Introduction
Spinocerebellar ataxia type 14 (SCA14) is a rare
autosomal dominant neurodegenerative disorder caused by mutations
in the PRKCG gene that encodes protein kinase Cγ (PKCγ).
SCA14 accounts for 1-4% of autosomal dominant cerebellar ataxias
overall, although the prevalence varies among cohorts and is
possibly underestimated due to challenges with detection (1).
The clinical presentation of SCA14 is variable, with
the age of onset ranging from early childhood to late adulthood,
typically between the third and fifth decades. The disease can be
subdivided into pure and complex phenotypes. The isolated variant
is marked by slowly progressive cerebellar ataxia with or without
brisk reflexes. By contrast, the complex variant includes ataxia
with other neurological features (1). In certain patients, episodic ataxia
is observed instead of progressive worsening, which is
contradictory to the assumption that SCA14 has a relentlessly
worsening course (2). Certain
patients initially manifest with task-specific dystonia, namely
writer's cramp or focal dystonia, and then develop frank ataxia
(3). Furthermore, certain patients
also experience sensory disturbances such as burning paresthesia
and proprioceptive loss, which suggest simultaneous peripheral
nervous system involvement along with cerebellar degeneration
(4). While it is uncommon,
Parkinsonism features have also been reported, which indicate
overlapping neurodegenerative processes (5). These findings establish the
importance of genetic testing in unexplained ataxia or movement
disorder presentation to prevent misdiagnosis.
Neuroimaging of SCA14 is mostly characterized by
diffuse cerebellar atrophy involving the vermis and cerebellar
hemispheres, worsening with disease severity. The brainstem is
typically spared, although mild pontine atrophy has occasionally
been reported. T2 hyperintensity of dentate nuclei is also
observed, as in other inherited ataxias (2). Advanced imaging, such as
fluorodeoxyglucose positron emission tomography, has been found to
reveal early microstructural as well as metabolic changes in the
cerebellum even before significant atrophy can be identified
(5). These findings indicate
certain imaging features that permit early diagnosis and
differentiation of SCA14 from other cerebellar ataxias.
The PRKCG gene codes for PKCγ, a
neuron-specific isoform with predominant expression in Purkinje
cells of the cerebellum. PKCγ contributes to regulation of synaptic
plasticity, intracellular signaling and motor coordination, mainly
via processes of long-term depression and postnatal synaptic
pruning. Disruption in these pathways results in Purkinje cell
dysfunction and cerebellar ataxia (6).
Genetic studies have revealed that most of the
disease-causing mutations in PRKCG are missense mutations, although
small deletions, insertions and splicing mutations have also been
reported (1). Most mutations are
found in the C1 and C2 regulatory domains, with the majority in C1,
which is essential for diacylglycerol (DAG) binding and membrane
translocation. Mutations in this region disrupt autoinhibitory
regulation, leading to increased basal kinase activity, impaired
protein degradation and persisting aberrant signaling. By contrast,
mutations in the catalytic domain are less common but have been
linked to more complex clinical features, further augmenting the
genetic and phenotypic heterogeneity of SCA14 (6-9).
The present case report describes a 68-year-old man
who carried a PRKCG mutation (c.424T>G; p.C142G). SCA14 with
PRKCG mutation has been reported in multiple countries; however,
the codon 142 variant is rare, previously documented only in two
families in Denmark and Japan (1,3). The
current report highlights features that are atypical in SCA14,
including late onset, slow progression and sensory impairment in
the present patient. The current study also discusses possible
mechanisms of disease progression, whether anticipation is present
or not, and differences from the pathogenic mechanisms observed in
triplet repeat SCAs. The present article emphasizes the importance
of genetic testing in undiagnosed ataxia and provides an overview
of genotype-phenotype associations, disease mechanisms and possible
targeted therapies.
