Introduction
Perivascular epithelioid cell tumors (PEComas) are a
rare group of mesenchymal neoplasms characterized by distinctive
histological and immunohistochemical features. This tumor family
was first described by Bonetti et al (1) in 1992, initially encompassing renal
angiomyolipoma and the clear cell ‘sugar’ tumor of the lung.
Subsequently, additional neoplasms were incorporated
into the PEComa family based on their characteristic co-expression
of melanocytic markers (HMB-45, Melan-A) and smooth muscle markers
(SMA), including lymphangioleiomyomatosis (2). The same research group later expanded
the category to include clear cell myomelanocytic tumors of the
falciform ligament (ligamentum teres) and other clear cell tumors
across multiple organs (3), such
as the urinary bladder, prostate, uterus, ovary, vulva, vagina,
lung, pancreas and liver (hepatic angiomyolipoma). Molecular
studies have established a strong link between PEComas and the
tuberous sclerosis complex (TSC), an autosomal dominant disorder
caused by mutations or deletions of the TSC1 (9q34) or TSC2
(16p13.3) genes (4,5). Clinically, TSC is associated with
intellectual disability, seizures, and various neoplasms, including
angiomyolipomas, subependymal giant cell astrocytomas, cutaneous
angiofibromas, cardiac rhabdomyomas, lymphangioleiomyomatosis, and
multifocal micronodular pneumocyte hyperplasia. Further research
has demonstrated that TSC1/TSC2 loss results in activation of
Rheb/mTOR/p70S6K signaling pathway, a key driver of tumorigenesis
in PEComas and the biological rationale for the use of mTOR
inhibitors in selected cases (6-8).
It is noteworthy, however, that PEComas do not occur more
frequently in patients with clinical TSC. Instead, most PEComas
harbor somatic mutations in TSC1 or, more commonly, TSC2 without
affected individuals meeting the diagnostic criteria for systemic
TSC. This distinction highlights the role of TSC gene alterations
as driver mutation in PEComa tumorigenesis, rather than as
manifestations of germline disease.
In 2005, Folpe et al (9) reported on 26 cases of soft tissue and
gynecologic PEComas, have proposed histological criteria for risk
classification into benign, uncertain malignant potential, and
malignant categories. Parameters associated with aggressive
behavior include tumor size >5 cm, infiltrative growth, high
nuclear grade, necrosis, and mitotic activity >1/50 high-power
fields. Surgical excision remains the standard treatment,
particularly for tumors with high-risk features.
Yamasaki et al (10) have reported the first description
of hepatic PEComa. in 2000. Since then, additional reports,
including malignant variants, have been described, with a total of
224 primary hepatic PEComas described across 75 publications
(11,12).
Here, we present an unusual case of hepatic PEComa
diagnosed in the hepatobiliary surgery center of the University
Hospital Münster, Germany. With a particular focus on
histopathological evaluation, this case highlights the importance
of distinguishing PEComas from morphologically overlapping hepatic
neoplasms, particularly hepatocellular carcinoma, in order to
ensure accurate diagnosis and appropriate treatment.
