells to
ferroptosis by elevating catalytic iron and accelerating lethal
lipid peroxide accumulation. Consistently, inducing ferritinophagy
(such as by caryophyllene oxide) increases free Fe²+ and
malondialdehyde levels (a lipid peroxidation product), whereas
NCOA4 knockdown or interfering with NCOA4-ferritin binding prevents
iron release and protects cells from ferroptotic damage (42). In addition, suppression of NCOA4 or
its regulator, polypyrimidine tract-binding protein 1 (PTBP1), can
reduce the labile iron pool and lipid peroxidation, blunting
ferroptotic death (43).
Therefore, NCOA4-driven ferritinophagic iron release is a pivotal
trigger of ferroptosis through labile iron accumulation and
subsequent ROS-mediated lipid peroxidation. In an in vitro
study, caryophyllene-oxide treatment (20 µM; 6 h) doubled the
extent of NCOA4-ferritin co-localization in Hep3B cells, raised
calcein-quenchable Fe²+ by 42% and increased
malondialdehyde by 2.5X. By contrast, NCOA4 knockdown abolished
iron release and rescued 60% cells from cell death (54).
Another study by Yang et al (57) identified a pathway involving PTBP1
that can regulate ferritinophagy and ferroptosis in HCC.
Specifically, PTBP1 can bind to NCOA4 mRNA to enhance its
translation, where the knocking down PTBP1 expression in
sorafenib-treated HCC cells led to lower NCOA4 levels, reducing
free iron and malondialdehyde levels whilst increasing glutathione
levels, suppressing ferroptosis (57). Simultaneously, PTBP1 depletion
lowered NCOA4 by 48 %, shrank the labile iron pool by 35 % and
suppressed erastin-induced ferroptosis in MHCC-97H xenografts
(57). However, ferritinophagy may
also help HCC cells survive under certain conditions by providing
iron for essential enzymes. A recent report on pancreatic cancer
has shown that NCOA4-mediated ferritinophagy is required for tumor
growth by supplying iron to the proliferating cells (58).
Induction of excessive ferritinophagy appears to be
a promising therapeutic approach for HCC, essentially pushing cells
into ferroptosis, which is a lethal oxidative form of cell death
that cancer cells cannot easily reverse. Various compounds, such as
artesunate, sorafenib and caryophyllene oxide, can induce
ferroptosis, at least partly by increasing free iron levels either
through ferritinophagy or direct iron import (54).
11. Fluidophagy in HCC
Fluidophagy is a relatively novel term, describing
the autophagic degradation of fluid-like, phase-separated
intracellular condensates (59).
Cells contain numerous non-membrane-bound organelles that behave as
liquid droplets. A number of biophysical studies have previously
shown that the ‘wetting’ properties of droplets on autophagosome
membranes are crucial (60-62).
This process of droplet autophagy has been named ‘fluidophagy’
(59,63). At present, fluidophagy is a
mechanistic insight into selective autophagy, rather than a
distinct pathway with unique regulators.
For HCC, the biophysical state of the cargo (solid
aggregates vs. liquid droplets) can affect autophagic clearance. In
particular, the NCOA4-ferritin complex, which can form liquid
condensates in the cytosol, helps concentrate ferritin into such
droplets, which are then autophagically consumed (63). In addition, the autophagic
degradation of p62/sequestosome 1 (SQSTM1) bodies is another
possible example of fluidophagy (59).
In HCC, fluidophagy likely underlies the removal of
certain protein aggregates or storage complexes from the cells. HCC
cells frequently accumulate aggregates of p62 and ubiquitinated
proteins (p62 is commonly elevated in HCC and is used as a marker
of autophagic flux) (11).
12. Therapeutic targeting
Dysregulated selective autophagy contributes to HCC
progression and resistance to therapy, making the components of
these pathways attractive therapeutic targets. Strategies to
modulate autophagy in cancer are two-fold: Inhibition (to prevent
tumor cells from recycling organelles and evading death) and
activation (to drive cancer cells into lethal self-digestion).
Inhibiting pro-survival autophagy
HCC has been a prime candidate for autophagy
inhibition trials. Chloroquine (CQ) and hydroxychloroquine, which
inhibit lysosomal acidification and late-stage autophagy, have been
tested in preclinical HCC models and in early clinical studies
(38,39). Combining CQ with chemotherapeutic
oxaliplatin led to more pronounced tumor suppression in HCC
xenografts compared with chemotherapy alone (38). Newer lysosome-targeted agents, such
as GNS561, show even greater potency. GNS561, a PPT1 inhibitor,
accumulates in the lysosomes and blocks autophagy at a late stage,
causing cancer cell death (12).