Case report
A 72-year-old male patient presented to Changhua
Christian Hospital (Changhua City, Taiwan) in November 2020 with
progressive gait instability that started when the patient was 40
years old. The patient had mild unsteadiness while walking, which
worsened over the years. On the first visit, a neurological
examination showed an unsteady wide-based gait and a positive
Romberg sign with worsening instability while the eyes of the
patient were closed. The patient also had bilateral mild intention
tremor and dysmetria on the finger-nose-finger test. The Scale for
the Assessment and Rating of Ataxia (SARA) score (10) of the patient was 8.5, with most
impairment in gait and stance. Over the next few months, the
patient developed mild dysarthria, slowed saccadic eye movements
and numbness in the lower limbs, which was more pronounced on the
left side. A sensory examination showed reduced light touch and
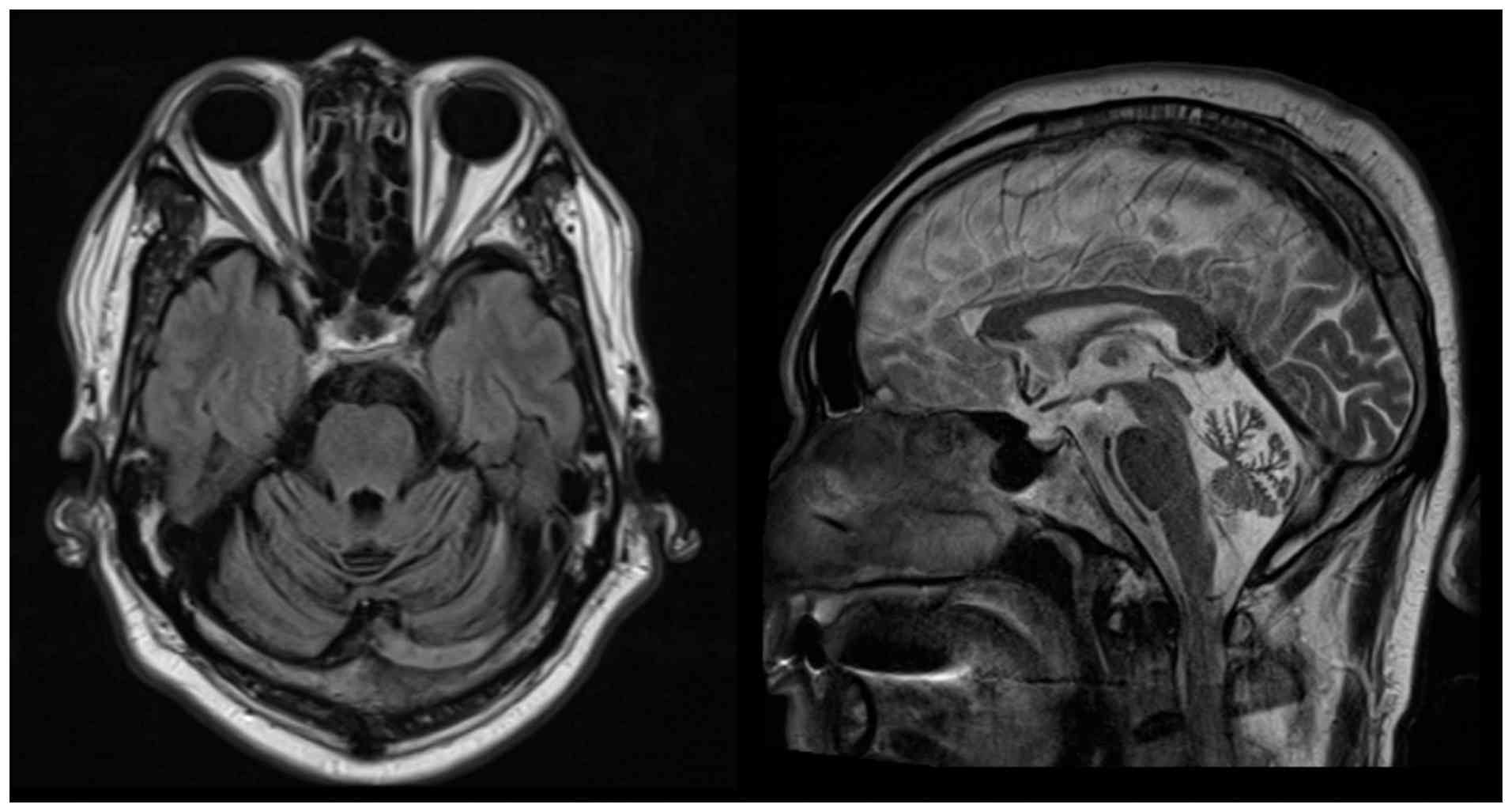
pinprick sensation in the left lower limb. Brain MRI showed severe
diffuse cerebellar atrophy with prominent cerebellar folia sulci
(Fig. 1).
