Introduction
Breast cancer (BC) represents a paramount global
health challenge, being the most commonly diagnosed cancer among
women worldwide with an estimated 2.30 million new cases in
2022(1). In the same year, it was
the leading cause of cancer-associated mortalities among women
globally and ranked first in incidence in 157 countries and in
mortality in 112 countries (2).
Human epidermal growth factor receptor (HER)-positive status is
observed in 15-20% of patients with BC (3) and is associated with aggressive
disease progression and poor prognosis (4).
Recent studies have shown that endoplasmic reticulum
(ER) stress and the unfolded protein response (UPR) are key in
oncogenic transformation, survival and cancer progression (5,6).
Cancer cells undergo chronic ER stress due to factors such as
hypoxia, nutrient deprivation, oxidative stress and genetic
mutations, adapting by activating UPR. However, severe ER stress
triggers programmed cell death by activating the pro-apoptotic
components of the UPR. The accumulation of misfolded proteins in
the ER activates three key sensors: PRKR-like ER kinase (PERK),
activating transcription factor 6 (ATF6) and inositol-requiring
enzyme 1 (IRE1), thereby initiating the UPR. While this response
initially promotes cell survival, prolonged or intense stress leads
to divergent outcomes, whereby PERK phosphorylates eukaryotic
translation initiation factor 2α (eIF2α), suppressing global
translation while inducing transcription factors such as activating
transcription factor 4 (ATF4) and CHOP. CHOP upregulates
pro-apoptotic genes, promoting cell death. Meanwhile, ATF6
activates ER chaperones, including glucose-regulated protein 78 kDa
(GRP78) and glucose-regulated protein 94 kDa, as well as X-box
binding protein 1 (XBP1) (5-7).
IRE1, through its endoribonuclease activity, mediates XBP1 splicing
or triggers apoptosis through the JNK pathway (7). This regulatory network highlights the
dual role of ER stress in maintaining cellular homeostasis or
inducing apoptosis, depending on the duration and intensity of the
stress signals.
The precise molecular mechanisms governing the
balance between ER stress-dependent pro-survival and pro-death
signals in BC pathogenesis remain incompletely understood. XBP1 is
highly expressed in estrogen receptor-positive BC (8), where estrogen activates the UPR
through GRP78 to enhance cell survival and promote tumor
progression (9,10). In triple-negative BC (TNBC), the
spliced form of X-box binding protein 1 (XBP1s)-hypoxia-inducible
factor 1-α complex has been shown to support poor prognosis
(11), while the IRE1α/XBP1s
pathway is activated by the MYC proto-oncogene to maintain cell
survival (12). Furthermore, the
PERK/eIF2α pathway initiates autophagy and redox control in TNBC
and persistent ER stress activates apoptotic mechanisms through
caspase-8, Noxa, CHOP and JNK (13-15).
In HER2-positive BC, activation of the PERK-ATF4-CHOP axis
increases cell sensitivity to apoptosis through upregulation of TNF
receptor superfamily member 10b and caspase-8(16), highlighting the complex interplay
between ER stress pathways and BC subtype-specific outcomes.
The majority of chemotherapeutic agents exert
therapeutic effects through either cell cycle inhibition or
activation of apoptotic pathways. Apoptosis, defined as a
programmed and regulated cell death mechanism, serves a key role in
maintaining homeostasis (17).
However, tumor cells develop resistance to chemotherapeutic agents
by suppressing apoptosis (18).
Therefore, triggering apoptotic processes in malignant cells
represents a potential therapeutic response. Furthermore, the
modulatory effect of the tumor microenvironment on ER
stress-induced apoptosis may be an important factor in the
emergence of adaptive mechanisms (19). Specifically, conditions within the
tumor microenvironment, such as hypoxia, nutrient deprivation and
stromal cell crosstalk, can activate compensatory pro-survival
signaling, thereby dampening the apoptotic response to ER stress
(19).
Metformin is a prescribed oral antidiabetic drug and
is a first-line medication for managing type 2 diabetes (20). Beyond its glucose-lowering effects,
this biguanide is a safe drug that interacts with numerous
oncogenic and tumor-suppressive pathways, such as AMPK-dependent
and -independent mechanisms, making it an attractive option for
cancer prevention and treatment (21). Studies have shown that metformin
use is associated with a 31% decrease in overall cancer risk
compared with other antidiabetic drugs and insulin. Although its
exact antitumor mechanisms are not fully understood, AMPK
activation and the inhibition of proliferative signaling pathways
may serve important roles (21,22).