In HCC models, GNS561 not only killed bulk tumor cells but was also
active against cancer stem cell populations (56). One previous clinical trial tested
hydroxychloroquine with the mTOR inhibitor everolimus in HCC
according to the ‘simultaneous activation and blockade’ strategy,
with disease control reaching 67% (with 6% partial responses) and
45% of patients had ≥6-month progression-free survival (14). Additionally, nutrient modulation,
such as ketogenic or fasting-mimicking diet to induce autophagy in
tumors, followed by autophagy inhibitor administration, is another
reported experimental approach aiming to catch cancer cells in a
vulnerable state (‘autophagy trap’) (15).
Targeting specific autophagy
pathways
A more tailored approach is to target regulators
unique to the selective autophagy types on which HCC depends. The
role of DRP1 in mitophagy in HCC has been proposed. DRP1-driven
mitophagy allows HCC cells to survive chemotherapy, since
inhibiting DRP1 with mdivi-1 was found to sensitize tumors to
cisplatin in mice (31). Another
proposed lysophagy target is CCDC50. Since CCDC50 knockdown causes
HCC cell death and tumor suppression by blocking lysophagy
(9), a drug that inhibits CCDC50
may mimic this tumor-suppressive effect. In the ER-phagy arena,
FAM134B and its regulator circFAM134B are potential targets to
enhance ferroptosis in tumors resistant to tyrosine kinase
inhibitors. An antisense oligonucleotide against circFAM134B was
found to decrease FAM134B levels, impairing ER-phagy and promoting
lenvatinib-induced ferroptosis (40). For lipophagy, one approach is to
restore BNIP3 function in HCC. BNIP3 is frequently silenced
epigenetically in cancers. Therefore, demethylating the BNIP3
promoter or delivering BNIP3 by gene therapy may reinstate
autophagic clearance of lipid droplets, potentially slowing tumor
growth in steatotic HCC (51).
Exploiting ferritinophagy and
ferroptosis
This is a promising novel direction. Ferroptosis
inducers (including sorafenib, sulfasalazine or experimental
agents, such as erastin and RSL3) can be combined with strategies
to increase ferritinophagy. Furthermore, various drugs, such as
caryophyllene oxide, may upregulate NCOA4 and drive ferritinophagy,
since they have shown efficacy in HCC models (54).
Precision considerations
Whether a given autophagy process is inhibited or
induced depends on the tumor context. Early-stage,
well-differentiated HCC may be more vulnerable to autophagy
induction to trigger cell death, whereas late-stage,
therapy-resistant HCC may require autophagy inhibition to remove
its survival crutch (4,5). Biomarkers are being investigated: For
instance, a ‘mitophagy gene signature’ stratifies patients with HCC
and correlates with immune microenvironment differences (16). The mitophagy gene signature is an
eight-gene score derived from core mitophagy regulators (e.g.,
optineurin and ATG12). By calculating a composite expression score
and splitting at the median, it divides patients with HCC into
high-risk and low-risk groups for survival. High scores indicate
aggressive disease and are associated with immunosuppressive tumour
microenvironments-characterised by increased infiltration of
certain immune cells (B cells, CD4/8 T cells, dendritic cells, NK
cells and macrophages) and altered cytokine gene expression. Thus,
the signature not only predicts prognosis but also reflects
differences in immune cell infiltration patterns (16).
13. Biomarkers of autophagy in HCC
Several autophagy-related biomarkers have been
documented to serve prognostic and predictive values for HCC
(Table III).
| Table IIIAutophagy-related biomarkers in
HCC. |
Table III
Autophagy-related biomarkers in
HCC.