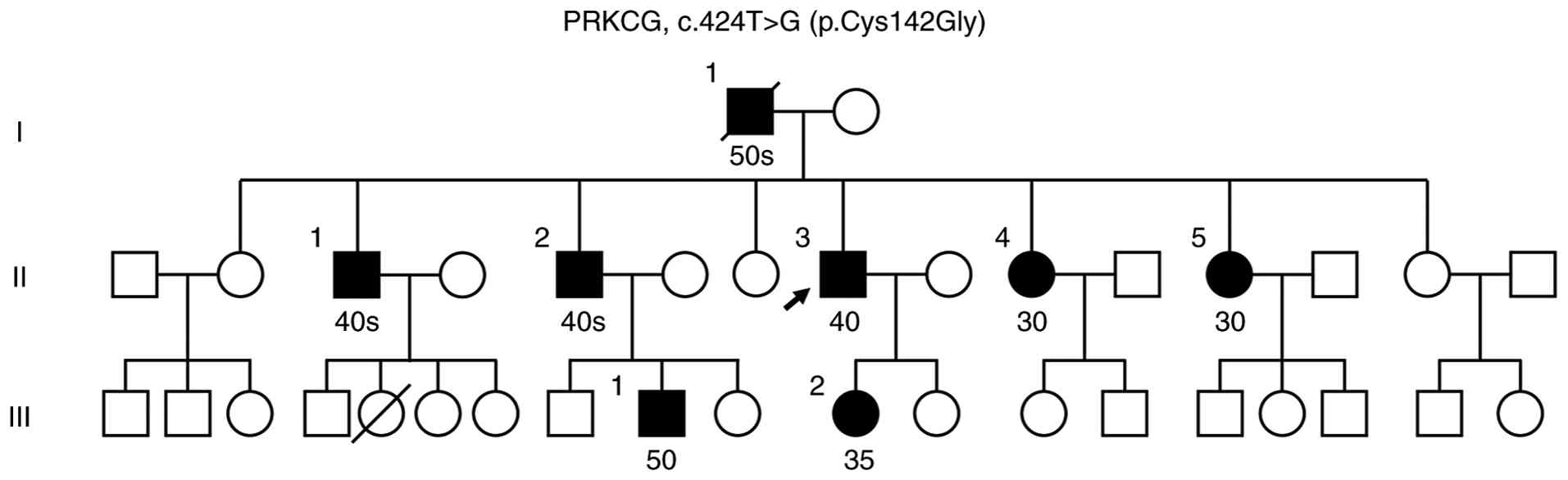
The family history of the patient was notable for an
autosomal dominant pattern of progressive gait ataxia (Fig. 2). The father of the patient had
developed similar symptoms in his 50s. Furthermore, two elder
brothers, two younger sisters, a daughter and a nephew all
developed unsteady gait between 35 and 50 years of age. Given the
strong family history, a genetic evaluation was pursued. An initial
genetic panel for SCA1, SCA2, SCA3 and SCA6 was negative. Given the
high suspicion of a hereditary ataxia, targeted sequencing was
performed using the Illumina TruSight One Sequencing Panel v1.1
(Illumina, Inc.), which enriches for the coding regions of 4,814
clinically relevant genes encompassing ~12 Mb of the human genome.
Genomic DNA was extracted from peripheral blood and quantified
using a Qubit fluorometer (Thermo Fisher Scientific, Inc.). Library
preparation was performed following the manufacturer's instructions
(document no. 15046431 v03). Briefly, 50 ng of input DNA underwent
Nextera transposome-mediated tagmentation to generate
adapter-tagged libraries. Indexed libraries were pooled and
hybridized with biotin-labeled oligonucleotide probes. The targeted
regions were enriched by streptavidin bead capture and a second
hybridization-capture cycle was performed to maximize on-target
specificity. Enriched libraries were sequenced on an Illumina
NextSeq 6000 platform using P3 Reagents (2x150 bp paired-end
reads), achieving a mean depth of ≥200x across targeted regions.