Case report
A 43-year-old man with no known comorbidities and in
good health (height: 188 cm, weight: 75 kg, BMI: 21.2 kg/m²)
presented with abdominal discomfort and right upper quadrant
tenderness. Initial laboratory tests were unremarkable. The
alpha-fetoprotein (AFP) was within the reference range (2.4 ng/ml;
reference <7 ng/ml). During admission, fluctuating elevations of
GPT and GOT were noted. Dynamic contrast-enhanced CT multi-slice
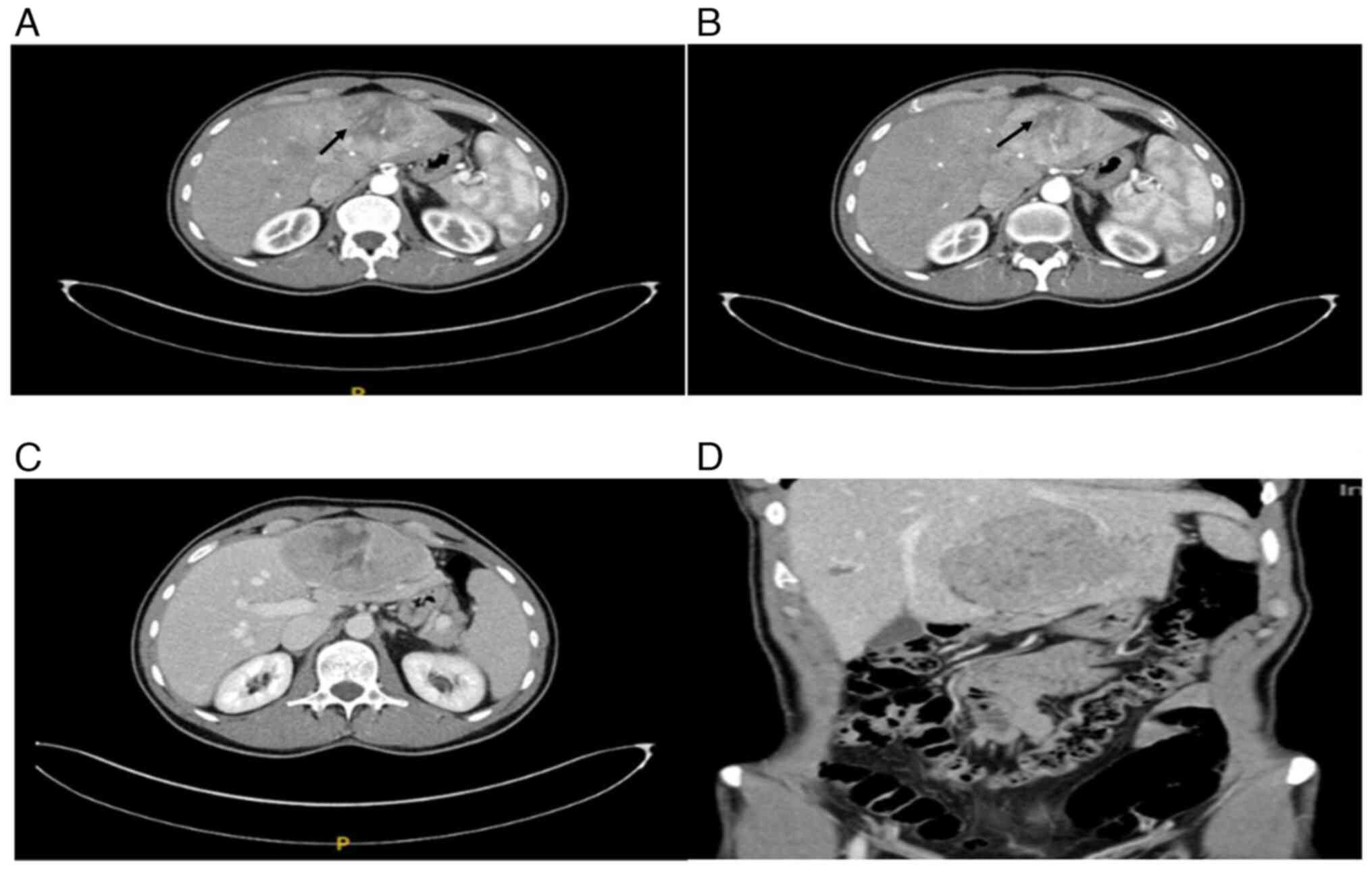
(MSCT) revealed a large tumor in the hepatic left lobe measuring
11.6x10.3x6.8 cm (Fig. 1A-D). The
lesion was inhomogeneous, peripherally hypervascular, and showed
partial washout in the portal venous phase. Based on imaging, a
liver adenoma was suspected, and the interdisciplinary tumor board
recommended resection.
