1. Introduction
Ferroptosis is a programmed cell death process
dependent on iron ions and reactive oxygen species (ROS), which is
primarily characterized by an imbalance in iron homeostasis,
inactivation of glutathione peroxidase 4 (GPX4) and lipid
peroxidation damage (1). Unlike
apoptosis, necrosis and autophagy, ferroptosis exhibits unique
metabolic and oxidative stress-driven characteristics (2). In previous years, ferroptosis has
been shown to serve a key role in numerous pathological processes,
including neurodegenerative diseases, cancer and
inflammation-related disorders (3). Within the context of spinal
degenerative diseases, increasing evidence suggests that
ferroptosis is a key contributor to intervertebral disc
degeneration (IDD) (4-6).
Mitochondria, the central organelles for cellular
energy metabolism and oxidative stress regulation, serve a key role
in ferroptosis (7). Firstly,
mitochondria serve as notable sites for iron storage and
utilization, and the disruption of iron homeostasis directly leads
to mitochondrial dysfunction (8).
Secondly, mitochondria generate marked amounts of ROS, which
promote ferroptosis through lipid peroxidation pathways (9). Furthermore, mitochondrial membrane
potential (ΔΨm) decline, increased mitochondrial outer membrane
permeability and mitochondrial DNA (mtDNA) damage have been
identified as key regulatory factors in ferroptosis (10,11).
Collectively, these mitochondrial dysfunction-associated events
contribute to ferroptosis-related nucleus pulposus cell injury and
extracellular matrix degradation in IDD, thereby suggesting that
targeting mitochondrial dysfunction may represent a potential
therapeutic strategy for IDD.
IDD is a leading cause of chronic lower back pain
with pathological mechanisms involving nucleus pulposus cell (NPC)
apoptosis, extracellular matrix (ECM) degradation, an imbalanced
inflammatory microenvironment and oxidative stress damage (12,13).
Recent studies have suggested that ferroptosis-related molecules
are abnormally expressed in the intervertebral disc tissues of
patients with IDD, with marked downregulation of GPX4, iron ion
accumulation and increased lipid peroxidation levels (14,15).
Additionally, research has demonstrated that interventions
targeting mitochondrial function, oxidative stress suppression or
iron homeostasis modulation may mitigate IDD progression (16). Consequently, therapeutic strategies
targeting mitochondria-mediated ferroptosis, including
antioxidants, iron chelators and mitochondrial protective agents,
have gained attention.
Ferroptosis appears to be uniquely associated with
age-related spinal degeneration. Unlike other forms of cell death,
such as apoptosis, necroptosis and autophagy-dependent cell death,
ferroptosis integrates imbalances in iron metabolism, oxidative
stress and mitochondrial dysfunction, all of which progressively
accumulate with age. Therefore, ferroptosis is not only a driver of
disc cell loss but also a key process linking aging biology to
spinal degenerative diseases (3,13).
This unique contribution underscores the necessity of developing
ferroptosis-targeted interventions for age-related IDD.
Overall, ferroptosis is a form of cell death that
has garnered attention regarding its potential role in IDD
pathology, with mitochondria being key regulators of this process.
Therefore, the present review explores the functional mechanisms of
mitochondria in ferroptosis and discusses therapeutic strategies
targeting mitochondria-mediated ferroptosis for IDD treatment,
aiming to provide novel insights for clinical intervention.
2. Molecular mechanisms of mitochondria and
ferroptosis
Core mechanisms of ferroptosis
Mitochondria serve a key role in ferroptosis by
acting as central regulators of cellular redox balance, iron
metabolism, lipid peroxidation and ROS production.
Ferroptosis involves three key processes consisting
of iron metabolism dysregulation, lipid peroxidation and an
imbalance in the antioxidant system. Within iron metabolism
regulation, iron is taken up by the transferrin receptor 1 (TfR1)
pathway and stored in ferritin, whereas ferroportin is responsible
for iron export. When ferritinophagy, mediated by nuclear receptor
coactivator 4 (NCOA4), is enhanced, the stored iron is released,
leading to the accumulation of intracellular free iron. This free
iron undergoes the Fenton reaction, generating hydroxyl radicals
(OH·) that induce ferroptosis (17,18).
Lipid peroxidation is a key process in ferroptosis.
Polyunsaturated fatty acids (PUFAs) are oxidized by acyl-CoA
synthetase long-chain family member 4 (ACSL4) and arachidonate
15-lipoxygenase (ALOX15), whereas GPX4 uses glutathione (GSH) to
inhibit this process. When GPX4 is inactivated, the excessive
accumulation of lipid peroxides disrupts membrane integrity,
ultimately resulting in cell death (19).
Furthermore, the antioxidant system serves an
important role in inhibiting ferroptosis. Solute carrier family 7
member 11 (SLC7A11, also known as xCT) maintains cellular
antioxidant capacity by synthesizing GSH, whereas the nuclear
factor erythroid 2-related factor 2 (Nrf2) signaling pathway
activates downstream genes, such as GPX4, SLC7A11 and heme
oxygenase 1 to suppress ferroptosis (20,21).
Additionally, the ferroptosis suppressor protein 1/coenzyme Q10
pathway functions independently of GPX4 to protect cells from
ferroptosis (22).
Role of mitochondria in
ferroptosis
Mitochondria serve as central regulators of cellular
iron metabolism, synthesizing iron-sulfur (Fe-S) clusters and heme
while maintaining iron homeostasis. Mitochondrial iron transport
proteins such as sideroflexin 1 import iron into the mitochondria,
whereas the ATP-binding cassette transporter ABCB7 facilitates iron
export. Excessive iron accumulation generates OH·, resulting in
oxidative damage and exacerbating ferroptosis (23,24).
Mitochondria are the primary sources of cellular
ROS. Superoxide anions (O2·-) are produced at
complexes I and III of the electron transport chain and are
converted to hydrogen peroxide (H2O2) by
superoxide dismutase 2 (SOD2). H2O2 further
reacts with Fe2+ to generate OH·, promoting lipid
peroxidation. In mitochondrial dysfunction, increased ROS levels
further inhibit GPX4 activity, accelerate PUFA oxidation and
ultimately induce ferroptosis (25-27).
The dysregulation of energy metabolism also serves a
notable role in ferroptosis. The tricarboxylic acid cycle in the
mitochondria regulates nicotinamide adenine dinucleotide phosphate
(NADPH) levels, which are key for GSH regeneration. When NADPH is
depleted, GSH levels decrease, leading to GPX4 inactivation and
promoting ferroptosis (26).
Moreover, reduced ATP synthesis impairs the function of SLC7A11,
hindering GSH synthesis and increasing cellular susceptibility to
ferroptosis (28).
Mitochondrial autophagy (mitophagy) is an important
protective mechanism that removes damaged mitochondria, and reduces
oxidative stress and iron accumulation. Moderate mitophagy lowers
the risk of ferroptosis; however, excessive mitophagy exacerbates
the cellular damage and promotes ferroptosis. For example,
PTEN-induced kinase 1 (PINK1)/Parkin-mediated mitophagy, while
removing damaged mitochondria, can lead to excessive mitochondrial
loss, impairing cellular energy metabolism and indirectly enhancing
ferroptotic signaling (29,30).
Additionally, mitophagy mediated by NIX/BCL2-interacting protein 3,
which is activated under hypoxic or metabolic stress, may influence
ferroptosis by regulating iron homeostasis (31).
Stage-specific dynamics of
mitochondrial dysfunction and ferroptosis in IDD
Emerging evidence has suggested that mitochondrial
dysfunction and ferroptosis are not uniformly engaged across the
course of IDD but instead display stage-dependent features
(32,33).
Early IDD (radiographically mild degeneration or
early symptomatic discs) is characterized by subtle bioenergetic
stress with a mild decline in ΔΨm and increased mitochondrial ROS
(MitoROS), preceding overt cell loss and ECM collapse. In NPCs,
SOD2-dependent redox imbalance sensitizes cells to lipid
peroxidation, whereas compensatory mitophagy is detectable but
often insufficient to fully restore mitochondrial quality control
(34). In parallel, iron-handling
disturbances (such as labile iron accumulation and
ferritinophagy-mediated iron release) initiate a pre-ferroptotic
state, lowering the threshold for ferroptotic execution upon
inflammatory or mechanical insults (13,14).
