Introduction
Familial adenomatous polyposis (FAP) is an autosomal
dominant inherited syndrome that predisposes individuals to
colorectal cancer (CRC), primarily due to germline mutations in the
adenomatous polyposis coli (APC) gene located on chromosome
5q21-22. Affected individuals typically develop numerous colorectal
adenomatous polyps during adolescence or early adulthood, which
inevitably progress to CRC if left untreated (1-3).
APC-associated polyposis conditions include classic FAP,
attenuated FAP, and gastric adenocarcinoma and proximal polyposis
of the stomach (GAPPS), all of which are linked to germline
APC mutations but exhibit distinct clinical presentations
(4).
In FAP, APC mutations are most commonly
located within the gene's coding regions; mutations in the promoter
region are relatively rare. Germline point mutations in the
promoter 1B region of the APC gene have been identified as a
major cause of GAPPS, which is an autosomal dominant syndrome.
GAPPS is primarily characterized by extensive fundic gland
polyposis and a markedly increased risk of gastric cancer; however,
it does not involve colorectal polyposis, which is typically
observed in FAP. The present study reported a rare case involving a
pathogenic variant located in a noncoding upstream regulatory
region of APC, beyond the typical promoter 1B region, in an
individual who presented with clinical features characteristic of
classical FAP (5).
Case report
A 63-year-old man with no previous personal or
family history of CRC presented to Kaohsiung Medical University
Chung-Ho Memorial Hospital (Kaohsiung, Taiwan) in March 2023 with a
1-week history of abdominal pain and hematochezia. A colonoscopy
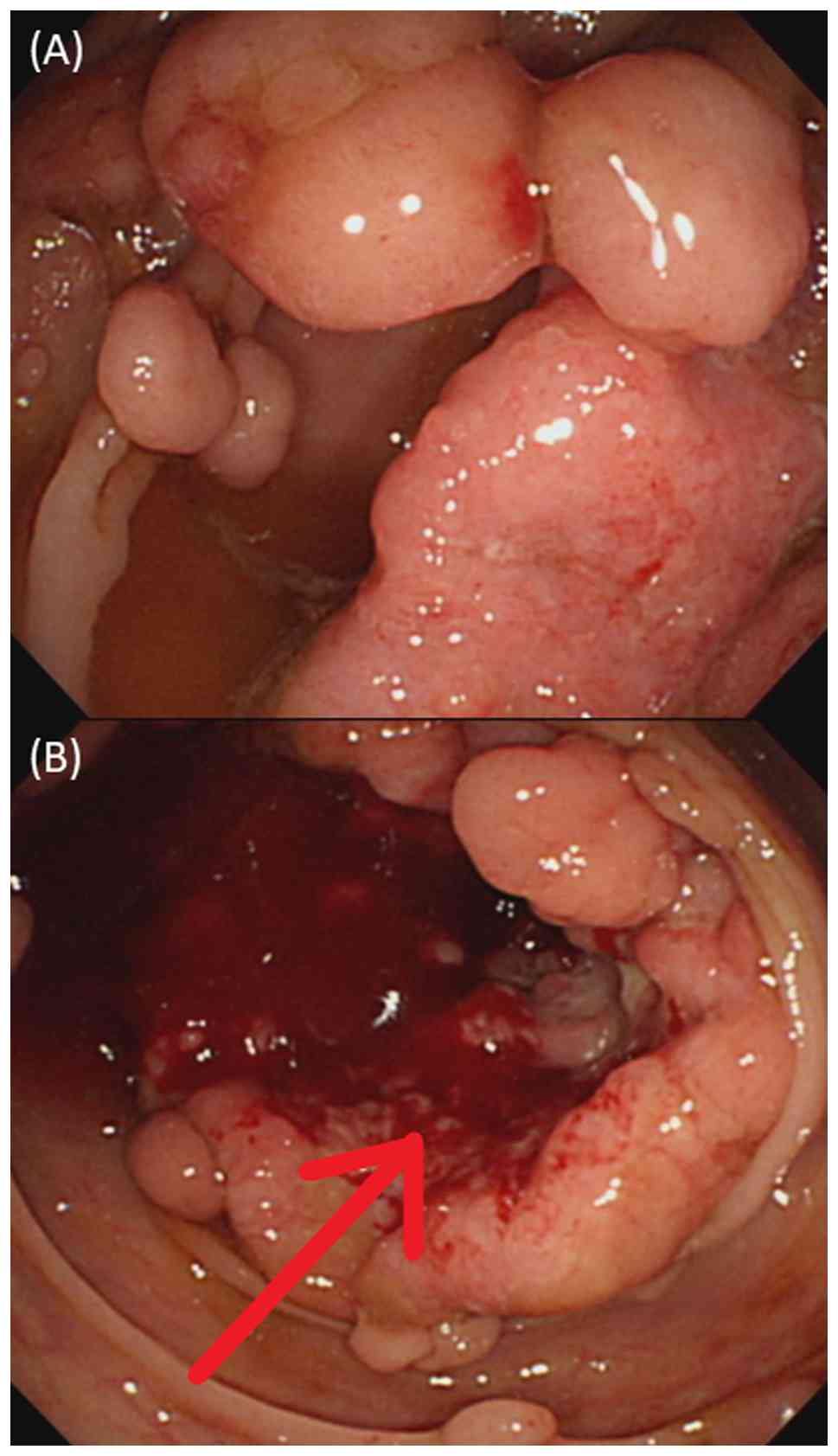
revealed >100 polyps distributed from the upper rectum to the
ascending colon (Fig. 1A), along
with a suspicious lesion near the hepatic flexure of the ascending
colon (Fig. 1B). A biopsy of the
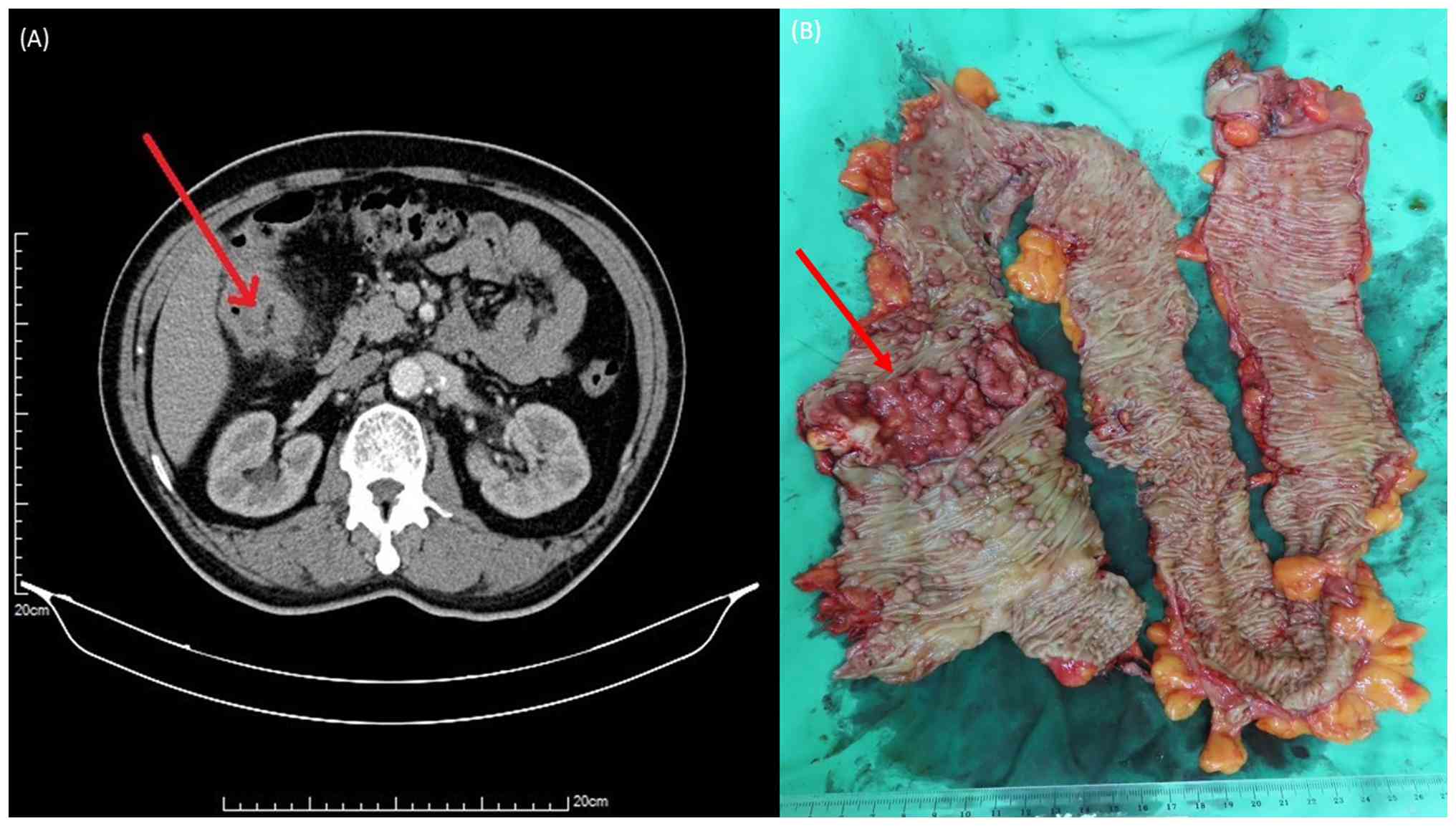
lesion confirmed adenocarcinoma. Contrast-enhanced abdominal
computed tomography revealed a colonic malignancy at the hepatic
flexure with pericolic infiltration, and the clinical stage was
cT4bN1M0 (Fig. 2A).