The patient underwent robotic-assisted left
hemihepatectomy. Postoperative recovery was uneventful, and he was
discharged on day 5 with negative resection margins.
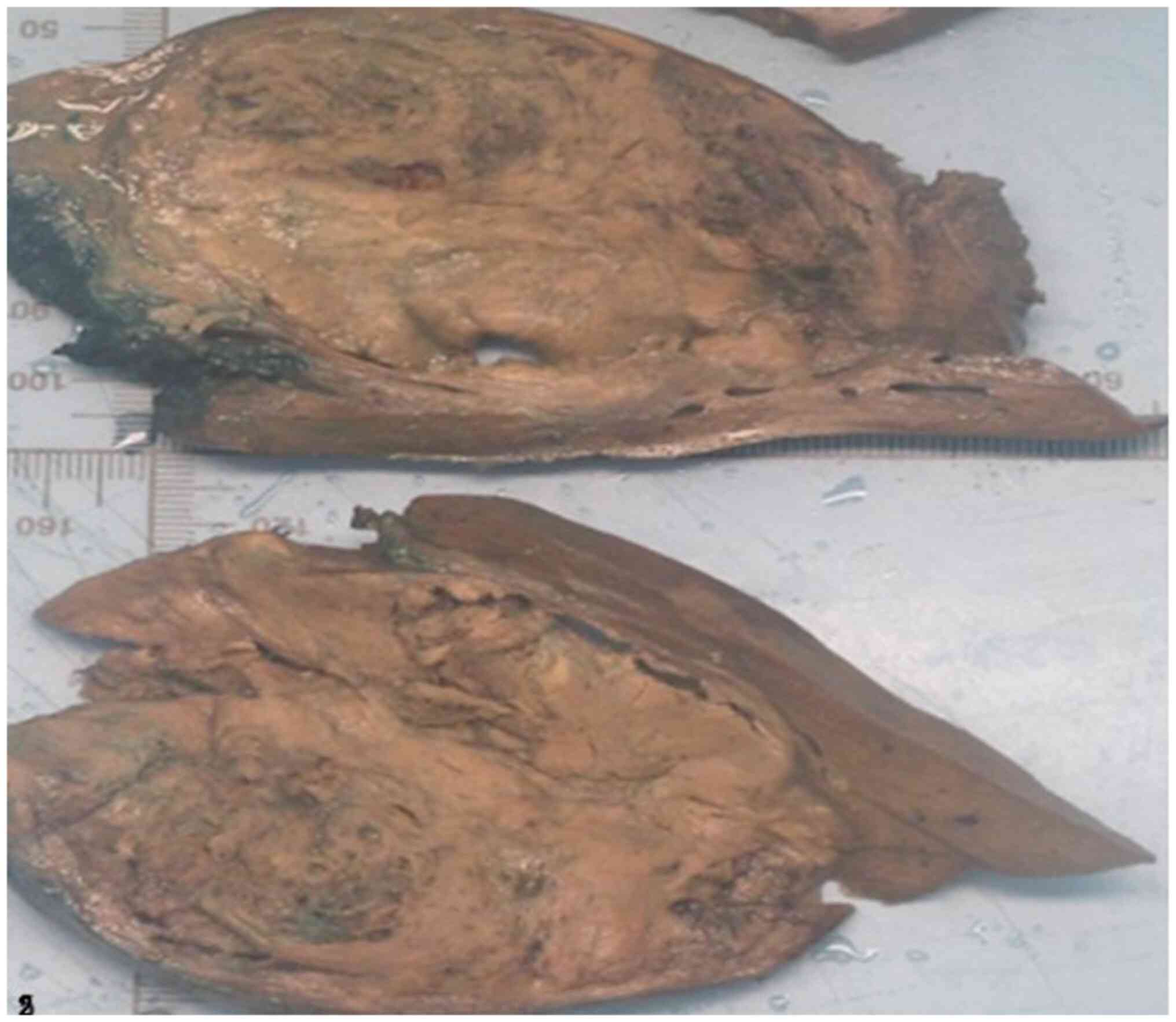
Gross pathology revealed a liver specimen weighing
604 g containing a lobulated, heterogeneous, pseudocapsulated tumor
measuring 12x8x5.5 cm, located 2 mm from the resection margin
(Fig. 2).