Sequence reads were aligned to the Genome Reference Consortium
Human Build 38 (GRCh38) reference assembly (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.26/).
This revealed a heterozygous missense mutation in PRKCG
(c.424T>G; p.C142G; Chr19q13; exon 5). To identify the PRKCG
(SCA14 c.424T>G) mutation, genomic DNA was amplified via
polymerase chain reaction (PCR) using specific primers (forward,
5'-AAGTTCCGCCTGCATAGCTA-3' and reverse, 5'-GGATCTCATCTGCTGTGGGA-3')
on an MJ Research Thermal Cycler (MJ Research PTC-200, Inc.). The
PCR program consisted of an initial denaturation at 95˚C for 5 min,
followed by 35 cycles of 95˚C for 1 min, 56˚C for 1 min and 72˚C
for 1 min, with a final extension at 72˚C for 5 min. The resulting
353 bp amplicons were subsequently analyzed by Sanger sequencing.
Purified amplicons were subjected to cycle sequencing in a total
reaction volume of 10 µl containing purified PCR product, BigDye
Terminator Ready Reaction Mix (BigDye™ Terminator v3.1 Cycle
Sequencing Kit; Thermo Fisher Scientific, Inc.), sequencing buffer
and a forward sequencing primer (5'-AAGTTCCGCCTGCATAGCTA-3'). Cycle
sequencing was performed with an initial denaturation at 95˚C for 1
min, followed by 25 cycles of denaturation at 95˚C for 10 sec,
annealing at 50˚C for 5 sec and extension at 60˚C for 4 min.
Following cycle sequencing, reaction products were purified by
ethanol/EDTA precipitation to remove unincorporated dye
terminators. Purified sequencing products were resuspended in Hi-Di
formamide, heat-denatured at 95˚C and immediately cooled on ice
prior to analysis. Capillary electrophoresis was performed using an
Applied Biosystems 3730xl DNA Analyzer (Thermo Fisher Scientific,
Inc.). All procedures were conducted according to the
manufacturer's instructions. Sequencing chromatograms were analyzed
using Sequencing Analysis software (version 5.4; Applied
Biosystems; Thermo Fisher Scientific, Inc.) and aligned to
reference sequences for variant identification. This confirmed the
diagnosis of SCA14.
The patient's younger sister, who had been
experiencing gait ataxia since she was 30 years old, also presented
with progressive wide-based unsteady gait and poor tandem walking.
Additionally, the sister had dysphonia, episodic choking, impaired
lateral gaze with slow saccades, dysmetria in both upper and lower
limbs and ideomotor apraxia. A cognitive assessment showed poor
calculation abilities and mild language impairment. Brain MRI at
the age of 64 years showed cerebellar atrophy with focal gliotic
changes in the left high frontal parasagittal region. Additionally,
the younger sister and daughter of the patient were later diagnosed
with SCA14. Both of them had developed a mild unsteady gait in
their 30s. Brain MRI also demonstrated cerebellar atrophy in both
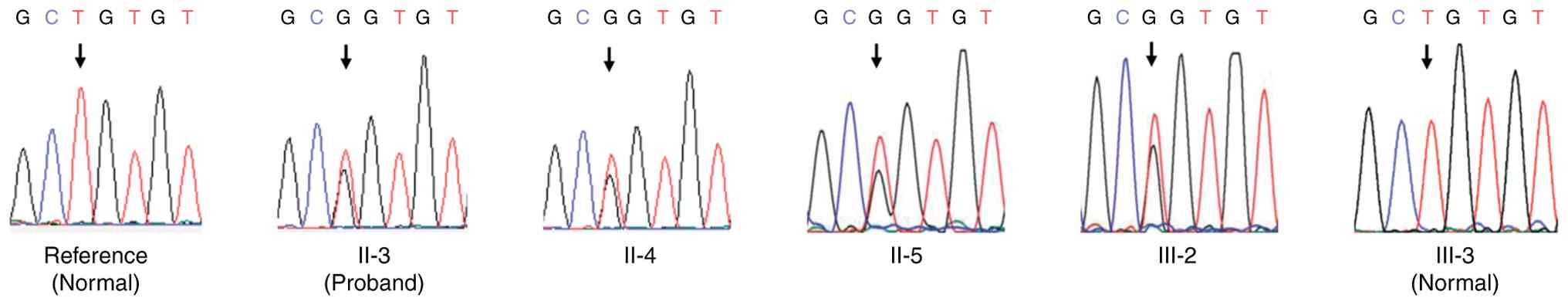
individuals. Genetic testing via Sanger sequencing verified that
all affected family members shared the identical PRKCG
mutation (c.424T>G; p.C142G) (Fig.