| Biomarker | Clinical
findings | (Refs.) |
|---|
| p62/SQSTM1 | • Accumulates in
autophagy-defective HCC | (18,65,66) |
| | • High levels
associate with larger tumors, venous invasion and poor
survival | |
| | • Acts as
oncoprotein through NRF2 activation | |
| LC3B | • Increased
expression associates with superior differentiation and smaller
tumors | (16,67) |
| | • Positive staining
predicts longer survival | |
| | •
Context-dependent: Sometimes indicates high autophagic flux in
advanced HCC | |
| Beclin-1 | • Frequently
downregulated in HCC (17q loss or low transcription) | (68-70) |
| | • Preserved
expression associates with favorable OS and DFS | |
| | • Low levels
associate with poor differentiation and vascular invasion | |
| BNIP3 | • Frequently
silenced in HCC, especially in fatty liver-associated cases | (44) |
| | • Low expression
associates with high lipid content and worse prognosis | |
| | • Higher expression
associated with lower microvascular invasion | |
| NCOA4 | • Emerging
biomarker with higher expression in some HCC tissues | (13,49) |
| | • May predict
response to ferroptosis-inducing therapy | |
| | • Prognostic value
unclear; correlation with iron load needed | |
| p62/LC3 ratio | • High p62/low LC3
indicates autophagy-deficient tumors with poor RFS | (66,67) |
| | • Low p62/high LC3
associated with less aggressive tumors | |
| | • Dual staining
used to gauge autophagy flux | |
| Autophagy gene | • 8-gene mitophagy
score (such as optineurin and ATG12) stratifies HCC prognosis | (72,74) |
| signatures | • Multiple
predictive signatures (6-13 genes) correlate with outcomes | |
| | • High autophagy
gene expression indicates aggressive disease | |
| Immune-autophagy
markers | • High LC3 in
surrounding tissue but low in tumor correlates with recurrence | (10,15) |
| | • Circulating HMGB1
may predict immunotherapy response | |
| | • Links autophagy
status to tumor microenvironment | |
p62/SQSTM1
p62/SQSTM1 frequently accumulates in HCC cells with
defective autophagy. High p62 levels are associated with larger
tumor size, venous invasion and poorer overall survival (17). In addition, p62 can serve as an
oncoprotein through nuclear factor erythroid 2-related factor 2
activation (18).
LC3B
Increased LC3B expression is associated with
increased differentiation and smaller tumor size. A meta-analysis
has previously indicated that positive LC3B immunostaining predicts
longer survival in patients with HCC (19).
Beclin-1
This protein is downregulated in HCCs due to the
monoallelic loss of 17q or low transcription. Patients with
preserved Beclin-1 expression tend to have a more favorable overall
survival and disease-free survival (20).
BNIP3
It is frequently silenced or have its expression
reduced in HCC, particularly in fatty liver-associated HCC. Low
BNIP3 expression is associated with high intracellular lipid levels
and poor prognosis (51).
NCOA4
Initial data have suggested higher NCOA4 mRNA levels
in HCC tissues compared with those in adjacent tissues. It has been
hypothesized that NCOA4-high tumors respond with superior efficacy
to ferroptosis-inducing therapies (51).
p62/LC3 ratio
The combination of high p62 and low LC3 levels in
HCC tumor tissues indicates autophagy-deficient tumors, which are
associated with shorter recurrence-free survival (64).
Autophagy gene signatures
Several prognostic signatures using
autophagy-related genes have been developed to predict recurrence
and overall survival (65). An
eight-gene mitophagy score containing optineurin, ATG12 and related
genes that stratifies patients into high- and low-risk groups and a
series of predictive signatures using 6-13 autophagy genes or gene
pairs (e.g., LC3B/p62/BNIP3). These signatures correlate with
recurrence or overall survival, and higher autophagy-gene
expression generally signifies more aggressive disease (65).
14. Clinical implications and future
directions
Understanding selective autophagy in HCC has
important clinical implications because HCC cells frequently co-opt
organelle-specific recycling to survive hypoxia, metabolic stress
and chemotherapy (51,66). The autophagy status may serve as a
stratification tool to identify patients who can benefit from
autophagy modulation. Tumors with high autophagic flux may be more
vulnerable to autophagy inhibition, whereas those with impaired
autophagy may benefit from strategies that overcome their
compromised degradation systems. Furthermore, late-stage agents,
such as hydroxychloroquine or the lysosomotropic inhibitor GNS561,
can be used to exploit this vulnerability by arresting lysophagy
and bulk flux. GNS561 was found to achieve durable disease control
in a previous phase I clinical study and is advancing to phase II
(30). Targeted inhibition of
pro-survival mitophagy may enhance drug efficacy. Mdivi-1, genetic
PINK1 or PRKN (the Parkin gene) knockdown was found to boost
sorafenib-induced apoptosis by >30 % in xenografts (67).
Conversely, activating death-linked pathways is
equally promising. Agents that stimulate NCOA4-dependent
ferritinophagy (such as caryophyllene oxide) can enlarge the
labile-iron pool, intensifying lipid peroxidation to synergize with
ferroptosis inducers, achieving tumor shrinkage without added
systemic toxicity (54,68). ER-phagy modulation offers
precision, where circFAM134B silencing can disable reticulophagy,
magnifying lenvatinib-triggered ferroptosis in resistant HCC models
(40). Finally, autophagy gene
signatures (LC3B, p62 and BNIP3) are emerging prognostic tools for
stratifying patients according to combination regimens (69). Collectively, these data support a
future in which pathway-specific autophagy modulators, paired with
existing systemic or locoregional therapies can be used to drive
personalized HCC management whilst sparing normal hepatocytes
(70,71).
