Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic,
progressive interstitial lung condition characterized by
radiological and histological features of usual interstitial
pneumonia (UIP), leading to progressive dyspnea and declining
pulmonary function, particularly in elderly patients (1). Although IPF is a rare disease, its
global incidence falls between 0.09-1.30 per 10,000, with a
prevalence of 0.33-4.51 per 10,000(2). Without treatment, the median survival
time after diagnosis is only 3-5 years (3).
Pulmonary function tests (PFTs) in IPF typically
reveal reduced forced vital capacity (FVC), total lung capacity and
diffusing capacity of the lungs for carbon monoxide (DLCO);
however, parameters may remain normal or only mildly impaired in
early disease (1). Notably,
pulmonary function testing alone is insufficient to predict
mortality, and prognostic indices such as the gender-age-physiology
(GAP) score have therefore been proposed as more reliable tools for
outcome prediction (4).
IPF is confined to the lungs and is pathologically
defined by the UIP pattern, best demonstrated on high-resolution
computed tomography (HRCT). Radiologically, UIP shows a subpleural
and basal predominance, with hallmark features including
honeycombing, traction bronchiolectasis, reticular abnormalities
and occasionally ground-glass opacities (1). Honeycomb lung, cystic lung diseases
and emphysema are radiologically similar; however, their
pathophysiological development is different. Honeycomb formation
results from the collapse of fibrotic alveolar septa and expansion
of terminal airways, leading to the formation of bronchiolar cysts.
Therefore, differentiating honeycombing from paraseptal emphysema
or fibrosis-associated cystic changes is important. When
considering the diagnosis of IPF, the current guidelines emphasize
the importance of radiology over pathology. Notably, honeycomb
pattern is an important radiological finding for IPF.
The pathogenesis of IPF involves aberrant
wound-healing responses. In normal repair, activated fibroblasts
and myofibroblasts deposit extracellular matrix (ECM) components,
primarily fibrillar collagen and fibronectin, to provide a
provisional scaffold that facilitates the proliferation and
differentiation of alveolar epithelial type II cells (5). Chronic injury and aging impair
re-epithelialization, driving abnormal mesenchymal activation and
disrupting epithelial regeneration. This dysfunctional interaction
between epithelial and mesenchymal compartments is central to IPF
progression (6). Current evidence
suggests that repetitive injury and dysregulated activation of key
profibrotic pathways, including TGF-β and WNT signaling, contribute
to myofibroblast expansion, cellular senescence and impaired
alveolar repair (6,7). Both pathways have therefore emerged
as key drivers of fibrogenesis and potential therapeutic targets in
IPF.
The approved antifibrotic agents pirfenidone and
nintedanib markedly attenuate the decline in FVC (1). However, neither therapy halts
structural remodeling nor induces reversal of fibrosis. Given the
limited efficacy of current treatments, novel therapeutic
strategies aimed at modulating fundamental fibrogenic pathways
remain urgently needed (8). A
previous study has shown that both canonical (WNT/β-catenin) and
non-canonical (such as WNT5A) signaling are implicated in IPF
pathogenesis (9). In IPF lungs,
WNT/β-catenin activity is increased and is associated with
epithelial injury, aberrant alveolar type II cell
reprogramming/senescence and impaired epithelial-mesenchymal
interaction, thereby fostering profibrotic remodeling (8). Non-canonical WNTs, particularly
WNT5A, are upregulated in IPF tissue and extracellular vesicles,
and can amplify TGF-β-driven responses and macrophage polarization,
processes that promote fibroblast activation and matrix deposition
across fibrotic organs (9,10).
Downstream WNT target genes, such as WNT1-inducible
signaling protein-1 (WISP1/CCN4), are elevated in IPF and
experimental fibrosis, whereas functional inhibition of WISP1
mitigates fibrotic progression, underscoring the translational
relevance of WNT-responsive programs (11,12).
Chronic activation of the WNT signaling pathway sustains epithelial
stress, promotes profibrotic fibroblast phenotypes and drives
maladaptive inflammation in IPF, thereby providing a strong
rationale for WNT-targeted therapeutic strategies, including
porcupine O-acyltransferase (PORCN) inhibitors, which are currently
under clinical investigation (13).
The primary aim of the present study was to
investigate the role of the WNT signaling pathway in mediating
inflammation and fibrosis during the pathogenesis of IPF. A
secondary objective was to identify potential therapeutic targets
within this pathway that could be exploited for novel treatment
strategies.
Therefore, WNT pathway activation was evaluated in
the peripheral blood of patients with IPF. Furthermore, the effects
of two selective WNT signaling inhibitors, ETC-159 and LGK-974,
were assessed within LL29 (AnHa) fibroblasts derived from a patient
with IPF. These experiments were designed to elucidate how
pharmacological blockade of WNT signaling influences fibrotic and
inflammatory responses. Collectively, this approach aimed to define
the contribution of WNT signaling to IPF pathogenesis and highlight
its potential as a therapeutic target.
Materials and methods
Study design
Designed as a prospective case-control study, the
present study was based on the diagnosis of IPF. This was
established through a multidisciplinary evaluation involving
radiologists, rheumatologists and pulmonologists, based on
radiological criteria consistent with IPF (1). Patients with confirmed IPF who
presented for routine outpatient follow-up at the Department of
Chest Diseases, Kütahya Health Sciences University Faculty of
Medicine (Kütahya, Turkey), were invited to participate. After
providing written informed consent, peripheral blood samples were
collected for subsequent analyses between June 2024 and April 2025.
The control group comprised individuals without a diagnosis of IPF
who attended the chest diseases outpatient clinic for unrelated
reasons and voluntarily consented to participate.
Ethics committee approval
Ethics approval for the present study was obtained
from the Kütahya Health Sciences University Faculty of Medicine
Non-Interventional Ethics Committee in May 2024 (approval no.
2024/06-23).
Sample size
Sample size calculation was performed using GPower
(version 3.1; Heinrich Heine University). An unpaired two-tailed
t-test was applied, assuming a medium effect size (d=0.5), α=0.05
and statistical power (1-β)=0.80. The analysis indicated a minimum
requirement of n=30/group. Based on this calculation, 33 patients
with IPF and 23 healthy controls were recruited. The slightly
unequal distribution was due to patient availability and ethical
considerations; however, the total sample size remained sufficient
to ensure adequate statistical power.
Inclusion criteria
Individuals >40 years of age with a radiological
or pathological diagnosis of IPF, no drug use known to cause lung
fibrosis, no history of lung infection resulting in fibrosis, and
both patients receiving or not receiving treatment for IPF were
included in the IPF group. The inclusion criterion for the control
group was defined as having no chronic illness.
Exclusion criteria
Individuals who did not give consent to participate
in the present study, individuals using long-term oxygen
concentrators and those with a history of lung infection resulting
in fibrosis were excluded from the present study.