Current research, including both in vivo and in vitro
studies, indicates that metformin may have anticancer effects in BC
treatment and may be considered a therapeutic option (23-25).
The literature presents conflicting results
regarding the effects of metformin, a potential anticancer agent in
BC treatment, on ER stress (26-28).
Modulation of the UPR is a potential strategy in cancer treatment
and enhancing pro-apoptotic signals of ER stress, in particular,
may allow selective targeting of cancer cells (29). Against this background, the present
study aimed to investigate the dose-dependent effects of metformin
on ER stress and apoptosis in SKBR3 cells, an aggressive
HER2-positive BC model.
Materials and methods
Cell culture
HER2+ BC SKBR3 cells (wild-type) was
obtained from the American Type Culture Collection and cultured in
25 cm2 flasks using McCoy's 5A modified medium (cat. no.
16600-082) containing 10% FBS (cat. no. 10500-064), 1% L-glutamine
(cat. no. 25030-081) and 1% 100U penicillin/0.1 mg streptomycin
(cat. no. 15140-122; all Gibco; Thermo Fisher Scientific, Inc.)
with 5% CO2 at 37˚C. The complete medium was removed
once the cells reached 70-80% confluence and cells were rinsed with
PBS. After PBS removal, the cells were detached using trypsin-EDTA
(0.25% trypsin and 0.02% EDTA; cat. no. 25200-056; Gibco, Thermo
Fisher Scientific, Inc.). A total of 5x103 cells/well
were transferred into an e-plate for a proliferation-cytotoxicity
assay. For gene expression and apoptosis analysis by flow
cytometry, cells were seeded in 25 cm2 flasks.
Study groups
For the real-time cell analyzer (RTCA) and gene
expression studies the following groups were used: Control and 5,
10 and 20 mM metformin. For Annexin V-FITC/PI analysis, only
control and 10 and 20 mM metformin groups were included. Only
complete medium was added for the control group. The administered
metformin doses were selected based on the literature (24,25).
RTCA
RTCA operates based on electronic impedance readings
obtained from sensor electrodes located beneath e-plates. As cells
attach to or detach from the surface electrodes, the change in
electronic impedance is mathematically expressed as cell index (CI)
values (30). Cells were cultured
in plates designed for the xCELLigence Real-Time Cell Analysis
system (Agilent Technologies, Inc.) at 5x103 cells/well
and incubated with 5% CO2 for 24 h at 37˚C. Furthermore,
the medium containing metformin (cat. no. D15095-9; Sigma-Aldrich;
Merck KGaA; 5, 10 and 20 mM) was added to different wells. Complete
medium without metformin was added to the control wells. The
e-plates were monitored for 6 days.
Reverse transcription-quantitative
PCR
Following 24 h of treatment with metformin (5, 10
and 20 mM) at 37˚C in a 5% CO2 incubator, the medium was
removed. Using the One Step-RNA Reagent (cat. no. BS410A Bio Basic
Inc.), RNA isolation was performed. The quality and quantity of
obtained RNA was evaluated by a NanoDrop 2000 spectrophotometer
(Thermo Fisher Scientific, Inc.). mRNA was converted to cDNA using
the iScript™ cDNA Synthesis kit (cat. no. 1708891; Bio-Rad
Laboratories, Inc.) according to the manufacturer's protocol. Using
the SsoAdvanced™ Universal SYBR Green Supermix (cat. no. 1725271;
Bio-Rad Laboratories, Inc.) GAPDH, GRP78, PERK, IRE1, ATF6 and CHOP
gene expression was measured through qPCR, using the Biorad CFX 96
system (Bio-Rad Laboratories, Inc.) with the following cycling
protocol: initial denaturation: 98˚C for 30 sec; 40 cycles of
[denaturation: 98˚C for 15 sec, annealing/extension: 60˚C for 30
sec]. Primer sequences were designed using the NCBI Primer-BLAST
tool. The relative gene expression was quantified using the
2-ΔΔCq method (31). The primer sequences of the genes
are detailed in Table I.
 | Table IPCR primer sequences. |
Table I
PCR primer sequences.
| Gene | Forward primer
(5'-3') | Reverse primer
(3'-5') |
|---|
| GAPDH |
GGAGCGAGATCCCTCCAAAAT |
GGCTGTTGTCATACTTCTCATGG |
| GRP78 |
ACAATCAAGGTCTATGAAGGTGAAAGAC |
CTCGAAGAATACCATTCACATCTATCTC |
| PERK |
AATCATAGCTCCTTCACCACAAA |
CATCTTCCACATCACAGTCTGTA |
| IRE1 |
CACAGTGACGCTTCCTGAAAC |
GCCATCATTAGGATCTGGGAGA |
| ATF6 |
AGCATGTTCCTGAGGAGTTGG |
AGGCTTATCTTCCTTCAGTGGC |
| CHOP |
GGAAACAGAGTGGTCATTCCC |
CTGCTTGAGCCGTTCATTCTC |
Annexin V-FITC/PI analysis
Early and late apoptotic cell populations were
detected using an Annexin V-FITC/PI apoptosis detection kit (cat.