Given the extensive colonic polyposis and confirmed
malignancy at the hepatic flexure, the patient underwent a subtotal
colectomy with end-to-end ileorectal anastomosis. Postoperative
histopathological analysis confirmed a moderately differentiated
adenocarcinoma of the hepatic flexure, which was staged as pT3N0M0
(Stage IIA), along with >100 tubulovillous adenomas observed
throughout the resected colon (Fig.
2B). Paraffin-embedded tissue sections were stained with
hematoxylin and eosin (H&E) according to a standard protocol.
Briefly, tissue samples were fixed in 10% neutral buffered formalin
at room temperature (~22˚C) for 8 h before being embedded in
paraffin. Sections 4-µm thick were cut using a microtome. H&E
staining was performed with hematoxylin at room temperature for 10
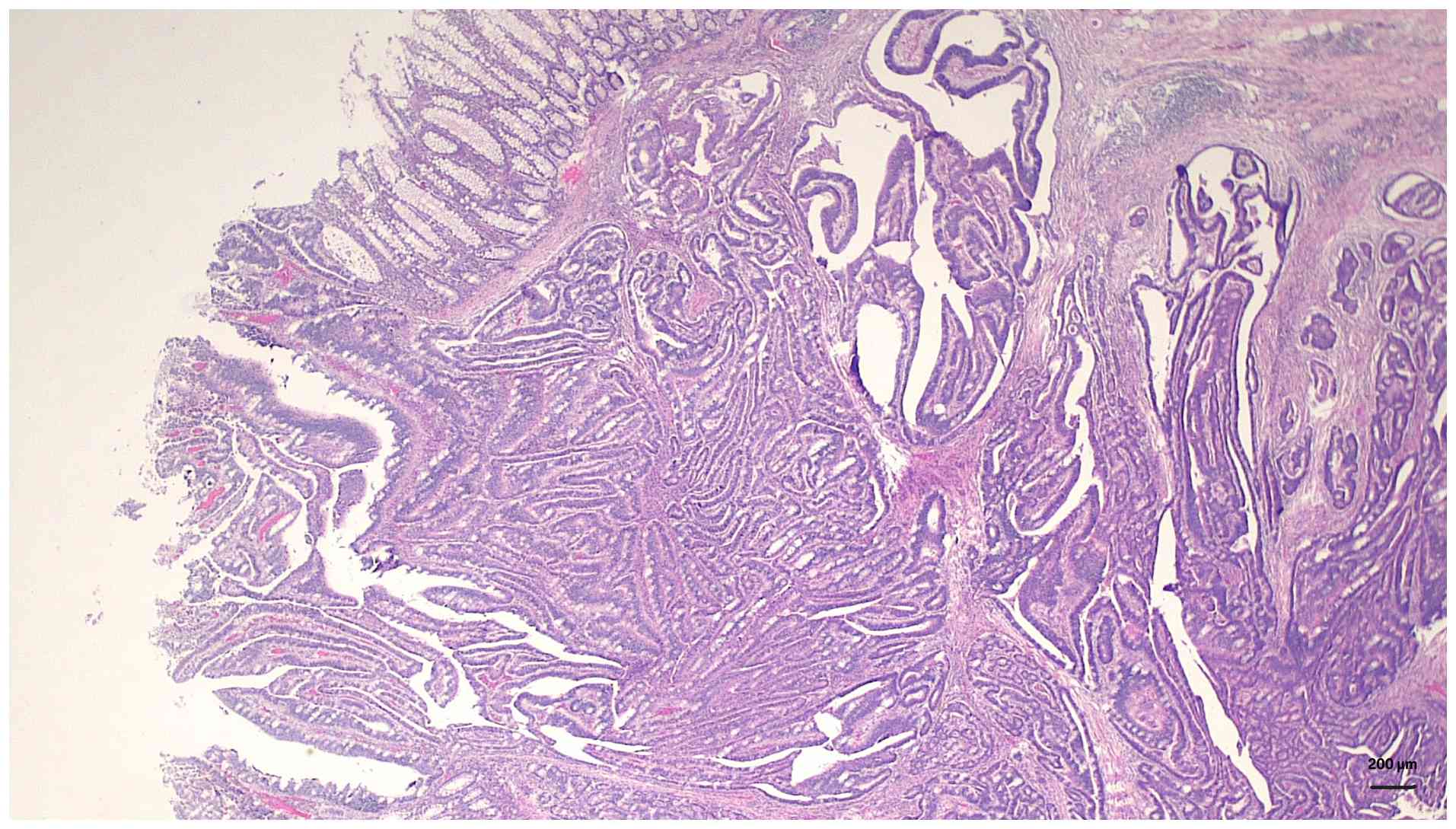
min followed by eosin for 2 min. Histopathological examination
revealed irregular dysplastic glands infiltrating into the
submucosa and muscularis propria, accompanied by a prominent
desmoplastic stromal reaction (Fig.
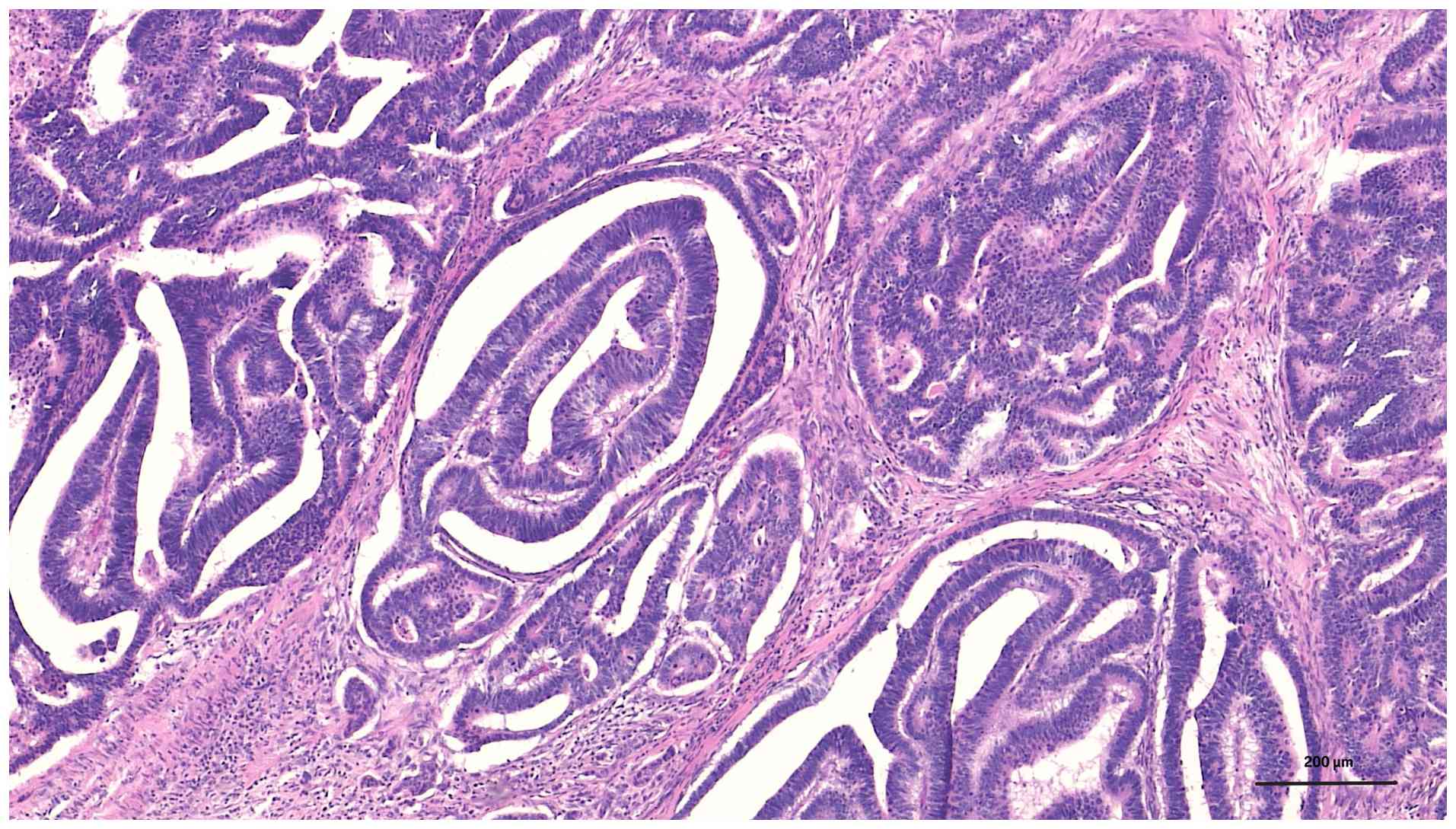
3). At higher magnification, the tumor was found to have an
atypical glandular structure with nuclear pleomorphism,