Microscopy (Fig.
3A-E) showed epithelioid, polyhedral cells with central to
eccentric nuclei and abundant eosinophilic cytoplasm, arranged in
nests and separated by a delicate capillary network. Central
necrosis (Fig. 3B; arrow) was
present, and, at the periphery, the tumor infiltrated its
pseudocapsule with vascular invasion (Fig. 3D and E; arrow). Nuclei displayed irregular
contours with focal nucleolar prominence, and the mitotic rate was
up to 6/10 HPF. Based on these features and the epithelioid
morphology, the initial histopathological differential diagnosis
was moderately differentiated hepatocellular carcinoma (HCC).
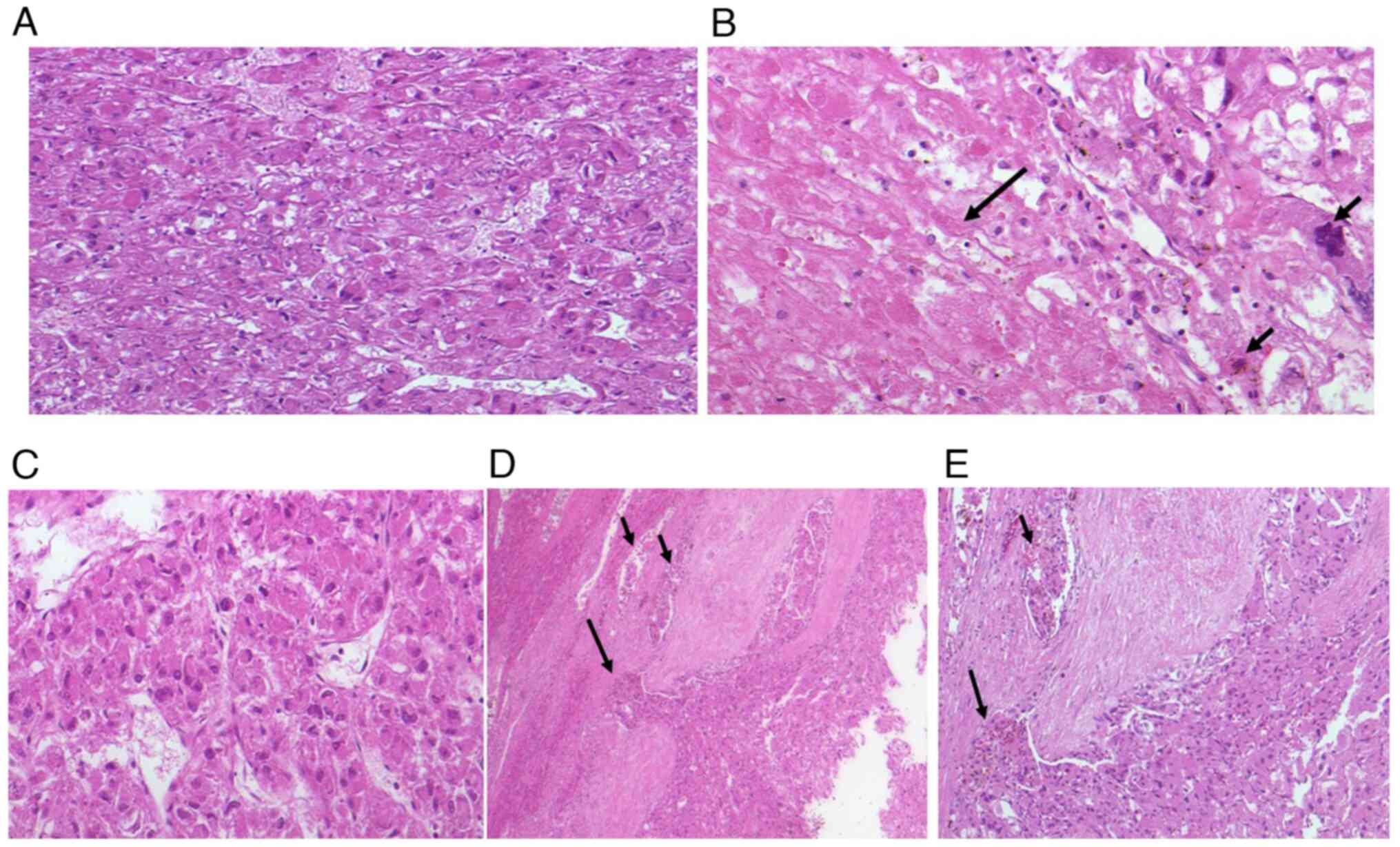 | Figure 3(A) Microscopic examination showed
epithelioid, polyhedral tumor cells with central to eccentric
nuclei and abundant eosinophilic cytoplasm, arranged in nests and
separated by a delicate capillary network (H&E staining;
magnification, x20). (B) Central necrosis (long arrow) was present.
Nuclei displayed irregular contours with focal nucleolar prominence
and abnormal mitotic figures (short arrow) (H&E staining;
magnification, x20). (C) Microscopic examination revealed
polyhedral epithelioid tumor cells with centrally to eccentrically