The concept of ‘synthetic lethality’ is particularly
relevant, identifying genetic or metabolic vulnerabilities in HCC
that, when coupled with autophagy manipulation, can lead to tumor
cell death (53,55,72,73).
HCC tumors with BNIP3 deficiency (and thus impaired
mitophagy/lipophagy) may be particularly sensitive to treatments
that increase lipid or oxidative stress (41,51,74).
Future directions in this field include the
following: i) Development of selective autophagy inhibitors, moving
beyond chloroquine to more targeted agents against specific
autophagy receptors or regulators (4); ii) biomarker-guided therapy, where
autophagy signatures can be used to guide treatment decisions (such
as which patients should receive autophagy inhibitors in addition
to conventional therapy) (75);
iii) Combined metabolic and autophagic targeting, by exploiting the
dependence of HCC on autophagy for metabolic adaptation by
simultaneously targeting metabolic pathways and autophagy (76); iv) local delivery technologies, by
developing liver-targeted delivery systems for autophagy modulators
to minimize systemic effects (77); and v) integration with
immunotherapy, by understanding how selective autophagy influences
tumor immune microenvironments and exploring combinations with
immune checkpoint inhibitors (78).
15. Limitations of the review
The present narrative review has several limitations
that readers should consider. No formal systematic review
methodology with structured database queries or predefined
inclusion/exclusion criteria was applied, which introduced
potential selection bias in the literature citations. Given the
breadth of the field, certain relevant studies may have been
inadvertently omitted, particularly since the primary focus was on
English-language publications in common databases, such as PubMed.
In addition, research on certain selective autophagy pathways in
HCC, such as nucleophagy and ribophagy, is limited, requiring
analogies to be drawn from other contexts or rely on preliminary
data. the majority of the evidence discussed is also only
preclinical, derived from cell lines or animal models rather than
from clinical trials. Although ongoing trials were mentioned, the
proposed therapeutic strategies using autophagy modulators lack
proven clinical efficacy for HCC treatment. These limitations
establish appropriate interpretive boundaries, ensuring the scope
of the present review is within the current preclinical landscape
whilst highlighting areas that require further investigation.
16. Conclusion
The selective autophagy pathways in HCC are emerging
as druggable nodes. By understanding whether a given type of
autophagy supports or hinders tumor growth, interventions can be
designed to tip the balance against cancer. The complexity of
autophagy in HCC highlights the need for personalized approaches.
Some tumors rely on specific autophagy pathways for survival,
whereas others may be suppressed by the same pathways. Therefore,
characterizing the autophagy dependence of individual tumors is
crucial for effective therapeutic interventions. The bidirectional
nature of autophagy, both tumor-promoting and tumor-suppressing,
necessitates careful consideration of the context, timing and
specific pathways when designing therapeutic strategies. As
understanding deepens, selective autophagy modulation promises to
become an important component of personalized HCC treatment. As
research progresses, a promising approach may be to first stress
cancer cells with conventional treatments and then disable their
selective autophagy defenses, thereby driving them into
irrecoverable failure.
Acknowledgements
Not applicable.
Funding
Funding: The present review was supported by a grant from Chang
Gung Memorial Hospital (grant no. CORPG8N0241).
Availability of data and materials
Not applicable.
Authors' contributions
CHH was involved in the conceptualization of the
review, literature search, writing of the manuscript and funding
acquisition. Data authentication is not applicable. The author has
read and agreed to the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Llovet JM, Kelley RK, Villanueva A, Singal
AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J and
Finn RS: Hepatocellular carcinoma. Nat Rev Dis Primers.
7(6)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ker CG: Hepatobiliary surgery in Taiwan:
The past, present, and future. Part I; biliary surgery. Formosan J
Surg. 57:1–10. 2024.
|
|
4
|
Alim Al-Bari A, Ito Y, Thomes PG, Menon
MB, García-Macia M, Fadel R, Stadlin A, Peake N, Faris ME, Eid N
and Klionsky DJ: Emerging mechanistic insights of selective
autophagy in hepatic diseases. Front Pharmacol.
14(1149809)2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nguyen TH, Nguyen TM, Ngoc DTM, You T,
Park MK and Lee CH: Unraveling the janus-faced role of autophagy in
hepatocellular carcinoma: Implications for therapeutic
interventions. Int J Mol Sci. 24(16255)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Klionsky DJ, Abdel-Aziz AK, Abdelfatah S,
Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abud
YP, Acevedo-Arozena A, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy (4th edition).
Autophagy. 17:1–382. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Vainshtein A and Grumati P: Selective
autophagy by close encounters of the ubiquitin kind. Cells.