3).
Discussion
To the best of our knowledge, the current case
report is the first report of the PRKCG c.424T>G
(p.C142G) mutation in a Han Chinese patient, expanding the
knowledge regarding both the geographic and ethnic distribution of
this rare variant. In contrast to typical SCA14 with gradually
progressive ataxia within the average age of onset (mean, 30.6
years; range, 3-66 years) (1), the
present patient had relatively delayed-onset symptoms with sensory
deficits of numbness and altered pinprick sensation reflecting
extra-cerebellar involvement. The SARA, an 8-item standardized and
widely used clinical scale to quantify the severity of ataxia,
ranges from 0 to 40, and a higher score indicates greater severity
(10). A prior cohort study of 17
patients with SCA14 reported a mean SARA score of 13.1 at the final
assessment, indicative of moderate severity (1). By comparison, the present patient's
score of 8.5 suggests a milder degree of ataxia and is consistent
with a low fall risk based on a functionality and balance study
(11). The severe cerebellar
atrophy observed in the current patient is uncommon, as in most
cases, cerebellar abnormalities are mild or moderate.
The present case is notable due to the rare location
of the mutation in PRKCG (c.424T>G; p.C142G), previously
mentioned in only one Danish family as a novel missense mutation
(1). Another case study from Japan
reported the same amino acid residue of the PRKCG gene
(c.424 T>A; p.C142S) (3).
Whereas the current patient had late-onset ataxia, pronounced
sensory impairment and cerebellar atrophy, the Japanese patient
(p.C142S) had focal dystonia with writer's cramp and milder
cerebellar involvement. By contrast, the Danish family presented
with a wider onset age (3-48 years) and a milder course.
Comparison of all reported PRKCG codon 142 mutation
cases (Table I), including the
present 4 Taiwanese patients, 6 Danish family members and 1
Japanese case, showed that all affected individuals had cerebellar
ataxia with limb involvement in a typically long disease duration,
implying a slowly progressive course. None of the patients
exhibited Parkinsonism and only a few had myoclonus, dystonia or
peripheral neuropathy. Oculomotor findings such as broken-up
pursuit and slowness of saccades were common, while dysarthria,
cognitive dysfunction and upper motor neuron signs were variable.
Notably, the sister of the current patient had a relatively earlier
onset as a complex phenotype, including mental decline, dysphasia,
oculomotor impairment and bulbar symptoms. This phenotypic
heterogeneity with the same target codon reflects the complexity of
genotype-phenotype associations in SCA14, and implies that certain
amino acid substitutions, individual modifiers or environmental
conditions are related to disease severity and expression.
 | Table IClinical comparison of PRKCG codon 142
variants in the Taiwanese family of the present study (n=4) and
previously reported Danish (n=6) and Japanese (n=1) families. |
Table I
Clinical comparison of PRKCG codon 142
variants in the Taiwanese family of the present study (n=4) and
previously reported Danish (n=6) and Japanese (n=1) families.