Sample collection
Patients diagnosed with IPF (radiologically or
pathologically) and a control group were informed about the present
study upon arrival at the outpatient clinic and a 1-ml blood sample
was collected from each of the patients who gave their consent. The
samples were stored at -80˚C after collection.
Laboratory stages
All laboratory assays [ELISA, quantitative PCR
(qPCR) and related wet-lab procedures] were performed under blinded
conditions. The researchers conducting the experiments were unaware
of the group allocation of the samples to minimize bias and ensure
objective data collection.
Clinical, radiological and laboratory parameters
were assessed to complement molecular analyses. Clinical parameters
included height, weight, age, PFT and Modified Medical Research
Council (mMRC) values (14). The
PFT values forced expiratory volume in 1 sec (FEV1), FVC and
FEV1/FVC were collected. The mMRC score was used to assess dyspnea
severity. PFT results and demographic data were obtained from
medical records. Radiologically, the presence of honeycomb and
septal thickening was determined by examining HRCT images at the
time of diagnosis. HRCT was used for radiological confirmation of
IPF and evaluation of fibrotic patterns, and fibrotic patterns were
recorded. Routine hemogram parameters were obtained from peripheral
blood samples as part of standard clinical evaluation.
Cell culture experiments
LL29 (AnHa) cells (CCL-134; American Type Culture
Collection) were used in the present study. These cells were
cultured in DMEM (cat. no. 11966; Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS (cat. no. A5256701; Gibco; Thermo Fisher
Scientific, Inc.) + 1% glutamine (cat. no. G8540; Sigma-Aldrich;
Merck KGaA) + 1% penicillin-streptomycin (cat. no. 15140122; Thermo
Fisher Scientific, Inc.) + 1% sodium. All procedures involving the
cells were performed in a laminar cabinet. The cells were incubated
at 37˚C in an incubator with 5% CO2. When the cells
reached 70% density, passaging was performed with EDTA solution
used to detach cells. The xCELLigence real-time cell analysis dual
purpose system (Agilent Technologies, Inc.) was used in the
IC50 and proliferation assay. The cells were seeded at a
concentration of 1x106 cells/ml in xCELLigence 16-well
E-plates. IC50 values were calculated using xCELLigence
real-time impedance measurements. The IC50 values of
ETC-159 and LGK-974 were 10 and 1 µM at 48 h, based on nonlinear
regression analysis of the dose-response curve.
Cells were maintained at 37˚C in a humidified
incubator containing 5% CO2. After 24 h, the culture
medium was removed and replaced with fresh medium supplemented with
ETC-159 (10 µM; cat. no. S6616; Selleck Chemicals) or LGK-974 (1
µM; cat. no. S7143; Selleck Chemicals), prepared at their final
working concentrations and incubated at 37˚C for 48 h. Untreated
cells were used as control cells. In all cell culture studies, both
treated and untreated cells were left to proliferate to
~3x106 and subsequently frozen at -80˚C until the
relevant experimental steps. Each ELISA and RT-qPCR measurement was
performed in triplicate to ensure technical reproducibility.
Reverse transcription-qPCR
experiments
RT-qPCR data were normalized to the expression of
b-actin, which served as the internal control. Relative gene
expression levels were calculated using the
2-ΔΔCq method (15). The mRNA expression levels of WNT
signaling genes were evaluated in blood samples and cells using
RT-qPCR. Briefly, total RNA was extracted from blood and cells
using the RNeasy Micro Kit (cat. no. 74004; Qiagen, Inc.),
following the manufacturer's protocol. Using 1 µg RNA, cDNA was
synthesized with the QuantiTect Reverse Transcription Kit (cat. no.
205311; Qiagen, Inc.), according to the manufacturer's protocol,
and was scaled to a final volume of 20 µl. Samples were stored at
-20˚C until qPCR. All cDNA samples were equally distributed in a
96-well plate and qPCR was performed using RT2
SYBR® Green Fluor qPCR Mastermix (cat. no. 330513;
Qiagen, Inc.). Plates were run on a StepOne Plus qPCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) under the
following thermocycling conditions: Initial denaturation at 95˚C
for 10 min, followed by 40 cycles at 95˚C for 15 sec and 60˚C for
30 sec. Melt curve analysis was performed for quality control.
Primer sequences are listed in Table
I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Sequence,
5'-3' |
|---|
| Human WNT-1 | F:
CTCCACGAACCTGCTTACAGA |
| | R:
GCTCGAGTACCAGTTGCAGA |
| Human WNT-2 | F:
CTACGACACCTCCCATGTCA |
| | R:
GGAACTTACACCCACACTTGGT |
| Human WNT-3a | F:
TCCTCAAGGACAAGTACGACAG |
| | R:
GTGCTTCTCCACCACCATCT |
| Human WNT-6 | F:
GCATCCTGCAACAGGACAT |
| | R:
AGTGATGGCGAACACGAAG |
| Human WNT-10a | F:
GTCCCATCTTCAGCAGAGGT |
| | R:
GATGGCGTAGGCAAAAGC |
| Human WNT-10b | F:
CTGGTGCTGCTATGTGCTCT |
| | R:
TCACCCACTCTGTAACCTTGC |
| Human α-SMA | F:
CTATGCCTCTGGACGCACAACT |
| | R:
CAGATCCAGACGCATGATGGCA |
| Human collagen type
I | F:
GATTCCCTGGACCTAAAGGTGC |
| | R:
AGCCTCTCCATCTTTGCCAGCA |
| Human β-actin | F:
CACCATTGGCAATGAGCGGTTC |
| | R:
AGGTCTTTGCGGATGTCCACGT |
| Human WNT-7a | F:
CGAAAGATCCTGGAGGAGAAC |
| | R:
ACGCCGTGGCACTTACAT |
| Human WNT-7b | F:
AAGCCCATGGAGACAGACC |
| | R:
CAGTAGTTGGGCGACTTCTCA |
| Human WNT-4 | F:
CAGAGGCAGGTGCAGATGT |
| | R:
ACCGAGTCCATGACTTCCAG |
| Human WNT-16 | F:
TGAAAGCATGACTGATGTCCA |
| | R:
AGGCTGGATGGAGTGGTTACT |
Measurement of inflammatory (IL-1β and
IL-6) and fibrotic (TGF-ß2) markers by ELISA
The Human IL-1β ELISA kit (cat. no. RE1074H; Reed
Biotech, Ltd.), Human IL-6 ELISA kit (cat. no. RE3186H; Reed
Biotech, Ltd.) and Human TGF-β2 ELISA kit (cat. no. RE3066H; Reed
Biotech, Ltd.) were used following the manufacturer's protocols.