no. E-CK-A211; Elabscience Bionovation Inc.), according to the
manufacturer's protocol. Following 24 h metformin treatment as
aforementioned, cells were harvested by trypsinization, washed in
PBS and stained for apoptosis analysis using an Annexin V-FITC/PI
assay. The staining procedure was performed as follows: Cells were
resuspended at a density of 5x105 cells per sample in
500 µl of 1X Annexin V Binding Buffer. Each sample was then stained
by adding 5 µl of Annexin V-FITC conjugate and 5 µl of PI directly
to the cell suspension. The mixture was gently vortexed and then
incubated at room temperature (~25˚C) in the dark for 15-20 min.
After the incubation,cells were analyzed immediately by flow
cytometry (BD FACSCalibur™; BD Biosciences) using the BD CellQuest™
acquisition and analysis software (version 5.2.1; BD
Biosciences).
Statistical analysis
Statistical analyses were performed using SPSS
(version 25; IBM Corp.) Data distribution normality was assessed
using the Shapiro-Wilk test. For parametric data, one-way ANOVA was
employed, followed by Tukey's post hoc test. Data are presented as
mean ± SEM. For non-parametric data the Kruskal-Wallis test was
used, followed by Dunn's test with Bonferroni correction for
multiple comparisons. All experiments were performed in
quadruplicate. P<0.05 was considered to indicate a statistically
significant difference.
Results
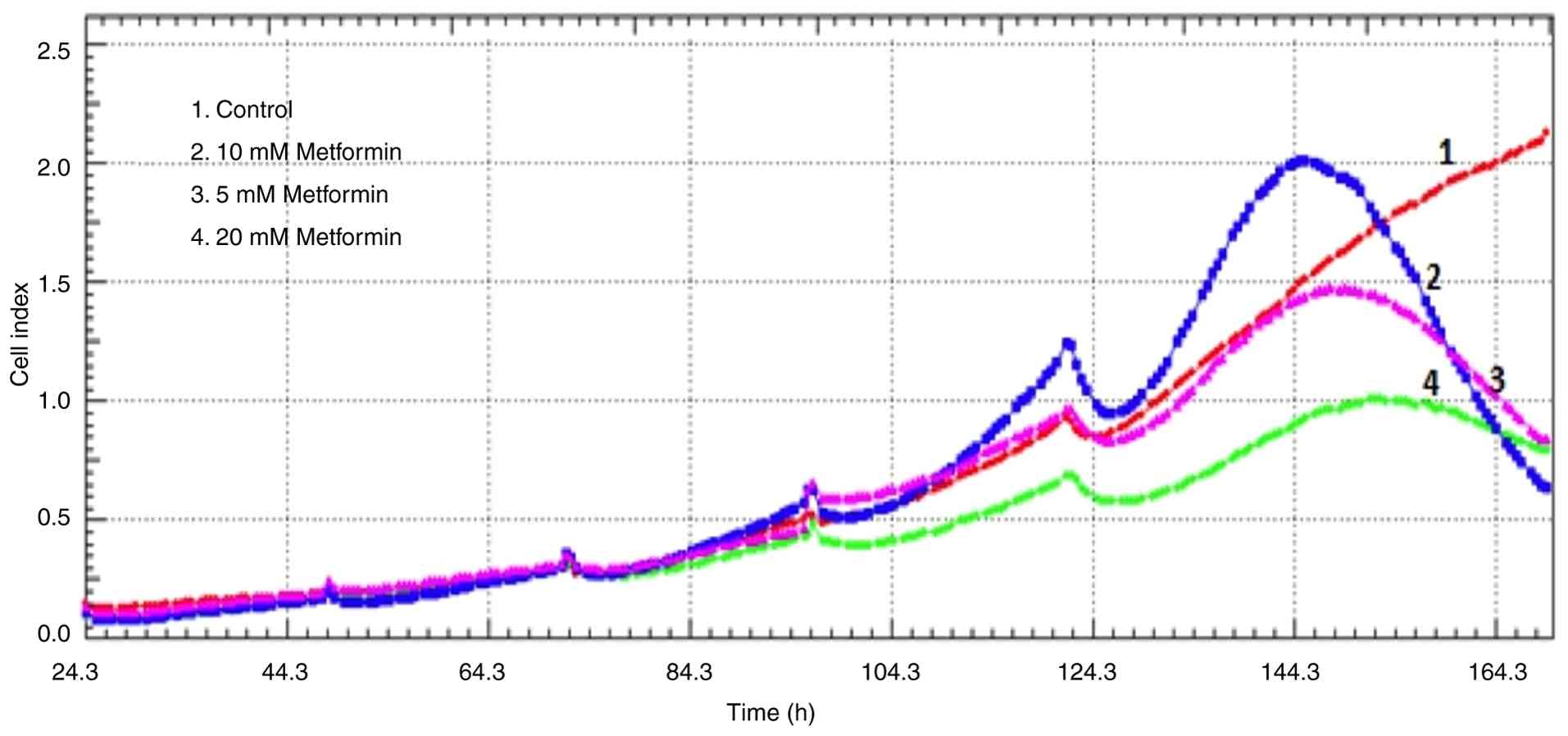
Metformin inhibits cell proliferation
in a dose-dependent manner
The anti-proliferative effects of metformin were
analyzed using RTCA via electrical impedance. While the CI in the
control group continued to increase, all doses of metformin (5, 10
and 20 mM) exhibited antiproliferative and cytotoxic activity by
day 6, as evidenced by a decrease in CI values based on electrical
impedance signals (Fig. 1).
Metformin induces dose-dependent
upregulation of ER stress marker genes
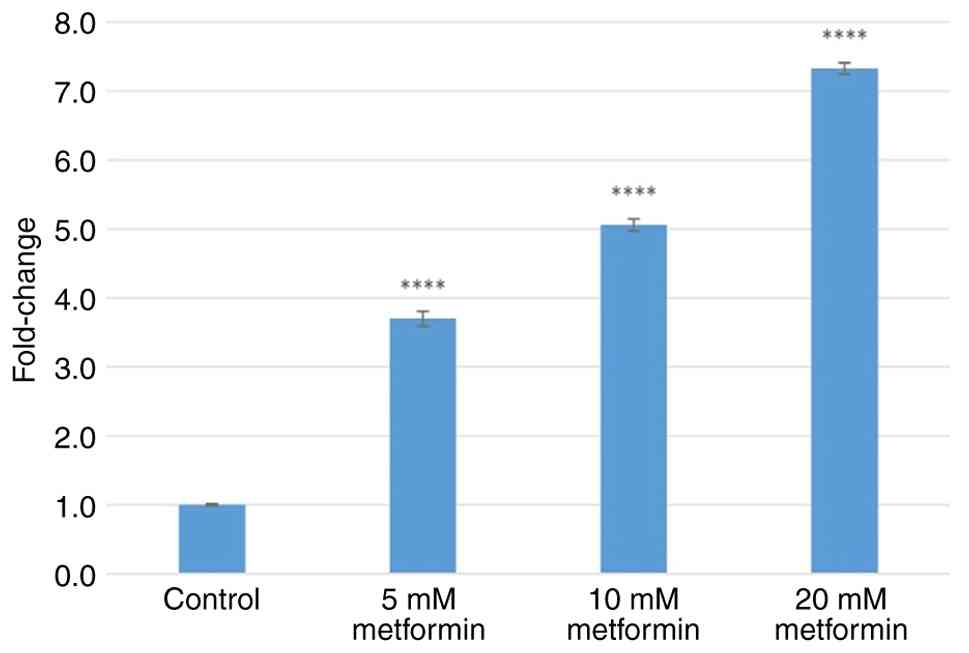
Treatment with 5, 10 and 20 mM metformin increased
GRP78 gene expression by 3.70±0.11-, 5.06±0.08- and 7.33±0.08-fold
(all P<0.0001), respectively (Fig.