During the intermediate stage, cumulative oxidative
injury amplifies lipid peroxidation through ACSL4/ALOX15-dependent
pathways, whereas GSH depletion limits GPX4 activity.
Concomitantly, impaired PINK1/Parkin-mediated mitophagy allows
damaged mitochondria and mtDNA to persist, reinforcing the
ROS-lipid peroxide feed-forward loops. Iron dysregulation becomes
more evident, with NCOA4-driven ferritinophagy exacerbating
oxidative injury and inflammatory cytokines (such as TNF-α and
IL-1β) suppressing SLC7A11/GPX4 expression, further priming
ferroptosis (14).
In advanced IDD, ferroptosis switches from a
permissive state to a dominant execution program in subsets of disc
cells. Profound ΔΨm collapse, extensive mitochondrial morphological
damage, iron overload and persistent mitophagy insufficiency
co-occur with ECM disintegration and cell depletion. At this stage,
mitochondria-directed interventions [such as mitochondria-targeted
coenzyme Q10 (MitoQ) and SkQ1] can still attenuate oxidative injury
but may require combination strategies (iron chelation, GPX4
preservation and mitophagy rebalancing) to achieve structural
benefits (35).
Together, these data support a temporal model in
which mitochondrial stress and iron abnormalities appear early,
intensify at the mid-stage and culminate in overt ferroptosis with
structural failure in late-stage IDD.
3. Ferroptosis and IDD
Within recent research, the role of ferroptosis in
IDD has gained increasing attention. The core pathological features
of IDD include ECM degradation, NP and annulus fibrosus cell death,
as well as oxidative stress and inflammatory responses.
Mitochondrial dysfunction and ferroptosis have been suggested as
key drivers of IDD progression, with mitochondria mediating this
process of ferroptosis in IDD (Fig.
1).
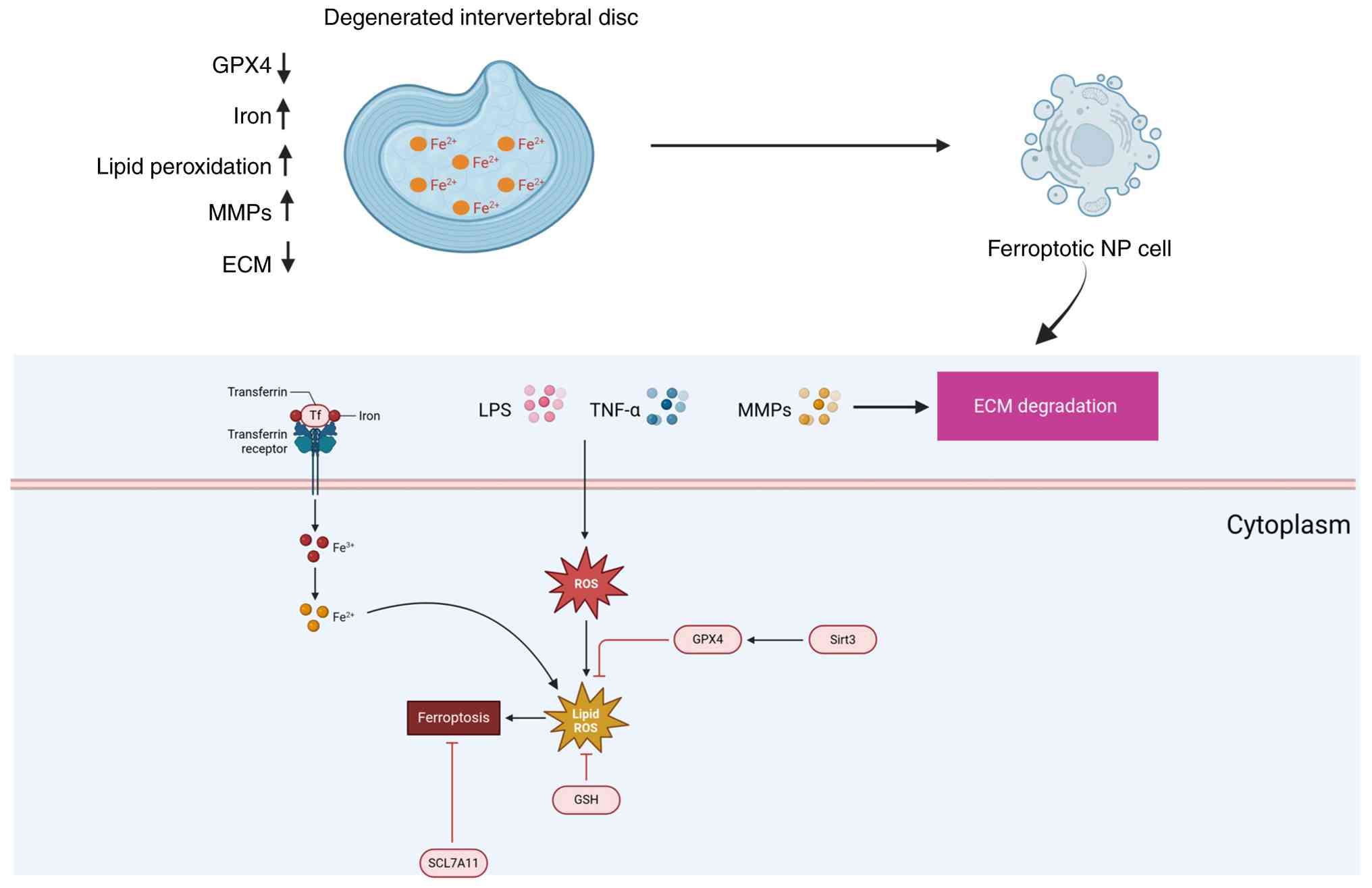 | Figure 1Mitochondria-mediated ferroptosis
promotes extracellular matrix degradation in intervertebral disc
degeneration. Mitochondrial dysfunction, triggered by iron overload
and oxidative stress, leads to the accumulation of MitoROS and the
activation of ferroptosis in nucleus pulposus cells (NPCs). Excess
iron accumulates through mechanisms such as TfR upregulation and
FTH1 downregulation, promoting lipid peroxidation and ROS
generation. These processes result in cell death and the subsequent
release of inflammatory mediators that exacerbate intervertebral
disc degeneration. Furthermore, mitochondrial dysfunction impairs
GPX4 activity, leading to reduced antioxidant capacity and
increased oxidative damage. This cascade of events ultimately
accelerates ECM degradation, contributing to the progression of
IDD. GPX4, glutathione peroxidase 4; ECM, extracellular matrix; NP,
nucleus pulposus; LPS, lipopolysaccharide; Tf, transferrin; Sirt3,
sirtuin 3; ROS, reactive oxygen species; GSH, glutathione; SCL7A11,
solute carrier family 7 member 11. |
Major mechanisms of IDD. The pathogenesis of
IDD is complex, involving a number of biological processes. A key
feature is ECM degradation, which results from the upregulation of
MMPs and ADAMTSs, leading to the breakdown of proteoglycans and
collagen, ultimately causing structural damage to the
intervertebral disc (36,37). Inflammatory activation serves a
notable role in this process, as M1 macrophage infiltration and the
release of pro-inflammatory cytokines, such as TNF-α, IL-1β and
IL-6, accelerate ECM degradation and promote cell death (38).
Oxidative stress is another key factor, whereby the
accumulation of ROS damages cellular components, particularly the
mitochondria. Oxidative damage triggers numerous forms of
programmed cell death, including apoptosis, pyroptosis and
ferroptosis (39). Within the
context of ferroptosis, increasing evidence has suggested that iron
metabolism dysregulation contributes to IDD pathology. Studies have
shown that ferroptosis-related genes, including GPX4, SLC7A11,
ferritin heavy chain 1 (FTH1) and ACSL4, exhibit abnormal
expression patterns in IDD tissues (40,41).
These changes indicate a diminished ability to counteract oxidative
damage and increased susceptibility to lipid peroxidation, further
exacerbating disc degeneration.
The interplay among ECM degradation, inflammatory
responses, oxidative stress and ferroptosis highlights the
multifactorial nature of IDD and underscores the importance of
targeting these mechanisms to develop potential therapeutic
strategies.