The optical density was measured spectrophotometrically at a
wavelength of 450 nm using the Multiskan™ Plate Reader
(Thermo Fisher Scientific, Inc.).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism software (version 10; Dotmatics). The Mann-Whitney U
test was used to compare two non-parametric groups. When more than
two groups were analyzed, the Kruskal-Wallis test was employed,
followed by Dunn's multiple-comparison post hoc test with adjusted
P-values. For correlation analysis, data distribution was assessed
using the Shapiro-Wilk normality test. Since the assumptions of
parametric correlation analysis were not met, Spearman's rank
correlation coefficient was applied to evaluate monotonic
associations between variables. Categorical variables were analyzed
using Fisher's exact test, where appropriate. Data are presented as
the mean ± SEM. At least three independent biological replicates
were assessed in all in vitro experiments. All tests were
two-tailed, and P<0.05 was considered to indicate a
statistically significant difference.
Results
General findings
A total of 33 patients with IPF (28 male patients
and 5 female patients,) and 23 control patients (20 male patients
and 3 female patients) were included in the present study (Table II). The mean age of the patients
with IPF was 68.91±1.55 years, the mean age of the control group
was 63.65±3.06 years. The mean BMI was 27.3±0.69 kg/m2
in the IPF group and 27.06±1.34 kg/m2 in the control
group. The mean cigarette pack/year history was 31.35±4.31 in the
IPF group and 28.04±4.77 in the control group. No statistically
significant differences were observed between the two groups
regarding the aforementioned parameters (all P>0.05).
| Table IIClinical, laboratory and radiological
characteristics of the IPF and control groups. |
Table II
Clinical, laboratory and radiological
characteristics of the IPF and control groups.
| Variable | IPF | Control | P-value |
|---|
| FEV1, l | 1.90±-0.11 | 2.12±-0.25 | >0.05 |
| FEV1, % | 73.55±-3.84 | 78.08±-9.05 | >0.05 |
| FVC, | 2.11±-0.13 | 2.62±-0.28 | >0.05 |
| FVC, % | 64.33±-3.45 | 68.28±-6.51 | >0.05 |
| FEV1/FVC, % | 87.91±-3.05 | 88.44±-4.58 | >0.05 |
| Neutrophil
count | 7.29±-0.88 | 5.76±-0.76 | >0.05 |
| Neutrophils, % | 64.45±-2.23 | 68.03±-2.66 | >0.05 |
| GAP score | 4.65±-0.28 | 3.08±-0.45 | 0.0076a |
| Age, years | 68.91±1.55 | 63.65±3.06 | 0.0916 |
| Sex,
male/female | 28/5 | 20/3 | - |
| BMI,
kg/m2 | 27.38±0.69 | 27.06±1.34 | 0.4086 |
| Cigarette
packs/year | 31.35±4.31 | 28.04±4.77 | 0.6209 |
| DLCO, % | 45.63±4.40 | 3.95±1.24 | 0.0128a |
| mMRC score | 2.16±0.20 | 0.78±0.95 |
<0.0001a |
| Alveolar septal
fibrosis, n (%) | | |
<0.0001a,b |
|
Yes | 30 (90.9) | 0 (0.0) | |
|
No | 3 (9.1) | 23 (100.0) | |
| Honeycombing, n
(%) | | |
<0.0001a,b |
|
Yes | 20 (60.6) | 0 (0.0) | |
|
No | 13 (39.4) | 23 (100.0) | |
Respiratory parameters and
radiological findings
The clinical, demographic and laboratory
characteristics of patients with IPF and control subjects are
summarized in Table II. There
were no significant differences between groups in terms of
pulmonary function parameters, including FEV1, FVC, FEV1/FVC ratio,
neutrophil count, neutrophil percentage, body mass index, smoking
history or age (all P>0.05).
The GAP score was significantly higher in the IPF
group compared with that in the control group (4.65±0.28 vs.
3.08±0.45; P=0.0076), indicating greater disease severity.
Similarly, DLCO values were significantly reduced in patients with
IPF (45.63±4.40 vs. 53.95±1.24%; P=0.0128), and mMRC dyspnea scores
were significantly higher in the IPF group (2.16±0.20 vs.
0.78±0.95; P<0.0001).
Alveolar septal fibrosis was observed in 90.9% of
patients with IPF (30/33), whereas none of the control subjects
exhibited this finding (0/23), indicating a highly significant
difference between groups (Fisher's exact test, P<0.0001).
Similarly, honeycombing was present in 60.6% of patients with IPF
but absent in controls (Fisher's exact test, P<0.0001).
WNT levels in patient samples
Comparison of the WNT gene levels in 33 patients
with IPF and 23 control patients revealed that WNT-2, WNT-4, WNT-6,
WNT-7a, WNT-7b, WNT-10a and WNT-10b levels were increased in
patients with IPF; however, there was no statistically significant
difference in WNT-1 or WNT-3a levels (Table III).
| Table IIIComparison of WNT levels of the
control and IPF groups. |
Table III
Comparison of WNT levels of the
control and IPF groups.