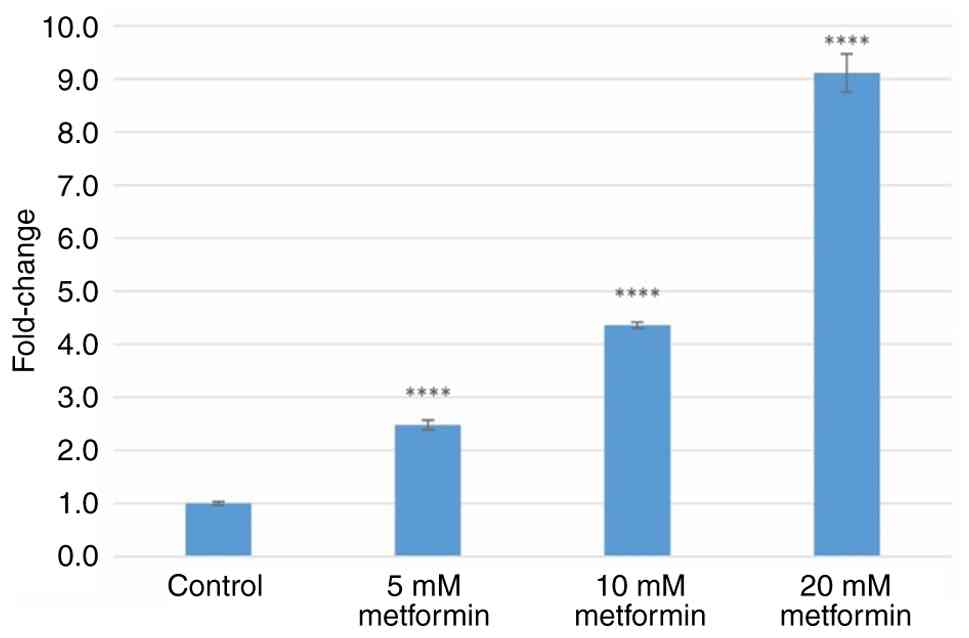
2). Treatment with 5, 10 and 20 mM metformin increased PERK
gene expression by 2.48±0.09-, 4.36±0.06- and 9.11±0.36-fold (all
P<0.0001), respectively (Fig.
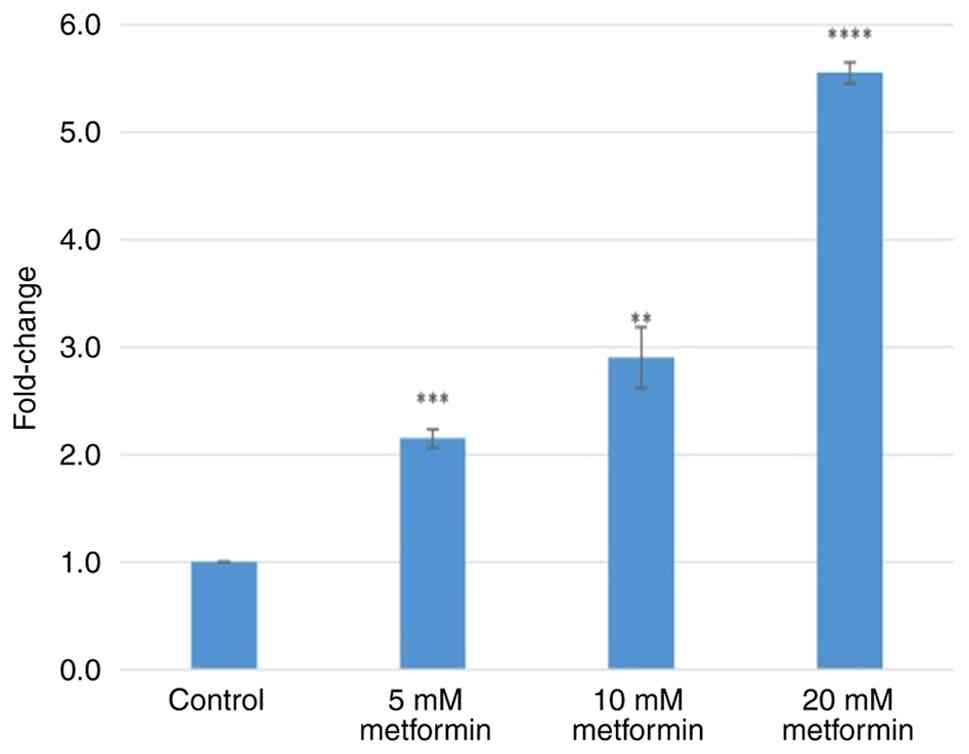
3). Treatment with 5, 10 and 20 mM metformin increased IRE1
gene expression levels by 2.15±0.08- (P=0.001), 2.90±0.03-
(P<0.001) and 5.55±0.10-fold (P<0.0001), respectively