Role of ferroptosis in IDD
progression
Recent studies have highlighted ferroptosis as a key
component of IDD progression (32,33).
Ferroptosis-related genes exhibit abnormal expression in IDD
tissues, with the downregulation of GPX4 and SLC7A11 reducing
antioxidant capacity and inducing ferroptosis. By contrast, the
upregulation of ACSL4, ALOX15, and NCOA4 promotes lipid
peroxidation and intracellular iron accumulation, exacerbating cell
death (42-45).
Elevated iron levels in IDD tissues further intensify oxidative
stress through the Fenton reaction, contributing to ECM degradation
and the loss of disc structural integrity. In vitro studies
have demonstrated that the iron overload induced by ferric ammonium
citrate triggers ferroptosis in NPCs and increases MMP expression,
accelerating ECM breakdown (5,33).
Conversely, ferroptosis inhibitors, such as iron chelators,
including deferoxamine (DFO), or GPX4 activators, have been shown
to mitigate IDD progression, suggesting a potential therapeutic
approach (4,46).
Additionally, inflammatory cytokines such as TNF-α
and IL-1β serve an important role in ferroptosis-mediated IDD.
These cytokines suppress the expression of GPX4 and SLC7A11,
leading to enhanced lipid peroxidation and increased sensitivity to
ferroptosis in IDD cells (42).
Lipopolysaccharide stimulation further amplifies this effect by
promoting ROS accumulation, making NPCs more vulnerable to
ferroptosis (47). In addition to
TNF-α and IL-1β, recent studies have suggested that IL-6
contributes to ferroptosis by impairing antioxidant defense and
altering iron metabolism (48,49).
These cytokines upregulate TfR1 and downregulate FTH1, thereby
promoting intracellular iron overload and ROS generation, which
directly sensitize NPCs to ferroptotic death (50). These findings indicate that the
inflammatory microenvironment not only accelerates ECM degradation
but also directly regulates ferroptosis pathways in IDD.
Although ferroptosis is considered an irreversible
form of programmed cell death, accumulating evidence has suggested
that its early stages are reversible or modifiable. Ferroptosis
inhibitors such as ferrostatin-1 and liproxstatin-1, iron chelators
(including DFO) and Nrf2 activators have demonstrated the ability
to restore GPX4 activity, scavenge lipid peroxides and rescue NPCs
from ferroptotic damage (51-55).
This suggests that ferroptosis can be effectively attenuated if the
intervention occurs before extensive mitochondrial collapse and
lipid peroxidation. However, once the oxidative injury surpasses a
specific threshold, ferroptosis becomes irreversible, ultimately
leading to disc cell death. These findings highlight the existence
of a therapeutic window in which ferroptosis-targeting strategies
may halt or even reverse the progression of IDD.
The role of mitochondrial regulation in ferroptosis
has also been demonstrated, particularly through the mitochondrial
deacetylase sirtuin 3 (Sirt3). Sirt3 expression is markedly reduced
in IDD tissues, and is associated with lower GPX4 levels and
increased ferroptosis. Experimental activation of Sirt3 using
nicotinamide riboside has been shown to restore GPX4 expression and
protect IDD cells from ferroptosis, further reinforcing the role of
mitochondria in IDD pathology (56).
Mitochondrial mediation of ferroptosis
in IDD
Mitochondria serve a pivotal role in IDD by
regulating oxidative stress, iron homeostasis and autophagy, all of
which are closely linked to ferroptosis. Mitochondria are a major
source of ROS and excessive MitoROS can damage lipid membranes and
promote lipid peroxidation, a key feature of ferroptosis (1). In IDD cells, ΔΨm decreases while
MitoROS levels increase, indicating mitochondrial dysfunction
(57,58). Antioxidants such as MitoQ and SkQ1
can reduce MitoROS accumulation and inhibit ferroptosis, suggesting
that targeting mitochondrial oxidative stress is a viable
therapeutic approach (59-61).
Mitochondria also serve as a central hub for
cellular iron metabolism and participate in the synthesis of Fe-S
clusters and heme (62). In IDD
tissues, FTH1 expression is reduced, thereby disrupting iron
homeostasis and increasing ferroptosis. Imbalances in mitochondrial
iron metabolism contribute to iron overload, exacerbating oxidative
damage and lipid peroxidation (1,33).
Additionally, mitophagy is important in the removal
of damaged mitochondria and the maintenance of cellular health.
However, IDD cells exhibit impaired PINK1/Parkin-mediated
mitophagy, thereby resulting in the accumulation of dysfunctional
mitochondria and further promotion of ferroptosis (63,64).
The pharmacological activation of mitophagy, particularly through
pathways such as PINK1/Parkin and Nrf2 signaling, mitigates
ferroptosis-induced cellular damage and slows the progression of
IDD by preserving NPC viability and reducing oxidative stress
(56,65).
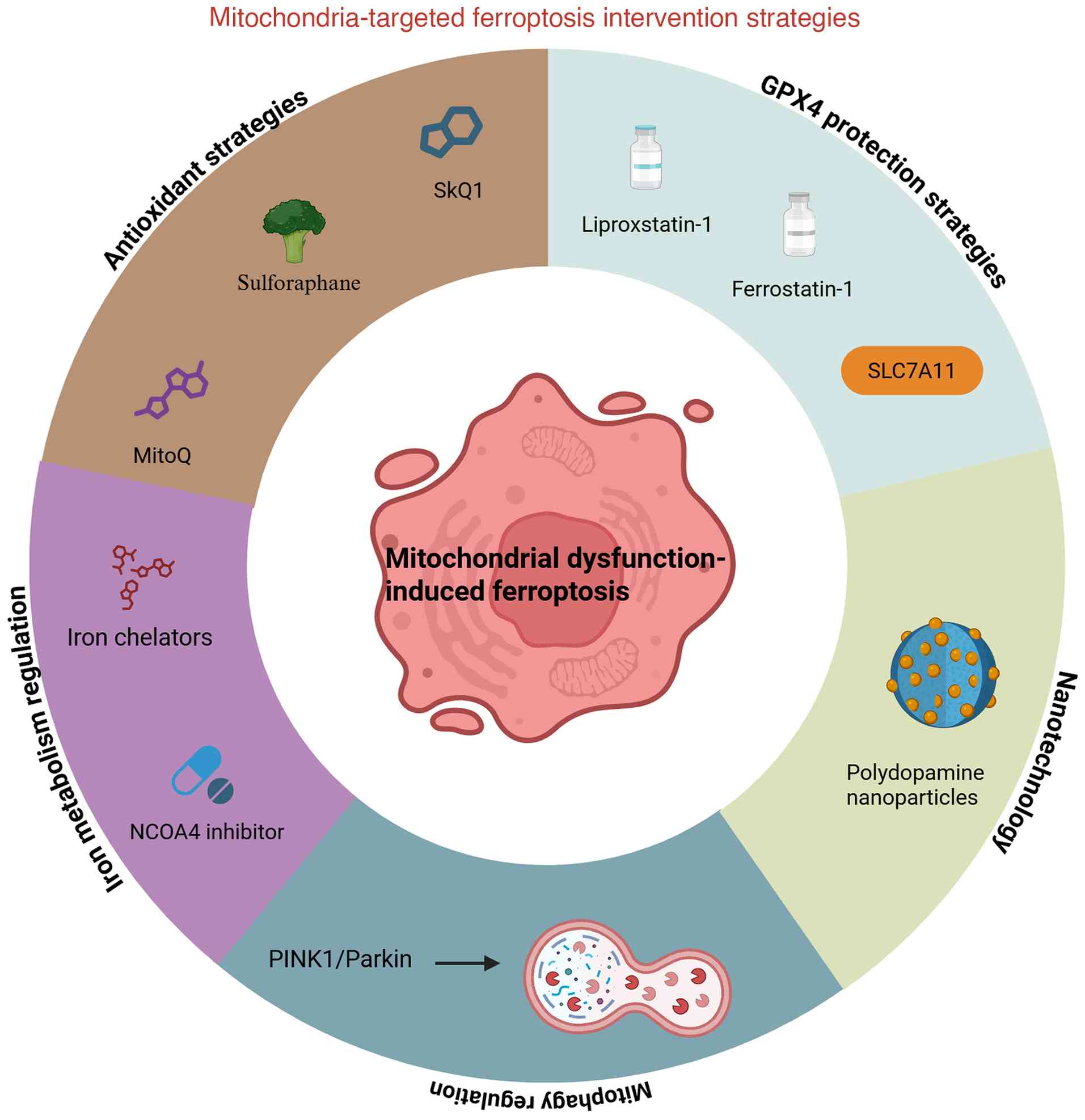
4. Mitochondria-targeted regulation of
ferroptosis as a therapeutic strategy for IDD
Through research into the mechanisms of ferroptosis,
mitochondria have been recognized as key regulatory centers, making
them potential therapeutic targets. Therefore, strategies for
mitochondria-mediated ferroptosis in IDD exhibit notable research
and application potential (Fig.