| Case | Country | PRKCG mutation | Age at onset,
years | Symptom at onset | Disease duration,
years | SARA score | Phenotype | Limb ataxia | Eye signs | Dysarthria | Parkinsonism | Dystonia | Myoclonus | Peripheral
neuropathy | Pyramidal
syndrome | Other | Cerebellar atrophy on
MRI |
|---|
| Present study | Taiwan | | | | | | | | | | | | | | | | |
|
II-3
(Proband) | | c.424T>G,
p.C142G | 40s | Mild unsteady
gait | 32 | 8.5 | Complex | + | Slowed saccadic eye
movements | + | - | - | - | +, Hypoesthesia at
LLE | - | Positive Romberg
sign, wide-based gait | Severe diffuse
cerebellar atrophy with prominent cerebellar folia sulci |
|
II-4 | | c.424T>G,
p.C142G | 30 | Mild unsteady
gait | 39 | 7 | Pure | + | Multiple directions
of nystagmus | + | - | - | - | - | - | - | Severe diffuse
cerebellar atrophy with prominent cerebellar folia sulci |
|
II-5 | | c.424T>G,
p.C142G | 30 | Gait ataxia | 34 | 8 | Complex | + | Impaired lateral gaze
with slow saccades | + | - | - | - | - | - | Episodic choking,
ideomotor apraxia | Cerebellar atrophy
with focal gliotic changes in left high frontal parasagittal
region |
|
III-2 | | c.424T>G,
p.C142G | 35 | Mild unsteady
gait | 8 | 3 | Pure | + | - | - | - | - | - | - | - | - | Atrophy of brainstem,
cerebellum, and pituitary gland |
| Chelban et al
(1) | Denmark | | | | | | | | | | | | | | | | |
|
Case 1 | | c.424T>G,
p.C142G | 39 | Gait and balance
impairment | 15 | 9.5 | Complex | + | Broken-up
pursuit | + | - | - | - | - | + | - | NA |
|
Case 2 | | c.424T>G,
p.C142G | 48 | Gait impairment | 6 | 10 | Complex | + | Unstable pursuit | + | - | - | - | - | + | Urge incontinence,
mild impairment of vibration sense | NA |
|
Case 3 | | c.424T>G,
p.C142G | 6 | Upper limbs postural
tremor precedes gait and balance impairment | 51 | 12 | Complex | + | Broken-up
pursuit | + | - | - | - | - | - | Urge incontinence,
decreased reflexes UL and LL, postural hand and head tremor | NA |
|
Case 4 | | c.424T>G,
p.C142G | 6 | Limb ataxia | 23 | NA | Pure | + | Slow pursuit | - | - | - | - | - | - | Increased patellar
reflexes | Mild |
|
Case 5 | | c.424T>G,
p.C142G | 30 | Balance
impairment/tremor/limb ataxia | 6 | 9 | Complex | + | Broken-up
pursuit | + | - | - | - | - | - | Increased reflexes
UL, postural hand tremor | NA |
|
Case 6 | | c.424T>G,
p.C142G | 3 | Limb ataxia | 1 | NA | Complex | + | - | + | - | - | Occasional
myoclonic truncal jerks | - | - | - | NA |
| Ito et al
(3) | Japan | c.424T>A,
p.C142S | 36 | Writer's cramp
precedes gait and limb ataxia for four years | 11 | 11 | Complex | + | Saccadic
pursuit | + | - | Writer's cramp | | - | - | Mild cognitive
impairment on memory, fluency, and language | Cerebellar atrophy
without apparent changes in the basal ganglia |
The PRKCG (c.424T>G; p.C142G) variant is
best categorized as ‘likely pathogenic’ according to the American
College of Medical Genetics and Genomics guideline from
2015(12). This was supported by
absence from population databases (PM2), presence in a known
functional hotspot (PM1), consistent deleterious predictions from
multiple computational predictors (PP3) and co-segregation in seven
family members across generations (PP1).
In addition, the present patient also meets PM5
based on a previously reported pathogenic missense variant,
p.C142S. Located within the C1 regulatory domain of PKCγ, C142S has
been shown to cause abnormal protein conformation, reduced kinase
activity and disrupted MAPK signaling in functional studies,
confirming its pathogenicity (8).
Clinical segregation data from a 2024 case report have also
confirmed the disease association (3). With the combination of PS3, PM1, PM2,
PP3 and PP1, the evidence supports the classification of p.C142S as
pathogenic, which fulfills PM5 for the p.C142G variant of the
current patient.
The PKCγ C1 domain possesses two zinc-finger motifs,
C1A and C1B, which are both functional DAG-binding modules and
contribute equally to ligand recognition and membrane association.
The C142G mutation with cysteine-to-glycine substitution undermines
zinc coordination and destabilizes the C1B fold due to the loss of
structural restraints. This leads to impaired DAG binding affinity,
membrane recruitment specificity and general protein stability that
promotes misfolding (13).