| WNT family gene
expression | Control | IPF, FC | P-value |
|---|
| WNT-1 | 1 | 0.99 | 0.9175 |
| WNT-2 | 1 | 1.68 | 0.0007a |
| WNT-3a | 1 | 1.31 | 0.5909 |
| WNT-4 | 1 | 1.37 | 0.0237a |
| WNT-6 | 1 | 1.64 | 0.0021a |
| WNT-7a | 1 | 2.27 |
<0.0001a |
| WNT-7b | 1 | 2.46 |
<0.0001a |
| WNT-10a | 1 | 2.55 | 0.0007a |
| WNT-10b | 1 | 1.78 |
<0.0001a |
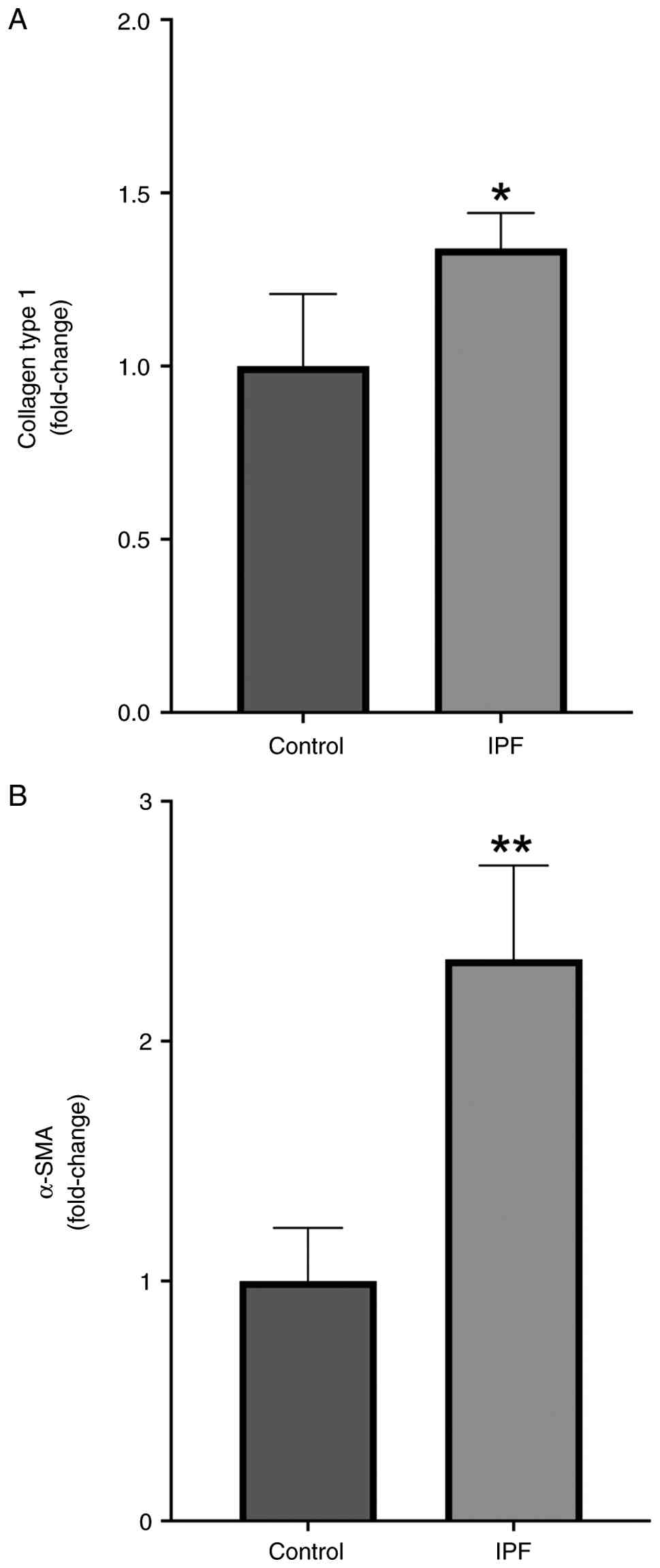
| α-SMA | 1 | 2.34 | 0.0018a |
| Collagen type
I | 1 | 1.33 | 0.0332a |
Fibrotic marker levels
Collagen type I and α-smooth muscle actin (α-SMA)
levels, which indicate fibrosis, were significantly higher in
samples from patients with IPF compared with those in the control
group (P=0.0332 and P=0.0018, respectively; Fig. 1).
Anti-fibrotic and anti-inflammatory
effects of WNT inhibitors
LL29 (AnHa) cells from a patient with IPF were
treated with WNT signaling pathway blockers (ETC-159 and LGK-974).
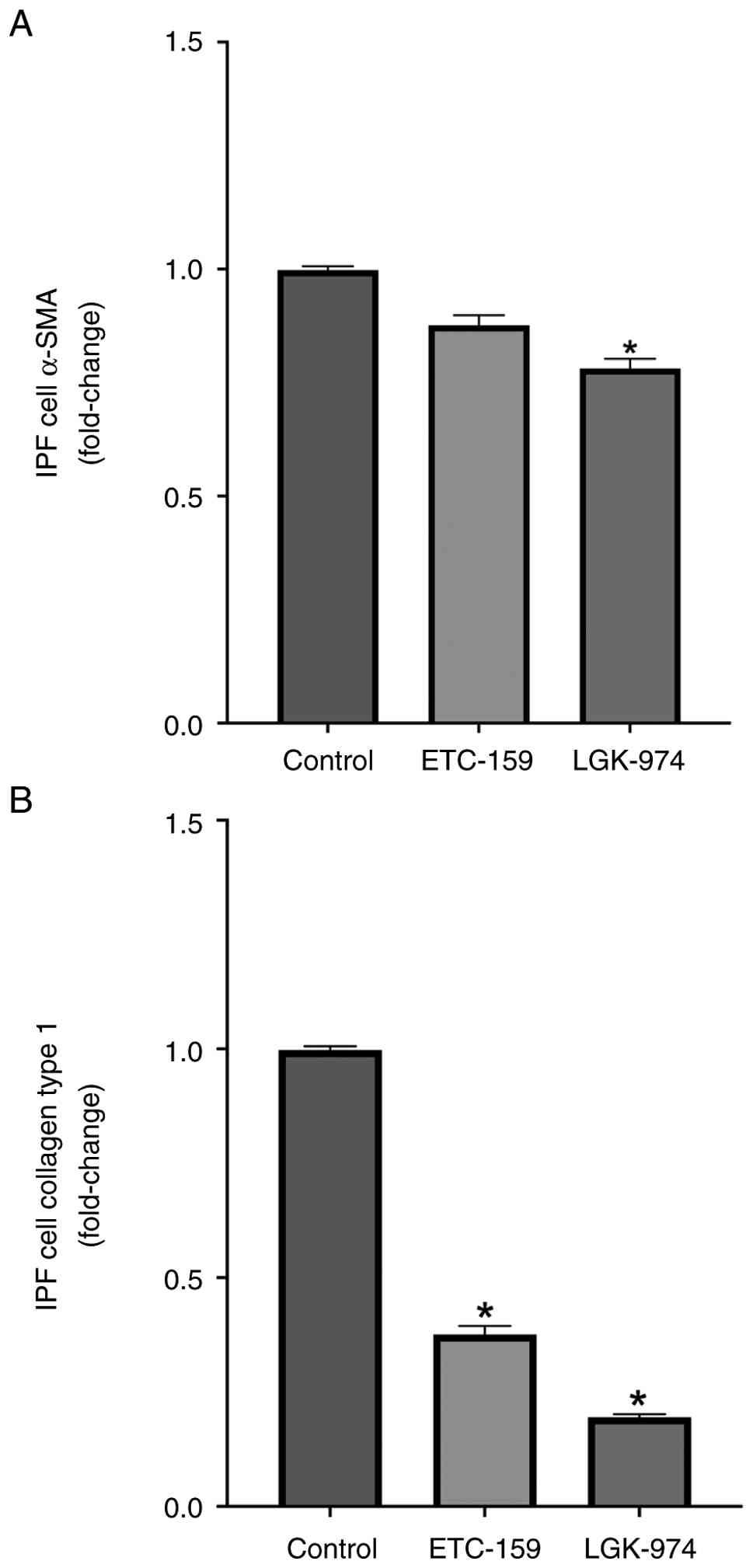
After treatment with ETC-159, there was no significant reduction in
α-SMA levels in the LL29 (AnHa) cells; however, there was a
significant reduction in collagen type I levels (P=0.02; Fig. 2). By contrast, after LGK-974
treatment, there was a statistically significant reduction in both
α-SMA and collagen type I levels in the LL29 (AnHa) cells (P=0.01
and P=0.01, respectively).