2).
Mitochondria-targeted antioxidant strategies.
Oxidative stress is a major driver of ferroptosis, with excessive
MitoROS accumulation leading to lipid peroxidation and the
exacerbation of IDD. Targeting mitochondria using antioxidant
strategies has emerged as a promising intervention for mitigating
these effects. One such approach involves the use of MitoQ, which
effectively reduces MitoROS production, inhibits lipid peroxidation
and disrupts ferroptosis signaling pathways (66). MitoQ alleviates oxidative damage in
intervertebral disc cells, enhances ECM synthesis and slows IDD
progression (67,68). Similarly, SkQ1, another
mitochondria-targeted antioxidant, exhibits potent free
radical-scavenging capacity and acts directly within mitochondria
to protect cells from ferroptosis-induced damage (69).
Furthermore, another key strategy involves
activation of the Nrf2 signaling pathway, which serves a notable
role in cellular antioxidant defense mechanisms (70). Compounds such as sulforaphane and
bardoxolone function as Nrf2 activators, enhancing antioxidant
capacity and increasing GPX4 activity, which leads to reduced lipid
peroxidation and prevents ferroptosis (71,72).
By boosting mitochondrial resilience against oxidative stress,
mitochondria-targeted antioxidant strategies offer a promising
option for therapeutic intervention in IDD. Although these
compounds have demonstrated protective effects under other
oxidative stress-related conditions, direct evidence for their
application in IDD remains limited. However, given the established
role of Nrf2 activation in IDD, as supported by existing research,
sulforaphane and bardoxolone may exert similar protective effects
in this context, yet further studies are required to validate the
efficacy of IDD treatment.
Translating mitochondria-targeted antioxidant
therapies into clinical applications faces a number of challenges.
For example, MitoQ and SkQ1 exhibit poor stability, limited
bioavailability and inefficient delivery to avascular disc tissues,
which may reduce their therapeutic efficacy in vivo
(73-75).
Innovative drug delivery systems, such as nanoparticle
encapsulation or hydrogel-based sustained-release platforms, are
currently being explored with the aim of overcoming these
challenges. However, clinical validation still remains lacking.
With regard to Nrf2 activators, such as sulforaphane and
bardoxolone, the majority of evidence is currently derived from
in vitro experiments or animal models of oxidative
stress-related disorders, including osteoarthritis and IDD
(72). Clinical trial data that
directly evaluate their efficacy in IDD are also lacking,
highlighting the gap between experimental findings and
translational applications. Therefore, further preclinical
optimization and early-phase clinical studies are required to
establish their safety, pharmacokinetics and therapeutic potential
in patients with IDD.
Iron metabolism-targeted drugs
Iron overload is an important trigger of ferroptosis
as excessive free iron catalyzes the Fenton reaction, leading to
increased ROS production and lipid peroxidation. Therefore,
regulating iron homeostasis is key in mitigating ferroptosis and
alleviating the progression of IDD. One widely studied approach
involves iron chelators, such as DFO and deferiprone, which can
effectively bind excess free iron, and reduce ROS generation and
lipid peroxidation (33,76,77).
DFO administration in IDD models helps alleviate oxidative stress,
lowering inflammation and protecting NPCs from apoptosis (33).
An additional emerging approach involves targeting
ferritinophagy, which regulates intracellular iron release by
mediating the degradation of ferritin. NCOA4 inhibitors have been
explored as potential therapeutic agents to prevent excessive
ferritin degradation, thereby reducing intracellular free iron
levels and minimizing the risk of ferroptosis (78,79).
These inhibitors may delay IDD progression by stabilizing iron
storage mechanisms and limiting iron availability for ROS
generation.
Iron chelators and ferritinophagy modulators exert
protective effects largely through mitochondrial mechanisms. These
agents suppress the Fenton reaction within the mitochondria by
reducing the mitochondrial labile iron pool, thereby limiting the
formation of OH· and preventing mitochondrial lipid peroxidation.
Moreover, restoration of mitochondrial iron balance preserves
electron transport chain function, stabilizes ΔΨm and decreases
MitoROS accumulation, all of which collectively attenuate
ferroptosis-induced mitochondrial injury in NPCs (80-82).
Therefore, iron metabolism-targeted interventions not only modulate
systemic iron homeostasis but also directly maintain mitochondrial
integrity, providing a mechanistic basis for their therapeutic
potential in IDD.
Regulation of mitophagy
Mitophagy, which is the selective degradation of
damaged mitochondria, serves a notable role in maintaining
mitochondrial homeostasis and regulating ferroptosis. Appropriate
mitophagy clears dysfunctional mitochondria, reduces MitoROS
accumulation and inhibits ferroptosis. However, excessive mitophagy
may lead to mitochondrial depletion and metabolic dysfunction,
further exacerbating IDD (81,83,84).
The PINK1/Parkin pathway is a primary mechanism
regulating mitophagy. Under oxidative stress, PINK1 accumulates on
the outer mitochondrial membrane, recruiting Parkin to promote the
ubiquitination and degradation of damaged mitochondria. This
process eliminates dysfunctional mitochondria, thereby reducing ROS
production and preventing ferroptosis-induced cellular damage
(85). However, maintaining a
balance in mitophagy activation is important as excessive
mitochondrial clearance may lead to energy metabolism disorders,
further aggravating IDD pathology (85).
A recent review has further elucidated the dynamic
interplay between mitophagy and ferroptosis in IDD, primarily
mediated by ROS as a key mediator and involving pathways such as
AMPK/mTOR and Nrf2/Kelch-like ECH-associated protein 1(86). Mechanical stress induces
ferroptosis in NPCs through the Piezo1 channel, while regulation of
mitophagy (through Sirt signaling and the PINK1/Parkin axis) can
alleviate ROS accumulation and mitochondrial damage. This previous
study proposed that interventions targeting these interacting
pathways (such as modulating ROS levels) may represent a new
direction for IDD treatment (86).
This underscores the importance of balancing mitophagy in IDD to
prevent excessive ferroptosis and provides a theoretical foundation
for developing comprehensive therapies.
Urolithin A, a natural compound derived from
polyphenols, is a mitophagy inducer with potential therapeutic
effects on IDD. By promoting mitochondrial quality control,
Urolithin A enhances mitochondrial function, reduces oxidative
stress and mitigates ferroptosis-related cell damage. Furthermore,
it can alleviate mitochondrial dysfunction and delay IDD
progression by maintaining mitochondrial integrity (85,87).
GPX4 protection strategies
GPX4 serves a central role in inhibiting ferroptosis
by reducing lipid peroxidation and preserving cell membrane
integrity. Strategies aimed at enhancing GPX4 activity or
modulating its associated pathways are promising therapeutic
approaches for IDD. For example, one effective method involves the
use of GPX4 activators, such as liproxstatin-1 and ferrostatin-1,
which mitigate ferroptosis-induced damage in NPCs. Preclinical
studies have indicated that these compounds markedly improve
intervertebral disc structure and suppress inflammatory responses
in animal models of IDD, highlighting their potential clinical
applications (47,88).
In addition, one alternative approach focuses on the
regulation of SLC7A11, an important component in GSH synthesis. As
GPX4 activity is heavily dependent on GSH availability,
upregulation of SLC7A11 enhances GPX4 function and prevents
ferroptosis. Inhibitors of erastin, which negatively regulates
SLC7A11, have been explored as potential therapeutic agents to
elevate intracellular GSH levels and protect intervertebral disc
cells from oxidative stress and lipid peroxidation (32,89).