9(2349)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate Parkin. PLoS Biol.
8(e1000298)2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Youle RJ and Narendra DP: Mechanisms of
mitophagy. Nat Rev Mol Cell Biol. 12:9–14. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang F, Denison S, Lai JP, Philips LA,
Montoya D, Kock N, Schüle B, Klein C, Shridhar V, Roberts LR and
Smith DI: Parkin gene alterations in hepatocellular carcinoma.
Genes Chromosomes Cancer. 40:85–96. 2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Feng J, Zhou J, Wu Y, Shen HM, Peng T and
Lu GD: Targeting mitophagy as a novel therapeutic approach in liver
cancer. Autophagy. 19:2164–2165. 2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Szargel R, Shani V, Elghani FA, Mekies LN,
Liani E, Rott R and Engelender S: The PINK1, synphilin-1 and SIAH-1
complex constitutes a novel mitophagy pathway. Hum Mol Genet.
25:3476–3490. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhou J, Feng J, Wu Y, Dai HQ, Zhu GZ, Chen
PH, Wang LM, Lu G, Liao XW, Lu PZ, et al: Simultaneous treatment
with sorafenib and glucose restriction inhibits hepatocellular
carcinoma in vitro and in vivo by impairing SIAH1-mediated
mitophagy. Exp Mol Med. 54:2007–2021. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Luo P, An Y, He J, Xing X, Zhang Q, Liu X,
Chen Y, Yuan H, Chen J, Wong YK, et al: Icaritin with
autophagy/mitophagy inhibitors synergistically enhances anticancer
efficacy and apoptotic effects through PINK1/Parkin-mediated
mitophagy in hepatocellular carcinoma. Cancer Lett.
587(216621)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang S, Wang Y, Cao Y, Wu J, Zhang Z, Ren
H, Xu X, Kaznacheyeva E, Li Q and Wang G: Inhibition of the
PINK1-parkin pathway enhances the lethality of sorafenib and
regorafenib in hepatocellular carcinoma. Front Pharmacol.
13(851832)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ma M, Lin XH, Liu HH, Zhang R and Chen RX:
Suppression of DRP1-mediated mitophagy increases the apoptosis of
hepatocellular carcinoma cells in the setting of chemotherapy.
Oncol Rep. 43:1010–1018. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Aman Y, Cao S and Fang EF: Iron out,
mitophagy in! A way to slow down hepatocellular carcinoma. EMBO
Rep. 21(e51652)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kim SJ, Khan M, Quan J, Till A, Subramani
S and Siddiqui A: Hepatitis B virus disrupts mitochondrial
dynamics: Induces fission and mitophagy to attenuate apoptosis.
PLoS Pathog. 9(e1003722)2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li Y and Ou JJ: Regulation of
mitochondrial metabolism by hepatitis B virus. Viruses.
15(2359)2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen YY, Wang WH, Che L, Lan Y, Zhang LY,
Zhan DL, Huang ZY, Lin ZN and Lin YC: BNIP3L-dependent mitophagy
promotes HBx-induced cancer stemness of hepatocellular carcinoma
cells via glycolysis metabolism reprogramming. Cancers (Basel).
12(655)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jia P, Tian T, Li Z, Wang Y, Lin Y, Zeng
W, Ye Y, He M, Ni X, Pan J, et al: CCDC50 promotes tumor growth
through regulation of lysosome homeostasis. EMBO Rep.
24(e56948)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Eapen VV, Swarup S, Hoyer MJ, Paulo JA and
Harper JW: Quantitative proteomics reveals the selectivity of
ubiquitin-binding autophagy receptors in the turnover of damaged
lysosomes by lysophagy. Elife. 10(e72328)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hoyer MJ, Swarup S and Harper JW:
Mechanisms controlling selective elimination of damaged lysosomes.
Curr Opin Physiol. 29(100590)2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang Y, Wang Z, Sun J and Qian Y:
Identification of HCC subtypes with different prognosis and
metabolic patterns based on mitophagy. Front Cell Dev Biol.
9(799507)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ding ZB, Hui B, Shi YH, Zhou J, Peng YF,
Gu CY, Yang H, Shi GM, Ke AW, Wang XY, et al: Autophagy activation
in hepatocellular carcinoma contributes to the tolerance of
Oxaliplatin via reactive oxygen species modulation. Clin Cancer
Res. 17:6229–6238. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Brun S, Bestion E, Raymond E, Bassissi F,
Jilkova ZM, Mezouar S, Rachid M, Novello M, Tracz J, Hamaï A, et
al: GNS561, a clinical-stage PPT1 inhibitor, is efficient against
hepatocellular carcinoma via modulation of lysosomal functions.