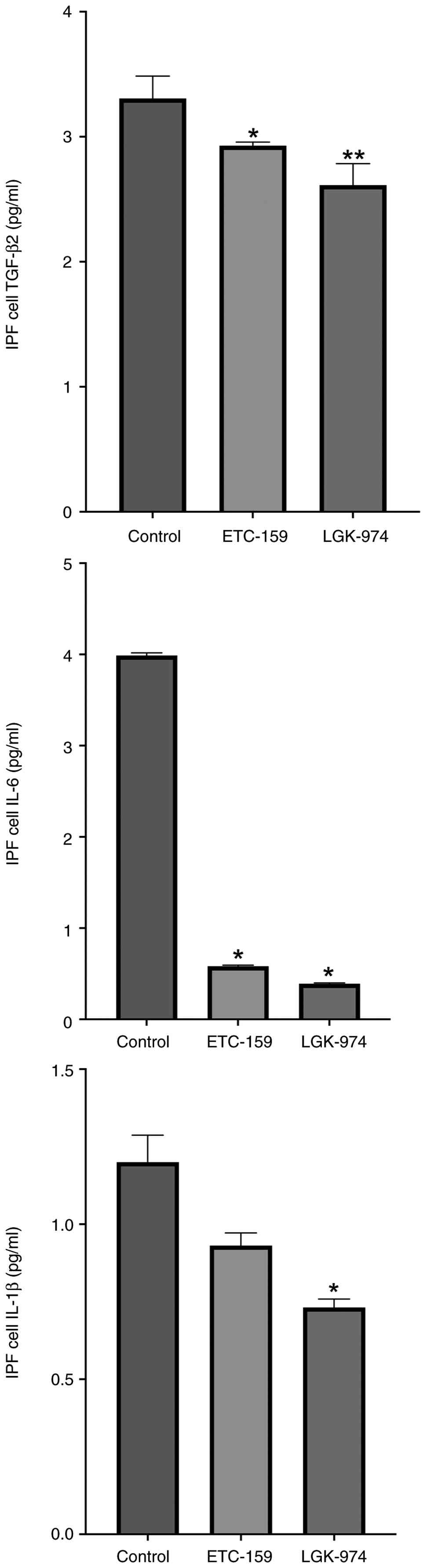
Furthermore, to determine the relationship between
the WNT signaling pathway and fibrosis, WNT inhibitors were applied
to LL29 (AnHa) IPF cells and the anti-fibrotic and
anti-inflammatory effects of WNT inhibitors were investigated. A
significant reduction in TGF-β2 levels was observed following
treatment with both ETC-159 and LGK-974 compared with in the
control group (P=0.0357 and P=0.0079, respectively), (Fig. 3) IL-6 levels were significantly
decreased after both ETC-159 and LGK-974 administration compared
with those in the control group (P=0.02 and P=0.02). In addition,
the administration of LGK-974 but not ETC-259 resulted in a
significant decrease in IL-1β levels compared with control
(P=0.03).
Effect of WNT inhibitors on LL29
(AnHa) cells
When WNT levels in LL29 (AnHa) cells treated with
WNT inhibitors were examined, it was found that the mRNA expression
levels of WNT-6, WNT-10a and WNT-10b were reduced in the treated
cells compared with those in the control group (Table IV). Additionally, the levels of
WNT-7a were significantly reduced upon ETC-159 treatment, however
there was no significant change upon LGK-974 treatment.
| Table IVEffects of ETC-159 and LGK-974 on the
expression levels of WNT genes and fibrotic markers. |
Table IV
Effects of ETC-159 and LGK-974 on the
expression levels of WNT genes and fibrotic markers.
| WNT family gene
expression (FC) | Control | ETC-159, FC | LGK-974, FC | P-value
ETC-159 | P-value
LGK-974 |
|---|
| WNT-1 | 1 | 0.52 | 0.62 | 0.05 | 0.05 |
| WNT-2 | 1 | 0.76 | 0.79 | 0.05 | 0.05 |
| WNT-3 | 1 | 0.61 | 0.75 | 0.05 | 0.05 |
| WNT-4 | 1 | 0.73 | 0.75 | 0.05 | 0.05 |
| WNT-6 | 1 | 0.16 | 0.28 | 0.02a | 0.02a |
| WNT-7a | 1 | 0.82 | 1.02 | 0.02a | 0.02a |
| WNT-7b | 1 | 1.18 | 1.00 | 0.10 | 0.10 |
| WNT-10a | 1 | 0.37 | 0.37 | 0.02a | 0.02a |
| WNT-10b | 1 | 0.46 | 0.52 | 0.02a | 0.02a |
| α-SMA | 1 | 0.82 | 0.85 | 0.01a | 0.01a |
| Collagen type
I | 1 | 0.17 | 0.38 | 0.02a | 0.01a |
Evaluation of WNT levels and collagen
markers together with clinical, radiological and laboratory
parameters
Correlation analysis revealed significant
associations between gene expression levels and clinical parameters
in patients with IPF (Table V).
WNT-3a expression showed a weak positive correlation with GAP score
and DLCO. In addition, collagen type I expression was correlated
with both FEV1 and FVC values. Neutrophil count and percentage were
positively correlated with WNT-1 expression. These findings
suggested that increased expression of fibrotic and WNT-related
genes may be associated with disease severity and impaired
pulmonary function in IPF.
| Table VCorrelations of clinical parameters
with gene expression levels in patients with idiopathic pulmonary
fibrosis. |
Table V
Correlations of clinical parameters
with gene expression levels in patients with idiopathic pulmonary
fibrosis.
| Correlation |
rs-value | P-value |
|---|
| GAP score vs.
WNT-3a | 0.3890 | 0.0450a |
| DLCO vs.
WNT-3a | 0.4602 | 0.0236a |
| FEV1 (l) vs.
collagen type I | 0.3190 | 0.0257a |
| FEV1 (l) vs.
WNT-3a | 0.3360 | 0.0192a |
| FVC (l) vs.
collagen type I | 0.3420 | 0.0247a |
| Neutrophil count
vs. WNT-1 | 0.4740 | 0.0490a |
| Neutrophil (%) vs.