located nuclei and abundant eosinophilic cytoplasm. The cells were
arranged in trabeculae, separated by a fine capillary network
(H&E staining; magnification, x20). (D) Microscopic examination
demonstrated tumor cell infiltration into blood vessels and
capillaries (short arrow) as well as penetration of the
pseudocapsule (long arrow) (H&E staining; magnification, x10).
(E) At higher magnification, peripheral tumor infiltration was
evident, with the long arrow indicating pseudocapsule penetration
and the short arrow indicating vascular invasion (H&E staining;
magnification, x20). |
Immunohistochemistry (IHC) demonstrated complete
negative for HepPar-1, Arginase-1, Glypican-3, and Glutamine
Synthetase. HSP-70 showed weak, non-specific positivity. The Ki-67
proliferation index was <5% and CD34 staining highlighted the
peritumoral capillary network.
To refine these unexpected findings, an extended IHC
panel was applied. The tumor was negative for β-catenin, Serum
Amyloid A (SAA), L-FABP, CD10, CK7, CK20, and pancytokeratin,
effectively excluding HCC and hepatocellular adenoma. Further
testing revealed no expression of SOX10, S100, PAX8, Chromogranin
A, Vimentin, CD117 (c-Kit), Desmin, H-Caldesmon, Myogenin, MyoD1,
OCT-4, and SALL4. BRG1 and INI-1 expression were preserved.