Autophagy. 18:678–694. 2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bi T, Lu Q, Pan X, Dong F, Hu Y, Xu Z, Xiu
P, Liu Z and Li J: circFAM134B is a key factor regulating
reticulophagy-mediated ferroptosis in hepatocellular carcinoma.
Cell Cycle. 22:1900–1920. 2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu Z, Ma C, Wang Q, Yang H, Lu Z, Bi T,
Xu Z, Li T, Zhang L, Zhang Y, et al: Targeting FAM134B-mediated
reticulophagy activates sorafenib-induced ferroptosis in
hepatocellular carcinoma. Biochem Biophys Res Commun. 589:247–253.
2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wu H, Liu Q, Shan X, Gao W and Chen Q: ATM
orchestrates ferritinophagy and ferroptosis by phosphorylating
NCOA4. Autophagy. 19:2062–2077. 2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wei X, Manandhar L, Kim H, Chhetri A,
Hwang J, Jang G, Park C and Park R: Pexophagy and oxidative stress:
Focus on peroxisomal proteins and reactive oxygen species (ROS)
signaling pathways. Antioxidants (Basel). 14(126)2025.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Dutta RK, Maharjan Y, Lee JN, Park C, Ho
YS and Park R: Catalase deficiency induces reactive oxygen species
mediated pexophagy and cell death in the liver during prolonged
fasting. Biofactors. 47:112–125. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ohshima K, Hara E, Takimoto M, Bai Y,
Hirata M, Zeng W, Uomoto S, Todoroki M, Kobayashi M, Kozono T, et
al: Peroxisome proliferator activator α agonist clofibrate induces
pexophagy in coconut oil-based high-fat diet-fed rats. Biology
(Basel). 13(1027)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Deosaran E, Larsen KB, Hua R, Sargent G,
Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J,
et al: NBR1 acts as an autophagy receptor for peroxisomes. J Cell
Sci. 126:939–952. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li Y, Jiang X, Zhang Y, Gao Z, Liu Y, Hu
J, Hu X, Li L, Shi J and Gao N: Nuclear accumulation of UBC9
contributes to SUMOylation of lamin A/C and nucleophagy in response
to DNA damage. J Exp Clin Cancer Res. 38(67)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Papandreou ME and Tavernarakis N:
Nucleophagy: From homeostasis to disease. Cell Death Differ.
26:630–639. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Cebollero E, Reggiori F and Kraft C:
Reticulophagy and ribophagy: Regulated degradation of protein
production factories. Int J Cell Biol. 2012(182834)2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu Y, Zou W, Yang P, Wang L, Ma Y, Zhang
H and Wang X: Autophagy-dependent ribosomal RNA degradation is
essential for maintaining nucleotide homeostasis during C. elegans
development. eLife. 7(e36588)2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wyant GA, Abu-Remaileh M, Frenkel EM,
Laqtom NN, Dharamdasani V, Lewis CA, Chan SH, Heinze I, Ori A and
Sabatini DM: NUFIP1 is a ribosome receptor for starvation-induced
ribophagy. Science. 360:751–758. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Xu F, Tautenhahn HM, Dirsch O and Dahmen
U: Blocking autophagy with chloroquine aggravates lipid
accumulation and reduces intracellular energy synthesis in
hepatocellular carcinoma cells, both contributing to its
anti-proliferative effect. J Cancer Res Clin Oncol. 148:3243–3256.
2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Xu C and Fan J: Links between autophagy
and lipid droplet dynamics. J Exp Bot. 73:2848–2858.
2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Berardi DE, Bock-Hughes A, Terry AR, Drake
LE, Bozek G and Macleod KF: Lipid droplet turnover at the lysosome
inhibits growth of hepatocellular carcinoma in a BNIP3-dependent
manner. Sci Adv. 8(eabo2510)2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Koutsifeli P, Varma U, Daniels LJ,
Annandale M, Li X, Neale JPH, Hayes S, Weeks KL, James S, Delbridge
LMD and Mellor KM: Glycogen-autophagy: Molecular machinery and
cellular mechanisms of glycophagy. J Biol Chem.
298(102093)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Gade TPF, Tucker E, Nakazawa MS, Hunt SJ,
Wong W, Krock B, Weber CN, Nadolski GJ, Clark TWI, Soulen MC, et
al: Ischemia induces quiescence and autophagy dependence in
hepatocellular carcinoma. Radiology. 283:702–710. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Xiu Z, Zhu Y, Han J, Li Y, Yang X, Yang G,
Song G, Li S, Li Y, Cheng C, et al: Caryophyllene oxide induces
ferritinophagy by regulating the NCOA4/FTH1/LC3 pathway in
hepatocellular carcinoma. Front Pharmacol.