WNT-1 | 0.5870 | 0.0100a |
Gene expression according to
radiological findings
Gene expression levels were further analyzed
according to radiological features of disease severity, including
alveolar septal fibrosis and honeycombing (Tables VI and VII). Because both parameters were
dichotomous variables, comparisons were performed using
Mann-Whitney U test.
| Table VIComparison of gene expression levels
according to alveolar septal fibrosis in patients with idiopathic
pulmonary fibrosis. |
Table VI
Comparison of gene expression levels
according to alveolar septal fibrosis in patients with idiopathic
pulmonary fibrosis.
| Gene | Alveolar septal
fibrosis (+) | Alveolar septal
fibrosis (-) | P-value |
|---|
| Collagen type
I | 1.405±0.1057 | 0.7106±0.0595 | 0.0165 |
| WNT-2 | 1.806±0.2860 | 0.5324±0.1907 | 0.0496 |
| WNT-4 | 1.461±0.1519 | 0.5756±0.0320 | 0.0016 |
| WNT-6 | 1.751±0.2629 | 0.6701±0.1557 | 0.0071 |
| WNT-7a | 2.431±0.2173 | 0.7674±0.0720 | 0.0004 |
| WNT-7b | 2.622±0.2659 | 0.9811±0.1656 | 0.0044 |
| WNT-10a | 2.750±0.3368 | 0.6435±0.1718 | 0.0486 |
| WNT-10b | 1.891±0.095 | 0.7848±0.1435 | 0.0004 |
| α-SMA | 2.532±0.4148 | 0.4982±0.2909 | 0.0159 |
| Table VIIComparison of gene expression levels
according to honeycombing in patients with idiopathic pulmonary
fibrosis. |
Table VII
Comparison of gene expression levels
according to honeycombing in patients with idiopathic pulmonary
fibrosis.
| Gene | Honeycombing
(+) | Honeycombing
(-) | P-value |
|---|
| WNT-2 | 2.347±0.3509 | 0.5865±0.0887 | <0.0001 |
| WNT-6 | 2.126±0.3516 | 0.8563±0.05 | 0.0002 |
| WNT-7a | 2.891±0.2435 | 1.130±0.1078 | <0.0001 |
| WNT-7b | 3.102±0.3295 | 1.411±0.1292 | 0.0002 |
| WNT-10b | 1.956±0.1447 | 1.432±0.1200 | 0.0154 |
Patients with alveolar septal fibrosis exhibited
significantly higher expression levels of fibrosis- and WNT
signaling-related genes compared with fibrosis-negative patients.
Specifically, the expression levels of collagen type I, WNT-2,
WNT-4, WNT-6, WNT-7a, WNT-7b, WNT-10a, WNT-10b and α-SMA were all
significantly increased in patients with alveolar septal fibrosis
(all P<0.05; Table VI).
Similarly, patients with radiological evidence of
honeycombing demonstrated significantly elevated expression of
WNT-2, WNT-6, WNT-7a, WNT-7b and WNT-10b compared with patients
without honeycombing (all P<0.05; Table VII). These findings indicated
that key fibrotic and WNT pathway-related genes are upregulated in
association with radiological markers of disease severity in
IPF.
Discussion
Within the present study, the majority of patients
were male and >65 years old, and there was no difference between
the descriptive characteristics of the IPF and control groups.
However, patients diagnosed with IPF had decreased functional
parameters compared with the control group. Subsequently, WNT-2,
WNT-4, WNT-6, WNT-7a, WNT-7b, WNT-10a and WNT-10b levels were
higher in patients diagnosed with IPF. Notably, fibrotic markers
(α-SMA and collagen type I) were upregulated in patients with IPF
compared with those in the control group. In addition, WNT
signaling pathway blockers were tested on cells from a patient with
IPF [LL29 (AnHa)] to assess their effects on WNT levels, as well as
their ability to reduce fibrosis and inflammation in these cells.
Wet-lab analyses were carried out under blinded conditions, which
helped reduce potential measurement bias despite the non-randomized
design.
IPF is an idiopathic condition resulting in impaired
lung function and mortality. In a study with 1,001 patients with
IPF, researchers examined how FVC and DLCO changed by looking at
both diagnosed patients and those who were tested for lung
function. As the absolute decline in FVC increased, mortality and
progression to lung transplantation increased (16). A previous study in children with
asthma indicated that WISP1, one of the WNT signaling genes, was
associated with FEV1 and FVC, whereas WNT inhibitory factor-1 was
associated with FVC and FEV1/FVC (17). WNT upregulation and β-catenin
increase have also been associated with decreased FEV1 and chronic
obstructive pulmonary disease (COPD) severity (18). A previous study assessed FEV1 and
FVC values in 15 patients with IPF, 32 patients with COPD and 30
healthy individuals. In patients with IPF, the FEV1 and FVC values
were 69%; in patients with COPD, FEV1 was found to be 49% and FVC
was 81%; and in the control group, FEV1 was 111% and FVC was 114%.
No significant differences were observed between the IPF, COPD and
control groups (19). Notably,
there are limited studies on IPF and PFTs. The present study
demonstrated that patients with IPF had lower FEV1 and FVC values
compared with those in the control group, although this was not
statistically significant.
WNT signaling and its effector β-catenin signaling
pathway are known to serve a role in mammalian lung development and
organogenesis (13). Additionally,
MMP-7, which is activated by β-catenin, has previously been
recognized as a key factor in the development of pulmonary fibrosis
(19). Furthermore, researchers
have observed the involvement of the WNT pathway in the
pathogenesis of some fibrosis-associated diseases. In a study of 20
patients with IPF, researchers found that β-catenin was present in
the nuclei of spindle cells that make up fibroblast foci in 16 out
of the 20 samples (20). In the
present study, alveolar septal thickness (90.9%) and/or honeycomb
appearance (60.6%) were present in a number of patients with IPF,
whereas they were not present in the control group. The present
study found that the levels of some WNTs were higher in patients
with alveolar septal thickness and honeycombing than those
without.
WNT-5A and WNT-5B levels have been reported to
increase in the lungs of aged mice (age, 8-12 weeks) independently
of β-catenin, compared with in young mouse lungs, and are
considered to be responsible for ageing (21). Previous study have shown an
increase in WNT-5A levels in IPF and experimental lung fibrosis,
which in turn contributes to the proliferation of primary human
lung fibroblasts (10). In a study
conducted in patients with scleroderma (n=85), a disease
characterized by fibrosis, WNT-1, WNT-10b, WNT-2 and WNT-6 gene
expression levels were increased compared with those in the healthy
control group (22). in the same
study, WNT-1 and WNT-2 were found to be higher in scleroderma with
organ involvement (22). In the
present study, with the exception of WNT-1 and WNT-3, other WNT
levels (WNT-2, WNT-4, WNT-6, WNT-7a, WNT-7b, WNT-10a and WNT-10b)
were increased in patients with IPF compared with those in the
control group.