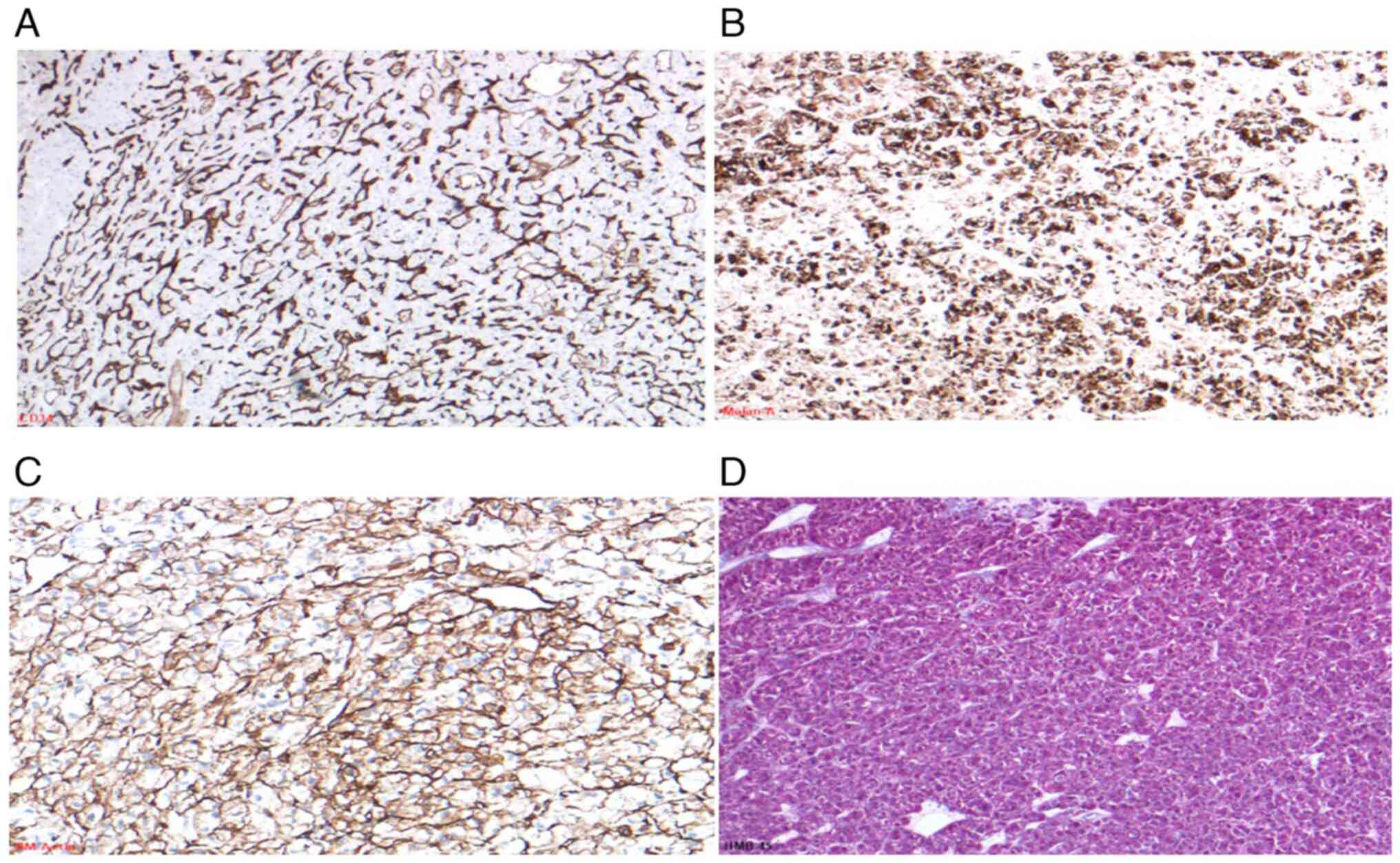
Notably, tumor cells showed strong, diffuse expression of Melan-A
and HMB-45, with focal weak positivity for SMA (Fig. 4A-D), consistent with a perivascular
epithelioid cell tumor (PEComa).
Molecular analysis supported this diagnosis. NGS
revealed truncating mutations in exons 10 and 27 of the TSC2 gene
(Table I). FISH analysis excluded
a TFE3 rearrangement at Xp11.23, ruling out TFE3-associated
PEComa.
 | Table IDetected mutations. |
Table I
Detected mutations.
| Gene | Reference
sequences | Exon | Alteration
(interpretation) | AF, % | Database ID |
|---|
| TSC1
(HGNC:12362) |
NM_000368/NP_000359 | -/- | -/- | -/- | -/- |
| TSC2
(HGNC:12363) |
NM_000548/NP_000539 | 10 | c.913G>T; p.
Gly305* (likely loss of function, likely pathogenic) | 11,3 | ClinVar: n.a. |
| TSC2
(HGNC:12363) |
NM_000548/NP_000539 | 27 | c.3093dup; p.