Mechanistically, GPX4 protection directly affects
mitochondrial homeostasis during ferroptosis. GPX4 localizes not
only in the cytosol but also within the mitochondria, where it
detoxifies the phospholipid hydroperoxides generated by MitoROS
(90,91). The upregulation or pharmacological
activation of GPX4 helps maintain mitochondrial membrane integrity,
prevents mtDNA oxidation and sustains ATP production during
oxidative stress (90).
Additionally, enhanced SLC7A11-GSH-GPX4 signaling ensures
sufficient antioxidant capacity to counteract MitoROS accumulation,
thereby interrupting the self-amplifying cycle of mitochondrial
oxidative injury and ferroptosis (91,92).
Thus, GPX4-targeted therapies protect cytoplasmic and mitochondrial
compartments, offering a dual-layer defense against ferroptotic
degeneration in IDD.
Mitochondria-targeted nanotherapy
With the rapid advancement of nanomedicine,
mitochondria-targeted nanotherapy has emerged as a promising
approach for the precise and efficient treatment of IDD by
modulating ferroptosis pathways and oxidative stress at the
mitochondrial level. A particularly notable strategy in this field
involves the use of polydopamine nanoparticles (PDA NPs), which
effectively inhibit oxidative stress-induced ferroptosis in NPCs
(93). PDA NPs exert their
protective effects by scavenging ROS, chelating Fe2+ to
mitigate iron overload and regulating iron storage proteins such as
FTH and TfR (93). In addition,
these nanoparticles colocalize with GPX4 around the mitochondria,
preventing its ubiquitin-mediated degradation, which in turn
enhances the clearance of phospholipid hydroperoxides and reduces
lipid peroxidation (94). In
vivo studies have further demonstrated that PDA NPs can
alleviate puncture-induced disc degeneration by suppressing
ferroptosis and restoring antioxidant defenses, offering a novel
therapeutic strategy for IDD that directly targets
ferroptosis-related damage at the mitochondrial level (94,95).
Furthermore, one recent study developed DFOM@-cerium (IV) oxide (CeO2)
nanoparticles, which reduce iron overload through the iron chelator
DFOM and scavenge ROS through CeO2 while simultaneously
achieving mitochondrial functional reprogramming. These
nanoparticles inhibit tert-butyl hydroperoxide- or erastin-induced
ferroptosis in vitro, restoring the expression of
mitochondrial respiratory chain complexes (such as NDUFB8 and
UQCRC2 subunits) and GPX4. Within an in vivo IDD rat model,
these nanoparticles outperformed single DFOM or CeO2
treatments, preserving intervertebral disc height and reducing ECM
degradation (96). This strategy
highlights the potential of nanotechnology in integrating iron
metabolism regulation and mitochondrial protection, offering a
novel option for the precise treatment of IDD.
Future research directions
Mitochondria-targeted regulation of ferroptosis
represents a promising approach for IDD treatment; however, notable
challenges remain before clinical application can be achieved.
Currently, the majority of research has used cellular and animal
models, with mature clinical trial data lacking. Future studies
should aim to focus on developing more representative animal
models, including larger animal experiments, to further mimic the
pathological characteristics of human IDD. Functional experiments
using gene editing (such as CRISPR/Cas9) and pharmacological
interventions may be key in validating the role of
ferroptosis-related genes, including GPX4, SLC7A1 and NCOA4, as
well as evaluating the effects of targeted therapies.
To optimize mitochondria-targeted therapies,
enhancing drug specificity remains a priority, as existing
antioxidants and iron chelators often exert systemic effects that
may disrupt normal cellular functions. The development of
mitochondria-targeted drug delivery systems should improve
selectivity and therapeutic efficiency while minimizing unintended
side effects. Additionally, long-term safety assessment of iron
chelators and GPX4 activators is important, as they may disrupt
normal iron metabolism and antioxidant balance. Furthermore, a
combination therapy strategy, integrating antioxidants with iron
chelators or leveraging mitochondria-targeted nanocarriers with
gene-editing technologies, could further enhance treatment
outcomes.
Despite this, a number of key knowledge gaps remain
unaddressed. Firstly, it remains unclear whether ferroptosis is an
initiating event or a secondary consequence of IDD progression.
Moreover, identifying the precise temporal dynamics of ferroptosis
during the early vs. late stages of disease is key. Secondly, the
threshold at which ferroptosis becomes irreversible remains
unclear, making it difficult to determine the optimal therapeutic
window for intervention. Thirdly, the current preclinical studies
lack standardized outcome measures, meaning more systematic in
vivo studies are required to validate the efficacy and safety
of mitochondria-targeted ferroptosis modulators. Addressing these
knowledge gaps remains important for accelerating clinical
translation.
Given the heterogeneity of patients with IDD, a
personalized treatment approach appears necessary. The expression
levels of ferroptosis-related molecules such as GPX4, ACSL4 and
SLC7A11 in intervertebral disc tissues or peripheral blood can
serve as biomarkers to guide therapeutic decisions. Advances in
artificial intelligence and big data analytics, particularly
single-cell sequencing and machine learning, may enable the
development of molecular classification systems for patients with
IDD, identifying those who would benefit the most from
mitochondrial-targeted interventions. Within this context,
precision medicine strategies should be emphasized to tailor
therapeutic regimens based on patient-specific molecular profiles,
genetic backgrounds and disease stages. Such individualized
approaches will enhance therapeutic efficacy and reduce the risk of
adverse effects from systemic treatments.
Nanomedicine and gene therapy represent another
frontier of IDD treatment, with potential advancements in
mitochondria-targeted nanoparticle design, such as TAT-PEG-MitoQ,
to enhance drug delivery efficiency while reducing systemic
toxicity. Gene editing tools, such as CRISPR/Cas9 and RNA
interference, enable the precise regulation of ferroptosis-related
genes in intervertebral disc cells, achieving long-term suppression
of ferroptosis. Moreover, bioengineered intervertebral disc
scaffolds loaded with mitochondria-targeted drugs enable controlled
local drug release during surgical implantation, thereby improving
treatment precision.
Finally, although the majority of the current
research on ferroptosis in IDD has focused on disease progression,
early intervention may exhibit a greater impact. Investigating the
role of ferroptosis-related molecules in the early stages of disc
degeneration and developing non-invasive diagnostic tools, such as
MRI combined with molecular imaging, could facilitate early
detection. Preventive strategies, including lifestyle modifications
such as exercise and antioxidant supplementation, may also help
mitigate the risk of ferroptosis in intervertebral disc cells,
reducing the likelihood of early IDD onset.
5. Conclusions
In summary, mitochondria-driven ferroptosis serves a
pivotal role in the pathogenesis of IDD by disrupting the redox
balance, promoting lipid peroxidation and impairing cellular
antioxidant defense. As the central hub for iron metabolism and ROS
production, mitochondria not only initiate ferroptotic signaling
but also serve as promising therapeutic targets. Increasing
evidence indicates that targeting mitochondrial dysfunction using
antioxidants, iron chelators, GPX4 activators and mitophagy
modulators effectively attenuates ferroptosis and preserves disc
cell viability. These findings underscore the importance of
mitochondrial health in disc homeostasis and provide novel insights
into precise strategies for treating age-related degenerative
spinal diseases. However, future studies focusing on
mitochondria-based interventions may pave the way for more
effective and targeted therapies for IDD.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the Naval Medical
University, fundamental research program of basic medicine, China
(grant no. 2023MS032).
Availability of data and materials
Not applicable.
Authors' contributions
YH and LL drafted the initial manuscript. XY, DQ and
JS performed the literature review, collected relevant references,
and assisted in figure conceptualization and design. YG conceived
the present study, provided critical intellectual input,
extensively revised the manuscript and supervised the entire
project. All authors critically reviewed the manuscript, and all
authors read and approved the final manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jing X, Wang W, He X, Liu X, Yang X, Su C,
Shao Y, Ge Z, Wang H and Cui X: HIF-2α/TFR1 mediated iron
homeostasis disruption aggravates cartilage endplate degeneration
through ferroptotic damage and mtDNA release: A new mechanism of
intervertebral disc degeneration. J Orthop Translat. 46:65–78.