(Fig. 4). Treatment with 5, 10 and
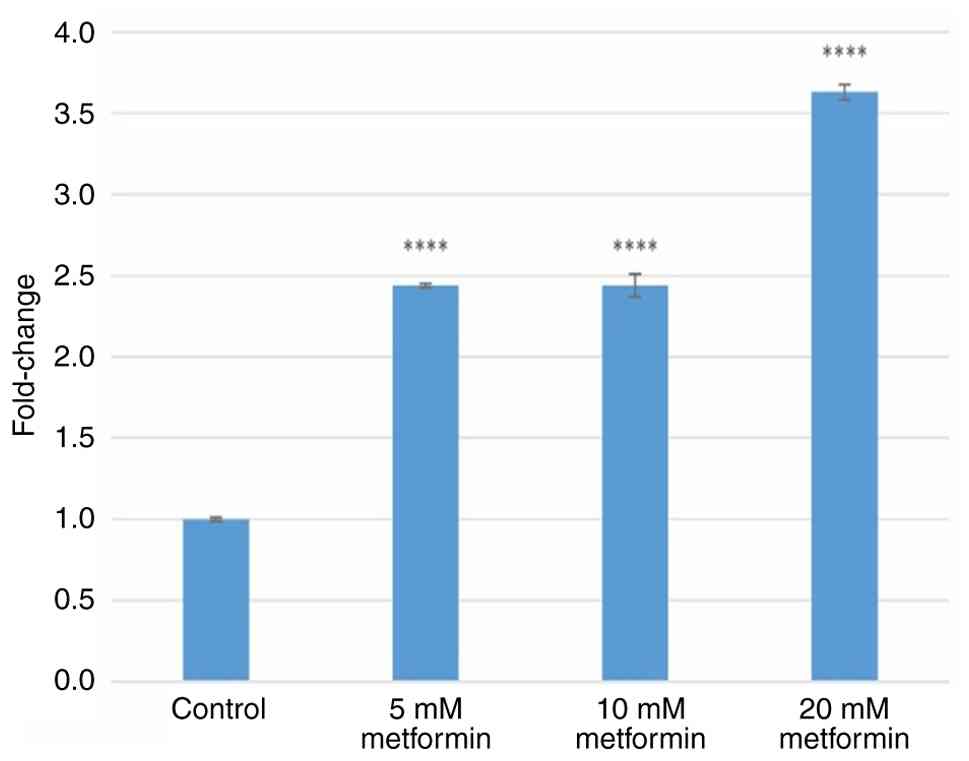
20 mM metformin increased ATF6 gene expression levels by
2.43±0.01-, 2.44±0.07- and 3.63±0.05-fold (all P<0.0001),
respectively (Fig. 5). Treatment
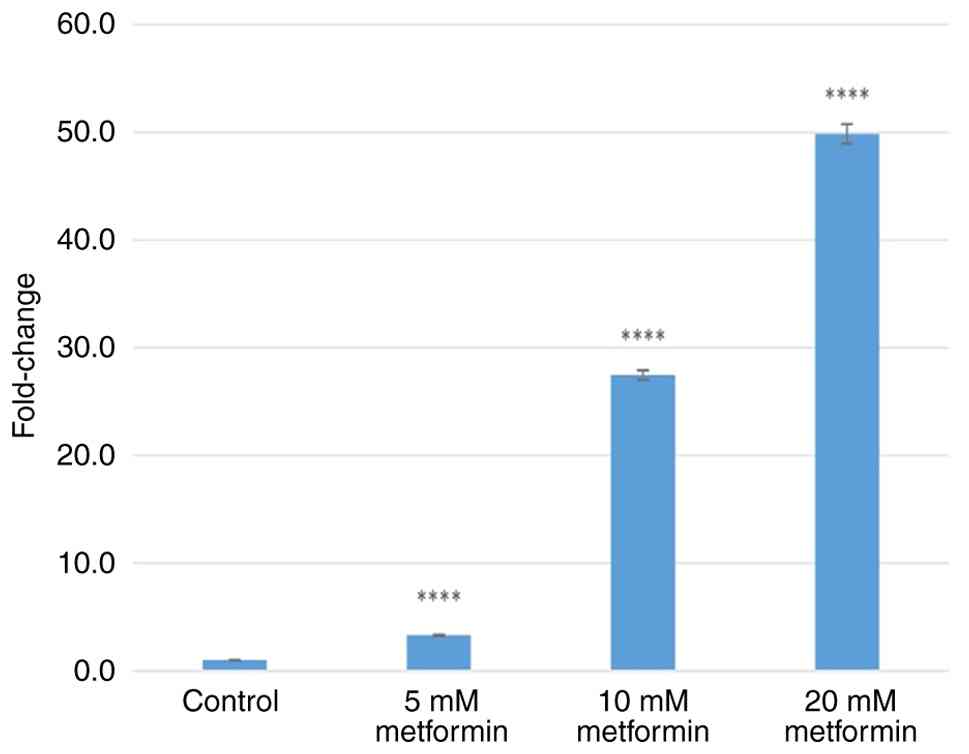
with 5, 10 and 20 mM metformin increased CHOP gene expression
levels by 3.31±0.06-, 27.47±0.44- and 49.85±0.9-fold (all
P<0.0001), respectively (Fig.