Arg1032Serfs*13 (likely loss of function, likely pathogenic) | 13,3 | ClinVar: 2910389 |
The final pathology report summarized the
histological and molecular features, highlighting adverse
prognostic indicators: large tumor size, infiltrative growth,
necrosis, and mitotic activity (7). The case was reviewed at the
multidisciplinary tumor board, and close clinical follow-up was
recommended. The patient has been treated surgically to date, and
follow-up was recommended. The most recent whole-body imaging,
performed five months after the left hemihepatectomy, showed no
metastases or residual lesions.
The Ethics Committee of University Hospital Münster
(Approval No. 2019-636-f-S) approves this case.
Discussion
Most hepatic PEComas reported to date are benign.
However, histological and clinical features predictive of
aggressive behavior have been defined. Yoo et al (12) have identified ‘worrisome features’,
including tumor size ≥7 cm, infiltrative borders, mitotic activity
>1/10 mm², necrosis, vascular invasion, and classification as
PEComa not otherwise specified (NOS). Risk stratification is based
on these features: high-risk if ≥3 are present, intermediate-risk
if 1-2 are present, and low-risk if none are identified. In the
present case, multiple adverse features indicated high-risk
disease.
Molecular findings further supported this
assessment. The identified truncating mutations in TSC2 are
typically loss-of-function variants that drive mTOR pathway
activation and are more frequently associated with aggressive
clinical behavior, whereas missense variants may have variable
consequences depending on their functional domains. Pan et
al (7) have demonstrated that
TSC2 alterations correlate with poor prognosis, highlighting the
biological relevance of mutation type.
Despite their malignant potential, PEComas generally
have a low recurrence (3.1%) and metastasis rate (2.7%), as
reported by Kvietkauskas et al (13). Complete surgical resection with
negative margins remains the standard treatment. In selected cases,
neoadjuvant mTOR inhibitors have been shown to reduce tumor size
and resection without complications (14).
Histological subtypes may also guide diagnostic
interpretation. Bennett et al (15) have emphasized that epithelioid
PEComas, such as in our patient, often lack strong smooth muscle
marker expression, in contrast to spindle cell-dominant PEComas,
which generally stain strongly positive. Recognition of this
pattern is essential to avoid misdiagnosis.
In summary, we report a rare case of hepatic PEComa
with multiple high-risk features and pathogenic TSC2 mutations.
This underscores the importance of a thorough immunohistochemical
and molecular workup in epithelioid liver tumors to differentiate
PEComa from morphologically similar entities such as hepatocellular
carcinoma Awareness of this diagnostic pitfall is crucial for
pathologists and clinicians to ensure accurate diagnosis,
appropriate patient management, and optimal therapeutic
decision-making.
Acknowledgements
The authors would like to thank Dr Kim Falkenberg
(Gerhard-Domagk-Institute of Pathology and Cytology, University
Hospital Muenster, D-48149 Muenster, Germany) for processing the
next-generation sequencing analysis, interpreting the results and
providing the data link, and Mrs. Petra Abbas (Muenster, Germany)
for proofreading.
Funding
Funding: No funding was received.
Availability of data and materials
The next-generation sequencing data generated in the
present study may be found in the Sequence Read Archive database
under accession number PRJNA1332571 or at the following URL:
https://www.ncbi.nlm.nih.gov/sra/PRJNA1332571. The
other data generated in the present study are not publicly
available due to data privacy and protection regulations but may be
requested from the corresponding author.
Authors' contributions
MA was involved in detection and diagnosis of the
rare case, performance and analysis of immunohistochemistry and
molecular pathology, development of the study concept, and
manuscript writing and final proofreading. BS participated in the
surgical procedure, was involved in case discussion, and critically
reviewed and proofread the manuscript, including validation of the
overall concept. MHM was involved in case discussion, patient
follow-up and proofreading of the manuscript. AP was involved in
case discussion, and critical review and reading of the manuscript,
with validation of the study concept. WH was involved in detection
and diagnosis of the rare case, contributed to the study concept,
analyzed immunohistochemistry and molecular pathology data, and
proofread the manuscript. EW was involved in detection and
diagnosis of the rare case, contributed to the study concept,
analyzed immunohistochemistry and molecular pathology data, and
proofread the manuscript. MA and WH confirm the authenticity of all
the raw data. All authors have read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
The Ethics Committee of University Hospital Münster
(approval no. 2019-636-f-S; Muenster, Germany) approved this
case.