The structural disturbance fundamentally alters the
regulatory mechanisms of PKCγ. Although the pathogenic process of
SCA14-associated PKCγ mutations is uncertain, post-mortem and
induced pluripotent stem cell studies have suggested a dual process
involving loss of PKCγ function at the plasma membrane in
combination with gain-of-function effects due to hyperactivated and
mislocalized PKCγ with impaired autophagy signaling (14). The latter mechanism may
incapacitate autoinhibitory regulation, partially enhancing basal
kinase activity rather than causing uncontrolled hyperactivation,
which leads to disruption of synaptic signaling cascades in
cerebellar Purkinje cells (7). The
mutant PKCγ-C142G protein probably exhibits gain-of-function
characteristics that activate MAPK pathways and affect downstream
effectors essential for synaptic plasticity (8,15).
This results in chronic, mild cellular stress that accumulates over
time without inducing rapid neuronal death. As opposed to
polyglutamine (polyQ) ataxias, in which the mutant proteins
aggregate into insoluble, highly toxic particles causing
neurodegeneration, PKCγ aggregation in SCA14 is limited and less
pathogenic in vivo (13).
Mutant PKCγ also leads to aberrant regulation of
calcium homeostasis and oxidative equilibrium, but these changes
may progress gradually with cumulative stressors. This overloads
protein quality control systems, including both
ubiquitin-proteasome and autophagy pathways, eventually causing
chronic activation of the unfolded protein response without
triggering immediate cell death (13,15).
Together, these mechanisms are responsible for the relatively slow
course of SCA14 compared with that of the more virulent polyQ
ataxias.
SCA14 is notable for its slow course. Most patients
preserve mobility and independence for decades and reach a normal
lifespan. Given the autosomal dominant inheritance of SCA14,
genetic counseling and analysis allows for early detection of
individuals in at-risk pedigrees. Additionally, the location of the
mutation and the consequent amino acid substitution may help to
predict the clinical manifestation and disease trajectory. Although
no disease-modifying therapy is currently available, early genetic
diagnosis provides prognostic clarity and reduces the psychological
burden on the patient and family. Phenotypic differences reflect
the individualized treatment strategies, including targeted
rehabilitation and symptom-based support, which are critical for
managing diverse clinical presentations and improving the quality
of life of patients.
The findings demonstrate the utility of genetic
testing in SCA14 diagnosis, particularly in atypical presentations,
and suggest the need for future functional studies to confirm its
pathogenic mechanism. Although the c.424T>G (p.C142G) mutation
is a recognized variant, this is the first SCA14 case reported in a
Han Chinese patient. Its identification not only expands the
knowledge regarding the ethnic and geographic spread of this
mutation but also broadens the understanding of the clinical and
genetic landscape of SCA14 in East Asian populations.
Acknowledgements
The authors extend their thanks to Mr. Wen-Hsien
(Bryce) Lin from The Genetics Generation Advancement Corporation,
Taiwan for his bioinformatics support and insightful guidance in
the analysis and interpretation of genetic variants.
Funding
Funding: The study was funded by a research grant from Changhua
Christian Hospital (Changhua, Taiwan) (grant no.
113-CCH-IRP-065).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HH, CJL and CSL conceived and designed the study. HH
and CJL analyzed and interpreted the data and drafted the
manuscript. WC and HC collected the samples and contributed to data
acquisition and analysis. WC and CSL confirm the authenticity of
all the raw data. All authors critically revised the manuscript and
have read and approved the final version.
Ethics approval and consent to
participate
All procedures were approved by the Independent
Ethics Committee of Changhua Christian Hospital (Changhua, Taiwan)
(CCH-IRB approval no. 251222; approval date: December 21, 2025).
Written informed consent was obtained from all participants,
including genetic testing.
Patient consent for publication
Written informed consent for publication was
obtained from the patients, including consent to publish anonymized
clinical details, genetic testing results and accompanying imaging
data (including brain MRI images).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chelban V, Wiethoff S, Fabian-Jessing BK,
Haridy NA, Khan A, Efthymiou S, Becker EBE, O'Connor E, Hersheson
J, Newland K, et al: Genotype-phenotype correlations, dystonia and
disease progression in spinocerebellar ataxia type 14. Mov Disord.