6).
Metformin triggers apoptosis in a
dose-dependent manner
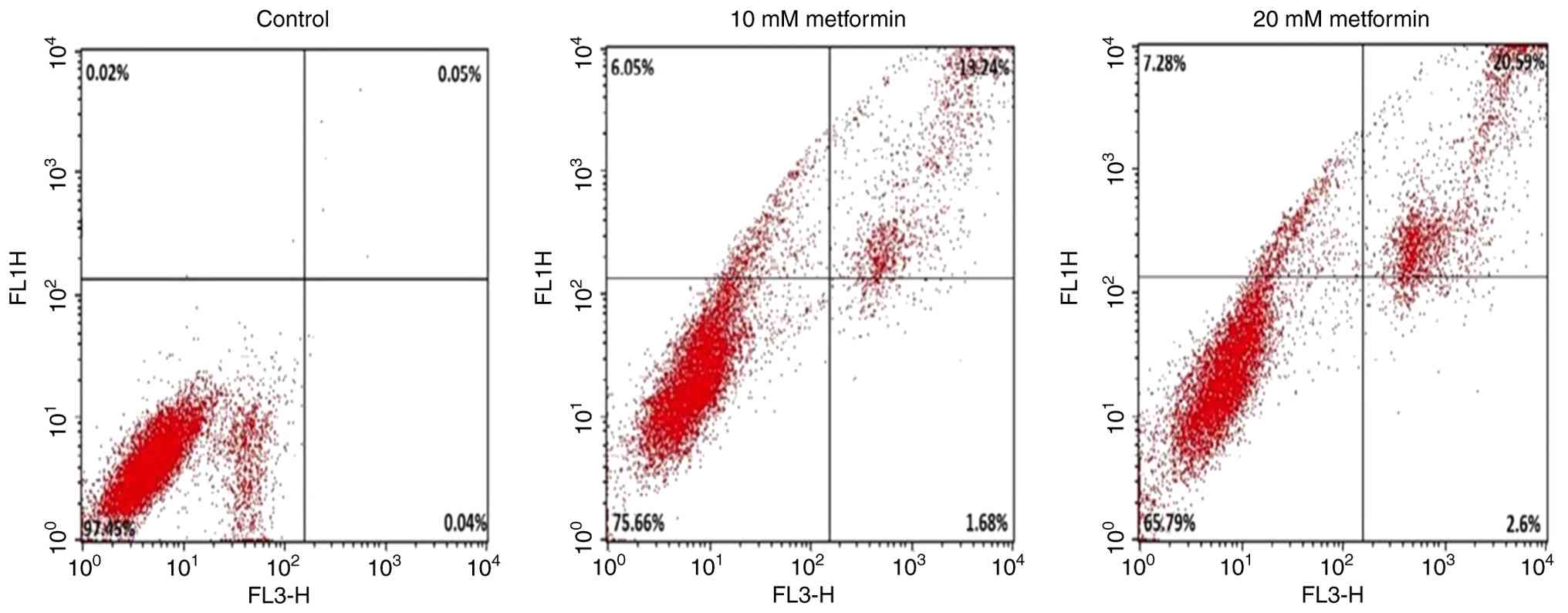
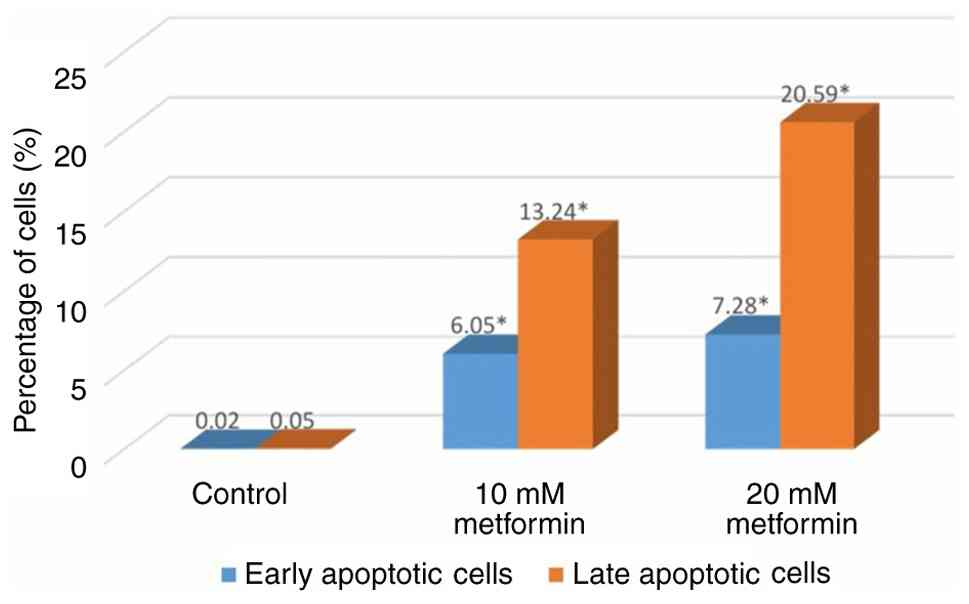
Treatment with 10 and 20 mM metformin significantly
increased early apoptotic cells to 6.05 and 7.28, as well as late
apoptotic cells to 13.24 and 20.59% (all P<0.001), respectively,
compared with the control group (Fig.