A previous study reported that a number of patients
with coronavirus disease 2019 (COVID-19) develop lung fibrosis
after the final stages of the disease (23). Fibrosis after COVID-19 has been
reported to be associated with TGF-β1 activation. The medication
pirfenidone helps reduce the buildup of inflammatory cells, stops
fibroblasts from multiplying and prevents the formation of excess
ECM. In addition, it has been observed that pirfenidone modulates
signaling pathways such as WNT and β-catenin, which serve a role in
the pathogenesis of pulmonary fibrosis after COVID-19, and it is
considered that these properties may alleviate fibrosis after
COVID-19(23).
In a study conducted with sputum biomarkers in IPF,
COPD and healthy volunteers, it was shown that TGF-β levels were
upregulated in patients with IPF compared with in healthy subjects.
IL-6 levels were also shown to be increased in patients with COPD
(19). It has been demonstrated
that deposition of collagen type I in the ECM is a characteristic
feature of IPF; α-SMA expression, a biomarker for myofibroblast
differentiation, is increased in fibrotic lung tissue; and TGF-β is
an important inducer in the initiation and maintenance of lung
fibrosis (24). In line with
previous research, the present study demonstrated that levels of
collagen type I and α-SMA were higher in the IPF group compared
with those in the control group.
One study has documented that the WNT/β-catenin
pathway is aberrantly activated in IPF, and serves a key role in
myofibroblast differentiation and activation (25). TGF-β acts on fibroblasts, with a
previous study showing that TGF-β interacts with non-canonical WNT
pathways (25). There are limited
antifibrotic drugs currently used in the treatment of IPF, and
these drugs are known to act through TGF-β and tyrosine kinases
(3). A previous study reported
that nintedanib, one of the therapies used to treat IPF, can
attenuate myofibroblast activation by inhibiting the expression of
genes downstream of WNT signaling (26). Therefore, inhibitors that act on
WNT pathways, which interact with the pathways of currently used
drugs, may be promising for the management of this rare, rapidly
progressive disease.
In a study using human-derived fibrotic lung
fibroblast cells (CCL-191), the PORCN inhibitors LGK-974 and
ETC-159 were used to examine their therapeutic effects on fibrosis.
CCL-191 cells treated with LGK-974 and ETC-159 exhibited decreased
levels of TGFβ-1, α-SMA, collagen type I, WNT-1, WNT-3a, WNT-10a
and WNT-10b. In the same study, WNT-6 was notably decreased only in
the group treated with ETC-159(27). In the present study, WNT-6, WNT-7a,
WNT-10a and WNT-10b levels were decreased when ETC-159 was
administered to LL29 (AnHa) cells, whereas WNT-6, WNT-10a, WNT-10b
levels were decreased when LGK-974 was administered.
Although the present in vitro findings
support the antifibrotic and anti-inflammatory potential of WNT
inhibition, translation into clinical practice must be approached
with caution. WNT signaling regulates key physiological processes
such as stem cell renewal, bone formation and epithelial
homeostasis (13). Thus, systemic
inhibition may carry risks of adverse effects, including
gastrointestinal toxicity, impaired tissue regeneration or
metabolic disturbances. An early-phase clinical trial of PORCN
inhibitors (including ETC-159 and LGK-974) reported dose-limiting
toxicities, underscoring the need for careful dosing and patient
selection (28). However, the
controlled microenvironment of cell culture does not fully capture
the complexity of IPF pathology in vivo. Therefore, while
the present results highlight WNT signaling as a promising
therapeutic target, further preclinical validation and clinical
safety studies are key before considering widespread application to
patients with IPF.
New treatment options are required for the
management of IPF, which is a rare disease with a short life
expectancy. Developing new treatment options is key for improving
patient outcomes. The elevated levels of fibrotic markers (α-SMA
and collagen type I) alongside pro-inflammatory cytokines (IL-6 and
IL-1β) in patients with IPF emphasize the interconnected roles of
fibroblast activation and chronic inflammation in disease
progression. These findings reinforce the concept that IPF
pathogenesis is not solely a fibrotic process but also driven by
sustained inflammatory responses. Clinically, such biomarkers may
serve as indicators of disease severity and progression, aiding in
patient stratification and monitoring. Moreover, their modulation
by WNT signaling suggests therapeutic potential in targeting
WNT-driven fibroblast activation while simultaneously attenuating
pro-inflammatory cytokine production. Together, these findings
suggest a dual therapeutic strategy capable of attenuating IPF
progression, consistent with emerging treatment paradigms that
combine antifibrotic and anti-inflammatory interventions to improve
clinical outcomes (28,29).
A limitation of the present study was the relatively
small sample size (33 patients with IPF and 23 controls), which
reduces statistical power and limits the generalizability of the
present findings. However, significant differences in WNT signaling
markers were observed, consistent with previous reports (8-11),
suggesting the robustness of the observed effects despite the
restricted cohort. Future studies with larger populations are
warranted to validate these results. In particular, multicenter
designs would improve external validity by including more diverse
patient groups, thereby strengthening the translational relevance
of targeting WNT signaling in IPF. As IPF is a disease of advanced
age, additional comorbidities were not addressed in detail,
potentially an additional limitation to the present study. However,
patients with a history of infections that could result in
fibrosis, such as COVID-19 were specifically excluded. An
additional limitation may be that IPF was studied in a single
center within the present study and since IPF is a rare disease,
collecting patients from a number of centers may have been more
desirable.
In conclusion, IPF is a rare disease with no
definitive cure and fibrosis is involved in its pathophysiology.
Both TGF-β and WNT signaling are responsible for the regulation of
myofibroblast differentiation and cellular senescence, which have
been proposed as targets for IPF therapy. Therefore, it is
considered that the present study, which investigated the impact of
WNT inhibitors, could potentially contribute to the treatment of
IPF. Future studies, particularly those involving human materials,
are needed to evaluate the efficacy of WNT inhibitors. Further
in vivo and in vitro studies are necessary for the
development of a new therapeutic product.
Acknowledgements
Not applicable.
Funding
Funding: Kütahya Health Sciences University Scientific Research
Projects Coordination Unit supported the present study (grant no.
TSA-2024-196).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
İK contributed to the investigation, methodology,
funding acquisition, data curation and conceptualization. Also, İK
contributed to writing, reviewing and editing the manuscript. AKS
contributed to formal analysis, methodology, investigation,
providing supervision, and reviewing and editing the manuscript. MG
contributed to providing supervision of wet-lab experiments. SEP,
FM, ÜTE and MD provided acquisition and interpretation of data. İK
and AKS confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Ethics approval for the present study was obtained
by the Kütahya Health Sciences University Faculty of Medicine
Non-Interventional Ethics Committee in May 2024 (approval no.
2024/06-23). Written informed consent was obtained from all
participants prior to their inclusion in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Raghu G, Remy-Jardin M, Richeldi L,
Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM,
Martinez FJ, et al: Idiopathic pulmonary fibrosis (an update) and
progressive pulmonary fibrosis in adults: An official
ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care
Med. 205:e18–e47. 2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Maher TM, Bendstrup E, Dron L, Langley J,
Smith G, Khalid JM, Patel H and Kreuter M: Global incidence and
prevalence of idiopathic pulmonary fibrosis. Respir Res.