hyperchromasia and loss of normal glandular polarity within a
fibrotic stroma, consistent with moderately differentiated
adenocarcinoma (Fig. 4).
Postoperatively, the patient received six cycles of
adjuvant chemotherapy with the FOLFOX-6 regimen, which consisted of
oxaliplatin (85 mg/m² on day 1), leucovorin (400 mg/m² on day 1)
and 5-fluorouracil administered as an intravenous bolus (400
mg/m2 on day 1) followed by continuous infusion (2,400
mg/m2 over 46 h), repeated every 2 weeks. After
completion of adjuvant chemotherapy, the patient was regularly
followed up at the outpatient clinic. Postoperative surveillance
was scheduled every 3 months, including physical examination and
measurement of serum carcinoembryonic antigen levels. In addition,
abdominal computed tomography and colonoscopic examinations were
performed annually. The most recent follow-up visit was in July
2025.
Owing to the extensive number of adenomatous polyps,
FAP was suspected, prompting genetic testing. Genomic DNA was
extracted from peripheral blood and analyzed externally by Invitae
Corporation, using the Invitae Multi-Cancer Panel according to the
manufacturer's instructions. This panel includes sequencing and
deletion/duplication analysis of 100 cancer-related genes,
including APC, CDH1 and RECQL4. Target regions
were enriched using a hybridization-based capture protocol and
sequenced on an Illumina next-generation sequencing platform
(Illumina, Inc.). All coding exons, ±20 base pairs of flanking
intronic regions, and selected clinically relevant noncoding
regions, including the APC promoter 1B, were analyzed.
Variants were identified by alignment to the GRCh37 reference
genome, and results were interpreted using clinically validated
transcripts. Copy number variants were assessed using a validated
read-depth-based algorithm, and variant classification was
conducted according to the American College of Medical Genetics and
Genomics guidelines. Confirmatory testing, when required, was
performed using orthogonal methods (6). Genetic testing revealed a
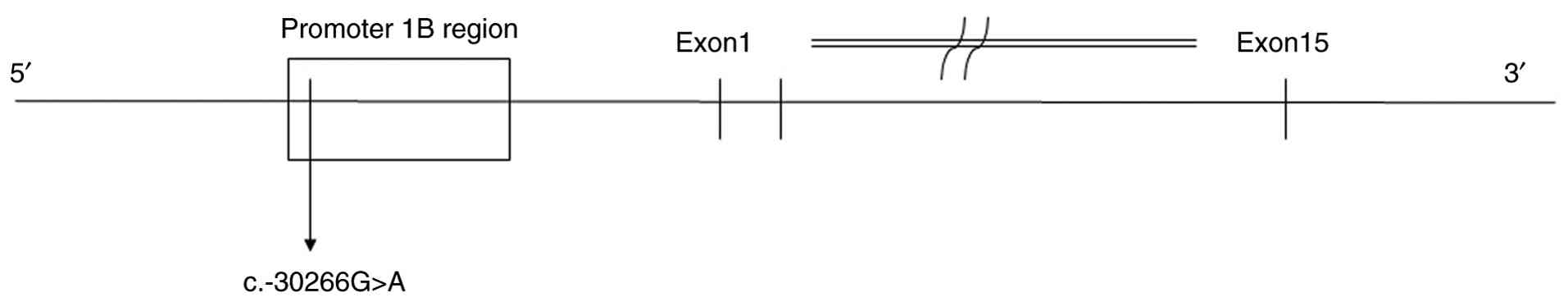
heterozygous pathogenic variant in APC (c.-30266G>A),
located in a noncoding upstream region beyond the typical promoter
1B (Fig. 5). This variant has been
predominantly associated with GAPPS, rather than classical FAP
(5). However, in the present case,
the patient exhibited a clinical phenotype consistent with FAP,
including extensive colorectal polyposis and progression to
CRC.