13(930958)2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wang G, Li J, Zhu L, Zhou Z, Ma Z, Zhang
H, Yang Y, Niu Q and Wang X: Identification of hepatocellular
carcinoma-related subtypes and development of a prognostic model: A
study based on ferritinophagy-related genes. Discov Oncol.
14(147)2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Subburayan K, Thayyullathil F,
Pallichankandy S, Cheratta AR, Alakkal A, Sultana M, Drou N, Arshad
M, Palanikumar L, Magzoub M, et al: Tumor suppressor Par-4
activates autophagy-dependent ferroptosis. Commun Biol.
7(732)2024.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Yang H, Sun W, Bi T, Wang Q, Wang W, Xu Y,
Liu Z and Li J: The PTBP1-NCOA4 axis promotes ferroptosis in liver
cancer cells. Oncol Rep. 49(45)2023.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Santana-Codina N, del Rey MQ, Kapner KS,
Zhang H, Gikandi A, Malcolm C, Poupault C, Kuljanin M, John KM,
Biancur DE, et al: NCOA4-Mediated ferritinophagy is a pancreatic
cancer dependency via maintenance of iron bioavailability for
iron-sulfur cluster proteins. Cancer Discov. 12:2180–2197.
2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Yang Z, Yoshii SR, Sakai Y, Zhang J, Chino
H, Knorr RL and Mizushima N: Autophagy adaptors mediate
Parkin-dependent mitophagy by forming sheet-like liquid
condensates. EMBO J. 43:5613–5634. 2024.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Agudo-Canalejo J, Schultz SW, Chino H,
Migliano SM, Saito C, Koyama-Honda I, Stenmark H, Brech A, May AI,
Mizushima N and Knorr RL: Wetting regulates autophagy of
phase-separated compartments and the cytosol. Nature. 591:142–146.
2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Mangiarotti A, Sabri E, Schmidt KV,
Hoffmann C, Milovanovic D, Lipowsky R and Dimova R: Lipid packing
and cholesterol content regulate membrane wetting and remodeling by
biomolecular condensates. Nat Commun. 16(2756)2025.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Mangiarotti A, Chen N, Zhao Z, Lipowsky R
and Dimova R: Wetting and complex remodeling of membranes by
biomolecular condensates. Nat Commun. 14(2809)2023.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Ohshima T, Yamamoto H, Sakamaki Y, Saito C
and Mizushima N: NCOA4 drives ferritin phase separation to
facilitate macroferritinophagy and microferritinophagy. J Cell
Biol. 221(e202203102)2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Cerda-Troncoso C, Varas-Godoy M and Burgos
PV: Pro-tumoral functions of autophagy receptors in the modulation
of cancer progression. Front Oncol. 10(619727)2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Brun S, Pascussi JM, Gifu EP, Bestion E,
Macek-Jilkova Z, Wang G, Bassissi F, Mezouar S, Courcambeck J,
Merle P, et al: GNS561, a new autophagy inhibitor active against
cancer stem cells in hepatocellular carcinoma and hepatic
metastasis from colorectal cancer. J Cancer. 12:5432–5438.
2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Shalhoub H, Gonzalez P, Dos Santos A,
Guillermet-Guibert J, Moniaux N, Dupont N and Faivre J:
Simultaneous activation and blockade of autophagy to fight
hepatocellular carcinoma. Autophagy Rep. 3(2326241)2024.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Qian R, Cao G, Su W, Zhang J, Jiang Y,
Song H, Jia F and Wang H: Enhanced sensitivity of tumor cells to
autophagy inhibitors using fasting-mimicking diet and targeted
lysosomal delivery nanoplatform. Nano Lett. 22:9154–9162.
2022.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Liu C, Wu Z, Wang L, Yang Q and Huang J
and Huang J: A mitophagy-related gene signature for subtype
identification and prognosis prediction of hepatocellular
carcinoma. Int J Mol Sci. 23(12123)2022.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Umemura A, He F, Taniguchi K, Nakagawa H,
Yamachika S, Font-Burgada J, Zhong Z, Subramaniam S, Raghunandan S,
Duran A, et al: p62, Upregulated during preneoplasia, induces
hepatocellular carcinogenesis by maintaining survival of stressed
HCC-initiating cells. Cancer Cell. 29:935–948. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Saito T, Ichimura Y, Taguchi K, Suzuki T,
Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T, et
al: p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular
carcinoma through Nrf2-dependent metabolic reprogramming. Nat
Commun. 7(12030)2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Meng YC, Lou XL, Yang LY, Li D and Hou YQ:
Role of the autophagy-related marker LC3 expression in
hepatocellular carcinoma: A meta-analysis. J Cancer Res Clin Oncol.