22(197)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Cottin V, Tomassetti S, Valenzuela C,
Walsh SLF, Antoniou KM, Bonella F, Brown KK, Collard HR, Corte TJ,
Flaherty KR, et al: Integrating clinical probability into the
diagnostic approach to idiopathic pulmonary fibrosis: An
international working group perspective. Am J Respir Crit Care Med.
206:247–259. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Barratt SL, Creamer A, Hayton C and
Chaudhuri N: Idiopathic pulmonary fibrosis (IPF): An overview. J
Clin Med. 7(201)2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chambers RC and Mercer PF: Mechanisms of
alveolar epithelial injury, repair, and fibrosis. Ann Am Thorac
Soc. 12 (Suppl 1):S16–S20. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chanda D, Otoupalova E, Smith SR,
Volckaert T, De Langhe SP and Thannickal VJ: Developmental pathways
in the pathogenesis of lung fibrosis. Mol Aspects Med. 65:56–69.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Schafer MJ, White TA, Iijima K, Haak AJ,
Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y,
et al: Cellular senescence mediates fibrotic pulmonary disease. Nat
Commun. 8(14532)2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Königshoff M, Balsara N, Pfaff EM, Kramer
M, Chrobak I, Seeger W and Eickelberg O: Functional Wnt signaling
is increased in idiopathic pulmonary fibrosis. PLoS One.
3(e2142)2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Newman DR, Sills WS, Hanrahan K, Ziegler
A, Tidd KM, Cook E and Sannes PL: Expression of WNT5A in idiopathic
pulmonary fibrosis and its control by TGF-β and WNT7B in human lung
fibroblasts. J Histochem Cytochem. 64:99–111. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Martin-Medina A, Lehmann M, Burgy O,
Hermann S, Baarsma HA, Wagner DE, De Santis MM, Ciolek F, Hofer TP,
Frankenberger M, et al: Increased vesicles mediate WNT5A signaling
in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
198:1527–1538. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Königshoff M, Kramer M, Balsara N, Wilhelm
J, Amarie OV, Jahn A, Rose F, Fink L, Seeger W, Schaefer L, et al:
WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in
mice and is upregulated in humans with idiopathic pulmonary
fibrosis. J Clin Invest. 119:772–787. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Berschneider B and Königshoff M: WNT1
inducible signaling pathway protein 1 (WISP1): A novel mediator
linking development and disease. Int J Biochem Cell Biol.
43:306–309. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Baarsma H and Königshoff M: ‘WNT-er is
coming’: WNT signalling in chronic lung diseases. Thorax.
72:746–759. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mahler DA and Wells CK: Evaluation of
clinical methods for rating dyspnea. Chest. 93:580–586.
1988.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Oldham JM, Neely Wojdyla DM, Gulati M, Li
P, Patel DC, Palmer SM and Todd JL: IPF-PRO Registry Investigators.
Changes in lung function and mortality risk in patients with
idiopathic pulmonary fibrosis. Chest. 168:415–422. 2025.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sharma S, Tantisira K, Carey V, Murphy AJ,
Lasky-Su J, Celedón JC, Lazarus R, Klanderman B, Rogers A,
Soto-Quirós M, et al: A role for Wnt signaling genes in the
pathogenesis of impaired lung function in asthma. Am J Respir Crit
Care Med. 181:328–336. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Carlier FM, Dupasquier S, Ambroise J,
Detry B, Lecocq M, Biétry-Claudet C, Boukala Y, Gala JL, Bouzin C,
Verleden SE, et al: Canonical WNT pathway is activated in the
airway epithelium in chronic obstructive pulmonary disease.
EBioMedicine. 61(103034)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guiot J, Henket M, Corhay JL, Moermans C
and Louis R: Sputum biomarkers in IPF: Evidence for raised gene
expression and protein level of IGFBP-2, IL-8 and MMP-7. PLoS One.
12(e0171344)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chilosi M, Poletti V, Zamò A, Lestani M,
Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, et
al: Aberrant Wnt/beta-catenin pathway activation in idiopathic
pulmonary fibrosis. Am J Pathol. 162:1495–1502. 2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wu X, van Dijk EM, Ng-Blichfeldt JP, Bos
IST, Ciminieri C, Königshoff M, Kistemaker LEM and Gosens R:
Mesenchymal WNT-5A/5B signaling represses lung alveolar epithelial
progenitors. Cells. 8(1147)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Koçak A, Harmancı D, Güner Akdoğan G and
Birlik M: Relationship of Wnt pathway activity and organ
involvement in scleroderma types. Int J Rheum Dis. 23:1558–1567.
2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Al-Kuraishy HM, Batiha GE, Faidah H,
Al-Gareeb AI, Saad HM and Simal-Gandara J: Pirfenidone and
post-Covid-19 pulmonary fibrosis: Invoked again for realistic
goals. Inflammopharmacology. 30:2017–2026. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ding H, Chen J, Qin J, Chen R and Yi Z:
TGF-β-induced α-SMA expression is mediated by C/EBPβ acetylation in
human alveolar epithelial cells. Mol Med. 27(22)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cohen ML, Brumwell AN, Ho TC, Garakani K,
Montas G, Leong D, Ding VW, Golden JA, Trinh BN, Jablons DM, et al:
A fibroblast-dependent TGF-β1/sFRP2 noncanonical Wnt signaling axis
promotes epithelial metaplasia in idiopathic pulmonary fibrosis. J
Clin Invest. 134(e174598)2024.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li X, Liu X, Deng R, Gao S, Yu H, Huang K,
Jiang Q, Liu R, Li X, Zhang L, et al: Nintedanib inhibits
Wnt3a-induced myofibroblast activation by suppressing the
Src/β-catenin pathway. Front Pharmacol. 11(310)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Koçak A, Gülle S and Birlik M: Porcupine
inhibitors LGK-974 and ETC-159 inhibit Wnt/β-catenin signaling and
result in inhibition of the fibrosis. Toxicol In Vitro.
104(105986)2025.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Shah K, Panchal S and Patel B: Porcupine
inhibitors: Novel and emerging anti-cancer therapeutics targeting
the Wnt signaling pathway. Pharmacol Res.
167(105532)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Rodon J, Argilés G, Connolly RM,
Vaishampayan U, de Jonge M, Garralda E, Giannakis M, Smith DC,
Dobson JR, McLaughlin ME, et al: Phase 1 study of single-agent
WNT974, a first-in-class porcupine inhibitor, in patients with
advanced solid tumours. Br J Cancer. 125:28–37. 2021.PubMed/NCBI View Article : Google Scholar
|