33:1119–1129. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
De Michele G, Galatolo D, Galosi S,
Mignarri A, Silvestri G, Casali C, Leuzzi V, Ricca I, Barghigiani
M, Tessa A, et al: Episodic ataxia and severe infantile phenotype
in spinocerebellar ataxia type 14: Expansion of the phenotype and
novel mutations. J Neurol. 269:1476–1484. 2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ito M, Sugiyama A, Higuchi Y, Takashima H,
Takahashi Y, Mizusawa H and Kuwabara S: Writer's cramps as an
initial symptom of spinocerebellar ataxia type 14. Intern Med.
63:2183–2186. 2024.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Nashi S, Singh R, Menon D, Arshad F,
Alladi S and Mahale RR: Sensory neuropathy in spinocerebellar
ataxia type 14: A novel phenotype. Ann Indian Acad Neurol.
26:591–593. 2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chen Y, Liu P, Cen Z, Liao Y, Lin Z and
Luo W: Early-onset Parkinson's disease with atypical molecular
imaging abnormalities in a patient carrying the de novo PRKCG
mutation. Parkinsonism Relat Disord. 95:100–102. 2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shirafuji T, Shimazaki H, Miyagi T, Ueyama
T, Adachi N, Tanaka S, Hide I, Saito N and Sakai N: Spinocerebellar
ataxia type 14 caused by a nonsense mutation in the PRKCG gene. Mol
Cell Neurosci. 98:46–53. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Pilo CA, Baffi TR, Kornev AP, Kunkel MT,
Malfavon M, Chen DH, Rossitto LA, Chen DX, Huang LC, Longman C, et
al: Mutations in protein kinase Cγ promote spinocerebellar ataxia
type 14 by impairing kinase autoinhibition. Sci Signal.
15(eabk1147)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Verbeek DS, Goedhart J, Bruinsma L, Sinke
RJ and Reits EA: PKC gamma mutations in spinocerebellar ataxia type
14 affect C1 domain accessibility and kinase activity leading to
aberrant MAPK signaling. J Cell Sci. 121 (Pt 14):2339–2349.
2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Grados M, Salehi M, Lotfi A, Dua S and Xie
I: A selective review of inhibitors of protein kinase C gamma: A
neuroplasticity-related common pathway for psychiatric illness.
Front Drug Deliv. 4(1364037)2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Schmitz-Hübsch T, du Montcel ST, Baliko L,
Berciano J, Boesch S, Depondt C, Giunti P, Globas C, Infante J,
Kang JS, et al: Scale for the assessment and rating of ataxia:
Development of a new clinical scale. Neurology. 66:1717–1720.
2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cruz GCD, Zonta MB, Munhoz RP, Mello NM,
Meira AT, Nunes MCA, Aranha NTG, Camargo CHF, Lopes Neto FDN and
Teive HAG: Functionality and disease severity in spinocerebellar
ataxias. Arq Neuropsiquiatr. 80:137–144. 2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Adachi N, Kobayashi T, Takahashi H,
Kawasaki T, Shirai Y, Ueyama T, Matsuda T, Seki T, Sakai N and
Saito N: Enzymological analysis of mutant protein kinase Cgamma
causing spinocerebellar ataxia type 14 and dysfunction in Ca2+
homeostasis. J Biol Chem. 283:19854–19863. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wong MMK, Hoekstra SD, Vowles J, Watson
LM, Fuller G, Németh AH, Cowley SA, Ansorge O, Talbot K and Becker
EBE: Neurodegeneration in SCA14 is associated with increased PKCγ
kinase activity, mislocalization and aggregation. Acta Neuropathol
Commun. 6(99)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Seki T, Takahashi H, Adachi N, Abe N,
Shimahara T, Saito N and Sakai N: Aggregate formation of mutant
protein kinase C gamma found in spinocerebellar ataxia type 14
impairs ubiquitin-proteasome system and induces endoplasmic
reticulum stress. Eur J Neurosci. 26:3126–3140. 2007.PubMed/NCBI View Article : Google Scholar
|