Given the presumed germline origin of this
pathogenic variant, cascade testing was recommended. Neither the
patient nor the patient's family had a history of CRC or
FAP-related conditions. After establishing the diagnosis, genetic
testing was recommended for all three of the patient's children -
two sons and one daughter. However, only the daughter underwent
testing, for whom the results of the analysis revealed the same
heterozygous pathogenic variant (c.-30266G>A) in the upstream
regulatory region of APC, located beyond the classical
promoter 1B region. At the time of drafting this manuscript, upper
endoscopy and colonoscopy of the daughter showed no evidence of FAP
or GAPPS, indicating the absence of phenotypic expression despite
the daughter's confirmed genetic predisposition (data not shown).
Despite being asymptomatic, regular surveillance was recommended.
The daughter was advised to return to the Outpatient Clinic
annually for clinical evaluation and endoscopic examination to
allow early detection of any adenomatous changes.
Discussion
GAPPS is a rare autosomal dominant hereditary
syndrome characterized by fundic gland polyposis and a markedly
increased risk of gastric adenocarcinoma. Germline point mutations
in the APC promoter 1B region, such as c.-191T>C and
c.-195A>C, have been shown to disrupt transcription factor
binding sites, resulting in reduced APC expression in
gastric mucosa. Iwatsuki et al (7) demonstrated the diagnostic
significance of these mutations, particularly in families without
classic colorectal polyposis. Similarly, Dixon et al
(8) reported a pathogenic
APC promoter 1B variant in a family meeting the criteria for
hereditary diffuse gastric cancer, with a retrospective analysis
revealing clinicopathological features consistent with GAPPS.
Overall, these findings support the inclusion of APC
promoter 1B in multigene panels for individuals or families with an
unexplained predisposition to gastric cancer, even in the absence
of overt polyposis or colonic involvement.
In the present case, the detected mutation
(c.-30266G>A) is located considerably farther upstream from the
transcription start site when compared with the classical
APC promoter 1B mutations, such as c.-191T>C and
c.-195A>C. Specifically, c.-30266G>A is situated ~30,266
bases upstream of the translation initiation codon (ATG), whereas
c.-191T>C and c.-195A>C are located only 191 and 195 bases
upstream, respectively. Given this substantial distance,
c.-30266G>A may lie outside the currently defined APC
promoter 1B region and may involve more distal regulatory elements
that have yet to be completely characterized. Despite the lack of
functional evidence, this variant may affect the expression of
APC by altering its transcription factor binding or local
chromatin structure. This positional difference suggests that
although these variants all affect the noncoding regulatory regions
of APC, they may modulate gene expression through different
mechanisms or act on different tissue-specific transcriptional
controls. Therefore, c.-30266G>A should be considered distinct
from the classical promoter 1B mutations described in the
literature. However, there is currently a lack of studies
investigating the pathogenic mechanism of this specific variant,
and further research is needed to elucidate its underlying
biological significance.