Patient consent for publication
The patient provided written consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bonetti F, Pea M, Martignoni G and Zamboni
G: PEC and sugar. Am J Surg Pathol. 16:307–308. 1992.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pea M, Martignoni G, Zamboni G and Bonetti
F: Perivascular epithelioid cell. Am J Surg Pathol. 20:1149–1153.
1996.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Martignoni G, Pea M, Reghellin D, Zamboni
G and Bonetti F: PEComas: The past, the present and the future.
Virchows Arch. 452:119–132. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
European Chromosome 16 Tuberous Sclerosis
Consortium. Identification and characterization of the tuberous
sclerosis gene on chromosome 16. Cell. 75:1305–1315.
1993.PubMed/NCBI View Article : Google Scholar
|
|
5
|
van Slegtenhorst M, de Hoogt R, Hermans C,
Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A,
Halley D, Young J, et al: Identification of the tuberous sclerosis
gene TSC1 on chromosome 9q34. Science. 277:805–808. 1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kwiatkowski DJ: Tuberous sclerosis: From
tubers to mTOR. Ann Hum Genet. 67:87–96. 2003.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Pan CC, Chung MY, Ng KF, Liu CY and Wang
JS: (2012). Constant allelic alteration of the TSC2 gene in
PEComas: A genetic hallmark? Modern Pathology, 25(3), 393–398.
|
|
8
|
Kenerson H, Folpe AL, Takayama TK and
Yeung RS: Activation of the mTOR pathway in sporadic PEComas.
Journal of Pathology. 211:445–453. 2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Folpe AL, Mentzel T, Lehr HA, Fisher C,
Balzer BL and Weiss SW: Perivascular epitheliod cell neoplasms of
soft tissue and gynecologic origin: A clinicopathologic study of 26
cases and review of the literature. Am J Surg Pathol. 29:1558–1575.
2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yamasaki S, Tanaka S, Fujii H, Matsumoto
T, Okuda C, Watanabe G and Suda K: Monotypic epithelioid
angiomyolipoma of the liver. Histopathology. 36:451–456.
2000.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fang SH, Zhou LN, Jin M and Hu JB:
Perivascular epithelioid cell tumor of the liver: A report of two
cases and review of the literature. World J Gastroenterol.
13:5537–5539. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yoo Y, Kim J and Song IH: Risk prediction
criteria for the primary hepatic perivascular epithelioid cell
tumour family, including angiomyolipoma: Analysis of 132 cases with
a literature review. Histopathology. 86:979–992. 2025.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kvietkauskas M, Samuolyte A, Rackauskas R,
Luksaite-Lukste R, Karaliute G, Maskoliunaite V, Valkiuniene RB,
Sokolovas V and Strupas K: Primary liver perivascular epithelioid
cell tumor (PEComa): Case report and literature review. Medicina
(Kaunas). 60(409)2024.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bergamo F, Maruzzo M, Basso U, Montesco
MC, Zagonel V, Gringeri E and Cillo U: Neoadjuvant sirolimus for a
large hepatic perivascular epithelioid cell tumor (PEComa). World J
Surg Oncol. 12(46)2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bennett JA, Braga AC, Pinto A, Van de
Vijver K, Cornejo K, Pesci A, Zhang L, Morales-Oyarvide V, Kiyokawa
T, Zannoni GF, et al: Uterine PEComas: A morphologic,
immunohistochemical, and molecular analysis of 32 tumors. Am J Surg
Pathol. 42:1370–1383. 2018.PubMed/NCBI View Article : Google Scholar
|