2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wang S, Zhang S, Li X, Leng C, Li X, Lv J,
Zhao S, Qiu W and Guo J: Development of oxidative stress- and
ferroptosis-related prognostic signature in gastric cancer and
identification of CDH19 as a novel biomarker. Hum Genomics.
18(121)2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chen J, Yang X, Feng Y, Li Q, Ma J, Wang L
and Quan Z: Targeting ferroptosis holds potential for
intervertebral disc degeneration therapy. Cells.
11(3508)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ma T, Du J, Zhang Y, Wang Y, Wang B and
Zhang T: GPX4-independent ferroptosis-a new strategy in disease's
therapy. Cell Death Discov. 8(434)2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chen S, Xiao J, Zhou S, Wumiti T, Zhao Z,
Zhao R, Pan Y, Wang Q, Ma Y, Wu L and Guo Y: The GPR30-Mediated
BMP-6/HEP/FPN signaling pathway inhibits ferroptosis in bone marrow
mesenchymal stem cells to alleviate osteoporosis. Int J Mol Sci.
26(2027)2025.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Richardson DR, Lane DJR, Becker EM, Huang
ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD and Ponka P:
Mitochondrial iron trafficking and the integration of iron
metabolism between the mitochondrion and cytosol. Proc Natl Acad
Sci USA. 107:10775–10782. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Berlingerio SP, Bondue T, Tassinari S,
Siegerist F, Ferrulli A, Lismont C, Cairoli S, Goffredo BM,
Ghesquière B, Fransen M, et al: Targeting oxidative stress-induced
lipid peroxidation enhances podocyte function in cystinosis. J
Transl Med. 23(206)2025.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chen Y, Guo X, Zeng Y, Mo X, Hong S, He H,
Li J, Fatima S and Liu Q: Oxidative stress induces mitochondrial
iron overload and ferroptotic cell death. Sci Rep.
13(15515)2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sumneang N, Siri-Angkul N, Kumfu S,
Chattipakorn SC and Chattipakorn N: The effects of iron overload on
mitochondrial function, mitochondrial dynamics, and ferroptosis in
cardiomyocytes. Arch Biochem Biophys. 680(108241)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wu J, Chen Z, Huang H and Wang H, Wang X,
Lu Z, Xu H, Ma X, Zeng F and Wang H: Custom-Made Ce-Mn bimetallic
nanozyme for the treatment of intervertebral disc degeneration by
inhibiting oxidative stress and modulating macrophage M1/M2
polarization. Biomater Res. 28(0118)2024.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shen J, Lan Y, Ji Z and Liu H: Sirtuins in
intervertebral disc degeneration: Current understanding. Mol Med.
30(44)2024.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li C, Fei C, Le S, Lai Z, Yan B, Wang L
and Zhang Z: Identification and validation of ferroptosis-related
biomarkers in intervertebral disc degeneration. Front Cell Dev
Biol. 12(1416345)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cui P, Sheng Y, Wu C and He D: Puerarin
modulates proliferation, inflammation and ECM metabolism in human
nucleus pulposus mesenchymal stem cells via the lncRNA LINC01535.
Heliyon. 10(e33083)2024.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tamagawa S, Sakai D, Nojiri H, Nakamura Y,
Warita T, Matsushita E, Schol J, Soma H, Ogasawara S, Munesada D,
et al: SOD2 orchestrates redox homeostasis in intervertebral discs:
A novel insight into oxidative stress-mediated degeneration and
therapeutic potential. Redox Biol. 71(103091)2024.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhu Q, Zhai J, Chen Z, Guo Z, Wang N,
Zhang C, Deng H, Wang S and Yang G: Ferritinophagy: Molecular
mechanisms and role in disease. Pathol Res Pract.
262(155553)2024.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen Z, Zheng N, Wang F, Zhou Q, Chen Z,
Xie L, Sun Q, Li L and Li B: The role of ferritinophagy and
ferroptosis in Alzheimer's disease. Brain Res.
1850(149340)2025.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhou Q, Zhang Y, Shi W, Lu L, Wei J, Wang
J, Zhang H, Pu Y and Yin L: Angiotensin II induces vascular
endothelial dysfunction by promoting lipid peroxidation-mediated
ferroptosis via CD36. Biomolecules. 14(1456)2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tan Q, Yang H, He Y, Shen X, Sun L, Du X,
Lin G, Zhou N, Wang N, Zhou Q, et al: Borna disease virus 1 induces
ferroptosis, contributing to lethal encephalitis. J Med Virol.
96(e29945)2024.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ding Y, Ye J, Liu Y, Zhang S, Xu Y, Yang Z
and Liu Z: Fucoxanthin ameliorates kidney injury by CCl4-Induced
via inhibiting oxidative stress, suppressing ferroptosis, and
modulating gut microbiota. ACS Omega. 10:7407–7421. 2025.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hadian K: Ferroptosis suppressor protein 1
(FSP1) and coenzyme Q10 cooperatively suppress ferroptosis.
Biochemistry. 59:637–638. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Munshi C, Paul T, Jas K, Bose M and
Bhattacharya S: Mitochondrial involvement in ferroptotic cell
death. Global Transl Med. 3(2208)2024.
|
|
24
|
Dietz JV, Fox JL and Khalimonchuk O: Down
the iron path: Mitochondrial iron homeostasis and beyond. Cells.
10(2198)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Fratta Pasini AM, Stranieri C, Girelli D,
Busti F and Cominacini L: Is ferroptosis a key component of the
process leading to multiorgan damage in COVID-19? Antioxidants
(Basel). 10(1677)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dong X, Li Y, Sheng X, Zhou W, Sun A and
Dai H: Mitochondria-related signaling pathways involved in breast
cancer regulate ferroptosis. Genes Dis. 11:358–366. 2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang X, Lu Y, Cheng X, Zhu X, Li D, Duan
H, Hu S, Xiao F, Du L and Zhang Q: Local multiple-site injections
of a plasmid encoding human MnSOD mitigate radiation-induced skin
injury by inhibiting ferroptosis. Curr Drug Deliv. 21:763–774.
2024.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li S, Lu Z, Sun R, Guo S, Gao F, Cao B and
Aa J: The role of SLC7A11 in cancer: Friend or foe? Cancers
(Basel). 14(3059)2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yang T, Yang Q, Lai Q, Zhao J, Nie L, Liu
S, Yang J and Chu C: AP39 inhibits ferroptosis by inhibiting
mitochondrial autophagy through the PINK1/parkin pathway to improve
myocardial fibrosis with myocardial infarction. Biomed
Pharmacother. 165(115195)2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu M, Liu S, Lin Z, Chen X, Jiao Q, Du X
and Jiang H: Targeting the interplay between autophagy and the Nrf2
pathway in Parkinson's disease with potential therapeutic
implications. Biomolecules. 15(149)2025.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Field JT and Gordon JW: BNIP3 and Nix:
Atypical regulators of cell fate. Biochim Biophys Acta Mol Cell
Res. 1869(119325)2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Fan C, Chu G, Yu Z, Ji Z, Kong F, Yao L,
Wang J, Geng D, Wu X and Mao H: The role of ferroptosis in
intervertebral disc degeneration. Front Cell Dev Biol.
11(1219840)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wang W, Jing X, Du T, Ren J, Liu X, Chen
F, Shao Y, Sun S, Yang G and Cui X: Iron overload promotes
intervertebral disc degeneration via inducing oxidative stress and
ferroptosis in endplate chondrocytes. Free Radic Biol Med.
190:234–246. 2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Song Y, Lu S, Geng W, Feng X, Luo R, Li G
and Yang C: Mitochondrial quality control in intervertebral disc
degeneration. Exp Mol Med. 53:1124–1133. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lyamzaev KG, Panteleeva AA, Simonyan RA,
Avetisyan AV and Chernyak BV: The critical role of mitochondrial
lipid peroxidation in ferroptosis: Insights from recent studies.
Biophys Rev. 15:875–885. 2023.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Mern DS and Thomé C: Collagen II
enrichment through scAAV6-RNAi-mediated inhibition of
matrix-metalloproteinases 3 and 13 in degenerative nucleus-pulposus
cells degenerative disc disease and biological treatment
strategies. Exp Biol Med (Maywood). 249(10048)2024.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lei Y, Zhan E, Chen C, Hu Y, Lv Z, He Q,
Wang X, Li X and Zhang F: ALKBH5-mediated m(6)A demethylation of
Runx2 mRNA promotes extracellular matrix degradation and
intervertebral disc degeneration. Cell Biosci.