Notably, although both the patient and the patient's
daughter carried the germline c.-30266G>A mutation, neither has
exhibited gastric fundic gland polyposis or gastric adenocarcinoma
to date. Disruptions in the upstream regulatory elements of
APC can reduce transcriptional activity and diminish APC
protein levels. Such promoter-level defects alter the gene's dosage
effect and may clinically present as classical FAP rather than
GAPPS, indicating the functional relevance of promoter integrity in
modulating disease phenotype. Similarly, rare cases of early-onset
CRC associated with APC germline mutations have been
reported, including a case involving a 10-year-old patient who
developed advanced CRC harboring both germline and somatic
APC mutations (9). These
findings underscore the phenotypic heterogeneity associated with
APC alterations, extending beyond the classical FAP and
GAPPS presentations. Accordingly, this case highlights the
phenotypic variability associated with APC noncoding
mutations and demonstrates the importance of considering a broader
clinical spectrum-including both FAP and GAPPS presentations - when
evaluating individuals carrying such variants. This discussion
systematically compares the current c.-30266G>A variant with
previously reported APC promoter 1B mutations and highlights
potential molecular and regulatory mechanisms underlying the
observed phenotypic differences.
Recent evidence reveals that deletions involving the
APC promoter 1B region may be an underrecognized cause of
classical FAP, particularly in patients who were initially negative
for coding-region mutations. Kadiyska et al (10) identified a novel deletion involving
the entire APC promoter 1B in a Bulgarian family with a
classical FAP phenotype, despite negative results from standard
sequencing and multiplex ligation-dependent probe amplification
(MLPA) assays. Performing an updated MLPA assay with probes
specific for promoter 1B, together with high-resolution array
comparative genomic hybridization and long-range polymerase chain
reaction, could enable accurate breakpoint characterization of this
deletion. This combined approach demonstrates how complementary
techniques can reliably detect and validate structural changes in
the noncoding regulatory regions of APC. Functional assays
confirmed the pathogenicity of this regulatory region alteration by
demonstrating significantly reduced APC expression. Similarly,
Azzopardi et al (11)
identified APC promoter 1B deletions in multiple unrelated
Italian families, indicating a founder effect; the affected
individuals consistently presented with classical FAP features,
including hundreds to thousands of colonic adenomas, but lacked
gastric polyposis, suggesting a phenotype distinct from GAPPS.
Overall, these findings demonstrate the clinical significance of
promoter 1B deletions in the pathogenesis of FAP and underscore the
importance of including this noncoding region in routine genetic
testing for patients with an FAP phenotype who are negative for
coding-region mutations in APC.
Yang et al (12) identified a rare point variant
(c.-190G>A) in the APC promoter 1B region in a family
exhibiting classical FAP features, despite negative results from
conventional gene panels and MLPA analysis. By using whole-exome
sequencing and functional dual-luciferase reporter assays, Yang
et al (12) demonstrated
that this noncoding variant significantly suppressed APC
transcriptional activity, supporting its pathogenicity. These
findings demonstrate the critical role of noncoding regulatory
regions, such as promoter 1B, in the genetic evaluation of FAP,
particularly in cases that test negative on standard diagnostic
panels.
Although the present case also involves a noncoding
regulatory variant upstream of APC, the c.-30266G>A
mutation is located considerably beyond the classical promoter 1B
region. Nevertheless, the patient's presentation with classical FAP
supports the notion that mutations in APC regulatory
elements, whether within or beyond promoter 1B, may contribute to
FAP development. Despite these observations, the exact molecular
mechanisms through which regulatory region mutations contribute to
FAP pathogenesis remain elusive and warrant further investigation.
As the present findings were derived from a single patient, they
should be interpreted with caution. Generally, the intrinsic
limitation of case reports is the absence of statistical power and
external validity. Therefore, the present findings should be
regarded as preliminary and hypothesis-generating, pending
confirmation in larger cohorts.
In conclusion, this rare case of FAP associated with
a point mutation in the noncoding upstream regulatory region of
APC, located beyond the classical promoter 1B region,
highlights the pathogenic role of distal regulatory elements. The
findings underscore the importance of including both promoter 1B
and adjacent upstream regions in extended genetic testing panels
for patients with unexplained FAP phenotypes. However, due to the
single-case nature of this report, the results should be
interpreted with caution and validated through larger studies.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by grants from the National
Science and Technology Council (NSTC) (grant nos. MOST
111-2314-B-037-070-MY3, NSTC 112-2314-B-037-050-MY3, NSTC
113-2321-B-037-006 and NSTC 113-2314-B-037-057) and the Ministry of
Health and Welfare (MOHW) (grant no. MOHW113-TDU-B-222-134014), and
funded by the health and welfare surcharge on tobacco products, and
the Kaohsiung Medical University Hospital (KMUH) (grant nos.