146:1103–1113. 2020.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Qiu DM, Wang GL, Chen L, Xu YY, He S, Cao
XL, Qin J, Zhou JM, Zhang YX and Qun E: The expression of beclin-1,
an autophagic gene, in hepatocellular carcinoma associated with
clinical pathological and prognostic significance. BMC Cancer.
14(327)2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Lin CW, Chen YS, Lin CC, Lee PH, Lo GH,
Hsu CC, Hsieh PM, Koh KW, Chou TC, Dai CY, et al: Autophagy-related
gene LC3 expression in tumor and liver microenvironments
significantly predicts recurrence of hepatocellular carcinoma after
surgical resection. Clin Transl Gastroenterol.
9(166)2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Cao J, Wu L, Lei X, Shi K and Shi L: A
signature of 13 autophagy-related gene pairs predicts prognosis in
hepatocellular carcinoma. Bioengineered. 12:697–707.
2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Wang S, Cheng H, Li M, Gao D, Wu H, Zhang
S, Huang Y and Guo K: BNIP3-mediated mitophagy boosts the
competitive growth of Lenvatinib-resistant cells via energy
metabolism reprogramming in HCC. Cell Death Dis.
15(484)2024.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Bassissi F, Jílková Z, Brun S, Courcambeck
J, Tracz J, Kurma K, Roth GS, Khaldi C, Chaimbault C, Quentin B, et
al: Abstract 5124: GNS561 a new quinoline derivative inhibits the
growth of hepatocellular carcinoma in a cirrhotic rat and human PDX
orthotopic mouse models. Cancer Res. 77 (Suppl 13)(5124)2017.
|
|
68
|
Miao Y, Yin Q, Ping L, Sheng H, Chang J,
Li W and Lv S: Pseudolaric acid B triggers ferritinophagy and
ferroptosis via upregulating NCOA4 in lung adenocarcinoma cells. J
Cancer Res Ther. 19:1646–1653. 2023.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Zhu J, Wang M and Hu D: Development of an
autophagy-related gene prognostic signature in lung adenocarcinoma
and lung squamous cell carcinoma. PeerJ. 8(e8288)2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Rahdan F, Abedi F, Dianat-Moghadam H, Sani
MZ, Taghizadeh M and Alizadeh E: Autophagy-based therapy for
hepatocellular carcinoma: from standard treatments to combination
therapy, oncolytic virotherapy, and targeted nanomedicines. Clin
Exp Med. 25(13)2024.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Zai W, Chen W, Han Y, Wu Z, Fan J, Zhang
X, Luan J, Tang S, Jin X, Fu X, et al: Targeting PARP and autophagy
evoked synergistic lethality in hepatocellular carcinoma.
Carcinogenesis. 41:345–357. 2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Qian M, Wan Z, Liang X, Jing L, Zhang H,
Qin H, Duan W, Chen R, Zhang T, He Q, et al: Targeting autophagy in
HCC treatment: Exploiting the CD147 internalization pathway. Cell
Commun Signal. 22(583)2024.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Macleod K: Abstract 4984: BNip3 suppresses
hepatocellular carcinoma (HCC) growth by limiting lipogenesis.
Cancer Res. 77 (Suppl 13)(4984)2017.
|
|
74
|
Zai W, Chen W, Liu H and Yuxuan H:
MO1-5-3-Compromised autophagy sensitizes hepatocellular carcinoma
to PARP inhibition. Ann Oncol. 30:vi91–vi2. 2019.
|
|
75
|
Sadagopan N and He AR: Recent progress in
systemic therapy for advanced hepatocellular carcinoma. Int J Mol
Sci. 25(1259)2024.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Byrnes K, Blessinger S, Bailey NT, Scaife
R, Liu G and Khambu B: Therapeutic regulation of autophagy in
hepatic metabolism. Acta Pharm Sin B. 12:33–49. 2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Allaire M, Rautou PE, Codogno P and
Lotersztajn S: Autophagy in liver diseases: Time for translation? J
Hepatol. 70:985–998. 2019.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Martini G, Ciardiello D, Paragliola F,
Nacca V, Santaniello W, Urraro F, Stanzione M, Niosi M, Dallio M,
Federico A, et al: How immunotherapy has changed the continuum of
care in hepatocellular carcinoma. Cancers (Basel).
13(4719)2021.PubMed/NCBI View Article : Google Scholar
|