14(79)2024.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Koroth J, Buko EO, Abbott R, Johnson CP,
Ogle BM, Stone LS, Ellingson AM and Bradley EW: Macrophages and
intervertebral disc degeneration. Int J Mol Sci.
24(1367)2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Chen X, Kang R, Kroemer G and Tang D:
Ferroptosis in infection, inflammation, and immunity. J Exp Med.
218(e20210518)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhu J, Sun R, Sun K, Yan C, Jiang J, Kong
F and Shi J: The deubiquitinase USP11 ameliorates intervertebral
disc degeneration by regulating oxidative stress-induced
ferroptosis via deubiquitinating and stabilizing Sirt3. Redox Biol.
62(102707)2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhou LP, Zhang RJ, Jia CY, Kang L, Zhang
ZG, Zhang HQ, Wang JQ, Zhang B and Shen CL: Ferroptosis: A
potential target for the intervention of intervertebral disc
degeneration. Front Endocrinol (Lausanne).
13(1042060)2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Liu XW, Xu HW, Yi YY, Zhang SB and Wang
SJ: Role of ferroptosis and immune infiltration in intervertebral
disc degeneration: novel insights from bioinformatics analyses.
Front Cell Dev Biol. 11(1170758)2023.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B,
Jiang LS and Jiang SD: Involvement of oxidative stress-induced
annulus fibrosus cell and nucleus pulposus cell ferroptosis in
intervertebral disc degeneration pathogenesis. J Cell Physiol.
236:2725–2739. 2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Sun K, Shi Y, Yan C, Wang S, Han L, Li F,
Xu X, Wang Y, Sun J, Kang Z and Shi J: Glycolysis-derived lactate
induces ACSL4 expression and lactylation to activate ferroptosis
during intervertebral disc degeneration. Adv Sci (Weinh).
12(e2416149)2025.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Xiang Q, Zhao Y and Li W: Identification
and validation of ferroptosis-related gene signature in
intervertebral disc degeneration. Front Endocrinol (Lausanne).
14(1089796)2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hu Y, Wang Y, Liu S and Wang H: The
potential roles of ferroptosis in pathophysiology and treatment of
musculoskeletal diseases-opportunities, challenges, and
perspectives. J Clin Med. 12(2125)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sun H, Guo J, Xiong Z, Zhuang Y, Ning X
and Liu M: Targeting nucleus pulposus cell death in the treatment
of intervertebral disc degeneration. JOR Spine.
7(e70011)2024.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Bin S, Xin L, Lin Z, Jinhua Z, Rui G and
Xiang Z: Targeting miR-10a-5p/IL-6R axis for reducing IL-6-induced
cartilage cell ferroptosis. Exp Mol Pathol.
118(104570)2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yang F, Liu X, Zhang Y, Qiu X, Lei S,
Zhang C, Zhang H, Duan Y, Hu X and Kang X: The
STAT3/S100A6/P53/SLC7A11 axis mediates intervertebral disc
degeneration by regulating ferroptosis in nucleus pulposus cells
and the metabolism of the extracellular matrix. Spine J: Oct 10,
2025 (Epub ahead of print).
|
|
50
|
Zhao Y, Ren P, Luo Q, Li X, Cheng X, Wen
Y, Wu X and Zhou J: Ferroptosis, pathogenesis and therapy in AS
co-depression disease. Front Pharmacol. 16(1516601)2025.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi
AA and Lei P: Ferroptosis: Mechanisms and links with diseases.
Signal Transduct Target Ther. 6(49)2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Lillo-Moya J, Rojas-Solé C,
Muñoz-Salamanca D, Panieri E, Saso L and Rodrigo R: Targeting
ferroptosis against ischemia/reperfusion cardiac injury.
Antioxidants (Basel). 10(667)2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Fan BY, Pang YL, Li WX, Zhao CX, Zhang Y,
Wang X, Ning GZ, Kong XH, Liu C, Yao X and Feng SQ: Liproxstatin-1
is an effective inhibitor of oligodendrocyte ferroptosis induced by
inhibition of glutathione peroxidase 4. Neural Regen Res.
16:561–566. 2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Zhang Q, Qu H, Chen Y, Luo X, Chen C, Xiao
B, Ding X, Zhao P, Lu Y, Chen AF and Yu Y: Atorvastatin induces
mitochondria-dependent ferroptosis via the modulation of
Nrf2-xCT/GPx4 axis. Front Cell Dev Biol. 10(806081)2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yan R, Lin B, Jin W, Tang L, Hu S and Cai
R: NRF2, a superstar of ferroptosis. Antioxidants (Basel).
12(1739)2023.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Wang Y, Cheng H, Wang T, Zhang K, Zhang Y
and Kang X: Oxidative stress in intervertebral disc degeneration:
Molecular mechanisms, pathogenesis and treatment. Cell Prolif.
56(e13448)2023.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ding F, Shao ZW, Yang SH, Wu Q, Gao F and
Xiong LM: Role of mitochondrial pathway in compression-induced
apoptosis of nucleus pulposus cells. Apoptosis. 17:579–590.
2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Lan T, Yang W, Yan B, Guo W and Zhang Y:
Melatonin attenuates intervertebral disc degeneration by restoring
mitochondrial homeostasis through PGC-1α signaling pathway. Cell
Mol Life Sci. 82(330)2025.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Lyamzaev KG, Panteleeva AA, Simonyan RA,
Avetisyan AV and Chernyak BV: Mitochondrial lipid peroxidation is
responsible for ferroptosis. Cells. 12(611)2023.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Sacks B, Onal H, Martorana R, Sehgal A,
Harvey A, Wastella C, Ahmad H, Ross E, Pjetergjoka A, Prasad S, et
al: Mitochondrial targeted antioxidants, mitoquinone and SKQ1, not
vitamin C, mitigate doxorubicin-induced damage in H9c2 myoblast:
Pretreatment vs. co-treatment. BMC Pharmacol Toxicol.
22(49)2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Song J, Sheng J, Lei J, Gan W and Yang Y:
Mitochondrial targeted antioxidant SKQ1 ameliorates acute kidney
injury by inhibiting ferroptosis. Oxid Med Cell Longev.
2022(2223957)2022.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Levi S and Rovida E: The role of iron in
mitochondrial function. Biochim Biophys Acta. 1790:629–636.
2009.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Wu Zl, Wang KP, Chen YJ, Song W, Liu Y,
Zhou KS, Mao P, Ma Z and Zhang HH: Knocking down EGR1 inhibits
nucleus pulposus cell senescence and mitochondrial damage through
activation of PINK1-Parkin dependent mitophagy, thereby delaying
intervertebral disc degeneration. Free Radic Biol Med. 224:9–22.
2024.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Key J, Sen NE, Arsović A, Krämer S, Hülse
R, Khan NN, Meierhofer D, Gispert S, Koepf G and Auburger G:
Systematic surveys of iron homeostasis mechanisms reveal ferritin
superfamily and nucleotide surveillance regulation to be modified
by PINK1 Absence. Cells. 9(2229)2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Pan C, Hou W, Deng X, Liu J, Chi R, Shang
X, Xu T and Hao X: The pivotal role of Nrf2 signal axis in
intervertebral disc degeneration. J Inflamm Res. 16:5819–5833.
2023.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Huang C, Santofimia-Castaño P, Liu X, Xia
Y, Peng L, Gotorbe C, Neira JL, Tang D, Pouyssegur J and Iovanna J:
NUPR1 inhibitor ZZW-115 induces ferroptosis in a
mitochondria-dependent manner. Cell Death Discov.
7(269)2021.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Kang L, Liu S, Li J, Tian Y, Xue Y and Liu
X: The mitochondria-targeted anti-oxidant MitoQ protects against
intervertebral disc degeneration by ameliorating mitochondrial
dysfunction and redox imbalance. Cell Prolif.