KMUH112-2R37, KMUH112-2R38, KMUH112-2R39, KMUH112-2M27,
KMUH112-2M28, KMUH112-2M29, KMUH113-2R31, KMUH113-2R32,
KMUH113-2R33, KMUH113-3M58, KMUH113-3M59, KMUH-S11303, KMUH-SH11309
and KMUH-SI11327), Kaohsiung Medical University Research Center
Grant (grant no. KMU-TC113A04) and National Tsing Hua University
(NTHU)-KMU Joint Research Project (grant no. NTHU-KMU-KT114P008).
In addition, this study was supported by the Grant of Taiwan
Precision Medicine Initiative and Taiwan Biobank, Academia Sinica,
Taiwan, R.O.C.
Availability of data and materials
The data generated in the present study may be found
in the Sequence Read Archive under accession number RJNA1338078 or
at the following URL: https://www.ncbi.nlm.nih.gov/sra/PRJNA1338078.
Authors' contributions
JYW and HLT were involved in the study conception
and design. CWH, WCS, TKC performed data collection. YCC
contributed by performing analysis and interpretation of results.
CHY drafted the manuscript. All authors reviewed the results and
approved the final version of the manuscript. JYW and YCC confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
The study was approved by the Institutional Review
Board of Kaohsiung Medical University Hospital (KMUHIRB)
[Kaohsiung, Taiwan; approval no. KMUHIRB-E(I)-20230267].
Patient consent for publication
The patient and the patient's daughter provided
written informed consent for publication of this case report and
any accompanying data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nieuwenhuis MH and Vasen HFA: Correlations
between mutation site in APC and phenotype of familial adenomatous
polyposis (FAP): A review of the literature. Crit Rev Oncol
Hematol. 61:153–161. 2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Herzig D, Hardimann K, Weiser M, Yu N,
Paquette I, Feingold DL and Steele SR: The American society of
colon and rectal surgeons clinical practice guidelines for the
management of inherited polyposis syndromes. Dis Colon Rectum.
60:881–894. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kudchadkar S, Ahmed S, Mukherjee T and
Sagar J: Current guidelines in the surgical management of
hereditary colorectal cancers. World J Gastrointest Oncol.
14:833–841. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yen T, Stanich PP, Axell L and Patel SG:
APC-associated polyposis. conditions.
|
|
5
|
Li J, Woods SL, Healey S, Beesley J, Chen
X, Lee JS, Sivakumaran H, Wayte N, Nones K, Waterfall JJ, et al:
Point mutations in exon 1B of apc reveal gastric adenocarcinoma and
proximal polyposis of the stomach as a familial adenomatous
polyposis variant. Am J Hum Genet. 98:830–842. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lincoln SE, Truty R, Lin CF, Zook JM, Paul
J, Ramey VH, Salit M, Rehm HL, Nussbaum RL and Lebo MS: A rigorous
interlaboratory examination of the need to confirm next-generation
sequencing-detected variants with an orthogonal method in clinical
genetic testing. J Mol Diagn. 21:318–329. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Iwatsuki M, Matsumoto C, Mimori K and Baba
H: The comprehensive review of gastric adenocarcinoma and proximal
polyposis of the stomach (GAPPS) from diagnosis and treatment. Ann
Gastroenterol Surg. 7:725–732. 2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Dixon K, Senz J, Kaurah P, Huntsman DG and
Schrader KA: Rare APC promoter 1B variants in gastric cancer
kindreds unselected for fundic gland polyposis. Gut. 70:1415–1416.
2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yeh YS, Chang YT, Ma CJ, Huang CW, Tsai
HL, Chen YT and Wang JY: First-decade patient with colorectal
cancer carrying both germline and somatic mutations in APC gene.
BMC Cancer. 17(849)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kadiyska TK, Todorov TP, Bichev SN,
Vazharova RV, Nossikoff AV, Savov AS and Mitev VI: APC promoter 1B
deletion in familial polyposis-implications for mutation-negative
families. Clin Genet. 85:452–457. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Azzopardi C, Vassallo E, Grech R and Mizzi
A: Adult colorectal intussusception. BMJ Case Rep: bcr2014205597,
2014.
|
|
12
|
Yang M, Zhu L, Zhu L, Xu D and Yuan Y:
Role of a rare variant in APC gene promoter 1B region in classic
familial adenomatous polyposis. Digestion. 102:527–533.
2020.PubMed/NCBI View Article : Google Scholar
|