53(e12779)2020.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Li D, Tao F and Jin L: Mitochondrial
dysfunction in intervertebral disc degeneration: From pathogenesis
to therapeutic target. Oxid Med Cell Longev: Nov 27, 2020 (Epub
ahead of print).
|
|
69
|
Song Y, Li S, Geng W, Luo R, Liu W, Tu J,
Wang K, Kang L, Yin H, Wu X, et al: Sirtuin 3-dependent
mitochondrial redox homeostasis protects against AGEs-induced
intervertebral disc degeneration. Redox Biol. 19:339–353.
2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Xiang Q, Zhao Y, Lin J, Jiang S and Li W:
The Nrf2 antioxidant defense system in intervertebral disc
degeneration: Molecular insights. Exp Mol Med. 54:1067–1075.
2022.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Jin X, Chen L, Yang Y, Tan R and Jiang C:
Adverse effects of Nrf2 in different organs and the related
diseases. Antioxid Redox Signal. 42:973–985. 2025.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Petrikonis K, Bernatoniene J,
Kopustinskiene DM, Casale R, Davinelli S and Saso L: The
antinociceptive role of Nrf2 in neuropathic pain: from mechanisms
to clinical perspectives. Pharmaceutics. 16(1068)2024.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Xiong Z, Liao Y, Zhang Z, Wan Z, Liang S
and Guo J: Molecular insights into oxidative-stress-mediated
cardiomyopathy and potential therapeutic strategies. Biomolecules.
15(670)2025.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Kong CG and Park JB: Apoptotic pathway in
intervertebral disc degeneration: From molecular pathways to
clinical interventions. Diagnostics (Basel).
15(1510)2025.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Shinn LJ and Lagalwar S: Treating
neurodegenerative disease with antioxidants: Efficacy of the
bioactive phenol resveratrol and mitochondrial-targeted MitoQ and
SkQ. Antioxidants (Basel). 10(573)2021.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Entezari S, Haghi SM, Norouzkhani N,
Sahebnazar B, Vosoughian F, Akbarzadeh D, Islampanah M, Naghsh N,
Abbasalizadeh M and Deravi N: Iron chelators in treatment of iron
overload. J Toxicol. 2022(4911205)2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Wang H, Zhou Z, Wu T, Fan Z, Jin Z, Cao Y,
Huangfu C, Wang Y, Liu X and Liu D: Deferoxamine improves
intervertebral disc degeneration by activating
HIF-1α/BNIP3-mediated mitophagy and inhibiting ferroptosis. Int
Immunopharmacol. 166(115583)2025.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Ma Z, Lu H, Feng X, Du T, Li J, Zhang Q,
Gu X, Shao Y, Jing X and Su C: Nrf2 protects against cartilage
endplate degeneration through inhibiting NCOA4-mediated
ferritinophagy. Int J Mol Med. 53(15)2024.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Wu J, Liu Q, Zhang X, Tan M, Li X, Liu P,
Wu L, Jiao F, Lin Z, Wu X, et al: The interaction between STING and
NCOA4 exacerbates lethal sepsis by orchestrating ferroptosis and
inflammatory responses in macrophages. Cell Death Dis.
13(653)2022.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Su C, Jing X, Liu X, Shao Y, Zheng Y, Liu
X and Cui X: Ferristatin II protects nucleus pulposus against
degeneration through inhibiting ferroptosis and activating HIF-1α
pathway mediated mitophagy. Int Immunopharmacol.
147(113895)2025.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Lu X, Lin Z, Li D, Gong Z, Ma T, Wu J,
Xiao W, Xu C, Guan Y, Yang S, et al: A novel mechanism of FBXW7 in
combating intervertebral disc degeneration: Mitigating ferroptosis
in nucleus pulposus cells through the regulation of mitophagy. Int
Immunopharmacol. 155(114668)2025.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Zhou Q and Ruan D: SIRT1-autophagy axis
may inhibit oxidative stress-induced ferroptosis in human nucleus
pulposus cells. Med Hypotheses. 159(110757)2022.
|
|
83
|
Wu T, Wang Y, Shen B, Guo K, Zhu Z, Liang
Y, Zeng J and Wu D: FBXO2 alleviates intervertebral disc
degeneration via dual mechanisms: Activating PINK1-Parkin mitophagy
and ubiquitinating LCN2 to suppress ferroptosis. Adv Sci (Weinh).
12(e06150)2025.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhang Y, Gao Y, Liu S, Liu S, Yang G, Rong
Y, Wu D and Gao Z: VMP1 attenuates ferroptosis and mitochondrial
dysfunction in nucleus pulposus cells through the
PINK1/Parkin-mediated mitophagy pathway. J Orthop Surg Res.
20(630)2025.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Chang B, Su Y, Li T, Zheng Y, Yang R, Lu
H, Wang H and Ding Y: Mito-TEMPO ameliorates sodium palmitate
induced ferroptosis in MIN6 Cells through PINK1/parkin-mediated
mitophagy. Biomed Environ Sci. 37:1128–1141. 2024.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Zhou Y, Mei Y, Wang H, Hao P, Song C, Liu
Z and Chen J: Targeting mitophagy and ferroptosis: A new direction
for the treatment of intervertebral disc degeneration. Tissue Cell.
98(103227)2026.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Lin J, Zhuge J, Zheng X, Wu Y, Zhang Z, Xu
T, Meftah Z, Xu H, Wu Y, Tian N, et al: Urolithin A-induced
mitophagy suppresses apoptosis and attenuates intervertebral disc
degeneration via the AMPK signaling pathway. Free Radic Biol Med.
150:109–119. 2020.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Li Z, Cheng P, Xi H, Jiang T, Zheng X, Qiu
J, Gong Y, Wu X, Mi S, Hong Y, et al: Tomatidine alleviates
intervertebral disc degeneration by activating the Nrf2/HO-1/GPX4
signaling pathway. Drug Des Devel Ther. 18:6313–6329.
2024.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Jin J, Chen Y, Chen X, Zhang Z, Wu Y, Tian
N, Wu A, Wang X, Shao Z, Zhou Y, et al: Beyond a ferroptosis
inducer: Erastin can suppress nutrient deprivation induced cell
death in the intervertebral disc. Spine J. 25:597–608.
2025.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Deng R, Fu L, Liang H, Ai X, Liu F, Li N,
Wu L, Li S, Yang X, Lin Y, et al: Inhibition of mitochondrial
complex I induces mitochondrial ferroptosis by regulating CoQH2
levels in cancer. Cell Death Dis. 16(254)2025.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Li J, Jia YC, Ding YX, Bai J, Cao F and Li
F: The crosstalk between ferroptosis and mitochondrial dynamic
regulatory networks. Int J Biol Sci. 19:2756–2771. 2023.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Liu X, Chen C, Han D, Zhou W, Cui Y, Tang
X, Xiao C, Wang Y and Gao Y: SLC7A11/GPX4 inactivation-mediated
ferroptosis contributes to the pathogenesis of triptolide-induced
cardiotoxicity. Oxid Med Cell Longev. 2022(3192607)2022.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Lei L, Yuan J, Yang Q, Tu Q, Yu H, Chu L,
Tang L and Zhang C: Curcumin-polydopamine nanoparticles alleviate
ferroptosis by iron chelation and inhibition of oxidative stress
damage. RSC Adv. 14:14934–14941. 2024.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Yang X, Chen Y, Guo J, Li J, Zhang P, Yang
H, Rong K, Zhou T, Fu J and Zhao J: Polydopamine nanoparticles
targeting ferroptosis mitigate intervertebral disc degeneration via
reactive oxygen species depletion, iron ions chelation, and GPX4
ubiquitination suppression. Adv Sci (Weinh).
10(e2207216)2023.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Zhang X, Xiang Y, Wang Q, Bai X, Meng D,
Wu J, Sun K, Zhang L, Qiang R, Liu W, et al: Regulation of iron
metabolism in ferroptosis: From mechanism research to clinical
translation. J Pharm Anal. 15(101304)2025.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Wu T, Teng Y, Song D, Yang Y, Shen H, Sun
X, Chen R, Zhao L, Zhong X, Yan Q, et al: A strategy targeting
ferroptosis for mitochondrial reprogramming and intervertebral disc
degeneration therapy. Theranostics. 15:9159–9178. 2025.PubMed/NCBI View Article : Google Scholar
|