7). These results demonstrated dose-dependent induction of
apoptosis (Fig. 8).
Discussion
Within the present study, the multifaceted
anticancer effects of metformin on the HER2-positive aggressive BC
cell line SKBR3 were investigated. RTCA revealed that metformin (5,
10 and 20 mM) exhibited notable antiproliferative activity on day
6. This proliferation inhibition was functionally associated with
dose-dependent increases in ER stress markers and apoptosis.
Administration of 5, 10 and 20 mM metformin led to a significant
dose-dependent increase in the gene expression of GRP78, PERK,
IRE1, ATF6 and CHOP. Percentages of early and late apoptotic cells
significantly increased in a dose-dependent manner in the 10 and 20
mM metformin groups. Collectively, these results support the
hypothesis that metformin induces ER stress-mediated apoptosis by
upregulating the ER chaperone GRP78 and key components of the UPR -
namely PERK, IRE1 and ATF6 - which in turn activates the
pro-apoptotic CHOP pathway. Despite the antitumor role of metformin
having been widely reported, its molecular mechanism in inhibiting
tumor progression remains unclear (21,32).
Both in vitro and in vivo studies demonstrate that
metformin exerts anticancer effects on BC through both
AMPK-dependent and -independent mechanisms. The AMPK-dependent
mechanisms include liver kinase B1-mediated AMPK phosphorylation,
inhibition of mTOR through AMPK, forkhead box O3 activation and the
regulation of histone deacetylases. AMPK-independent effects occur
through numerous pathways, including microRNA modulation, increased
oxidative stress, antifolate activity, inhibition of angiogenesis,
NF-κB activation by suppressing proinflammatory cytokines such as
IL-6 and IL-17, downregulation of specificity protein
(SP)-1/SP3/SP4 transcription factors, upregulation of caveolin-1
and the modulation of cell cycle regulatory proteins (21,32).
A potential anticancer mechanism of metformin may involve
activation of ER stress-induced apoptotic cascades. The literature
has consistently demonstrated the cytotoxic effects of metformin
across a number of BC cell lines (24-28,33-35).
In MCF-7 cells (BC cell line, ER positive), 10 µM metformin
markedly decreases viability and migration compared with controls
(27). Li et al (28) reported that 0.125 mg/ml metformin
promotes death in glucose-deprived MDA-MB-231 cells (BC cell line,
triple-negative). A number of studies using SKBR3 cells have shown
dose- and time-dependent effects (24,25,33-36).
For example, Chen et al (33) observed that 0.5-8.0 mM metformin
inhibits proliferation and induces apoptosis, while Neamati et
al (34) found that 30-100 mM
treatment increases the number of apoptotic cells by 48 h. Xu et
al (35) demonstrated that
20-120 µM metformin caused time-dependent proliferation, inhibition
and G1-phase arrest, with 96.25 µM treatment increasing
early and late apoptosis to 4.48 and 17.13%, respectively, at 48 h.
Ahmadpour et al (24) and
Amaral et al (25)
demonstrated that 10-80 and 0.01-5.00 mM metformin concentrations,
respectively, dose-dependently decrease viability at numerous
timepoints. Notably, metformin exhibits enhanced efficacy against
trastuzumab-resistant cells (36).
The present findings of increased early/late apoptosis and reduced
viability with 10-20 mM metformin treatment are consistent with the
aforementioned effects.
The dichotomous role of ER stress in promoting
cancer cell survival or triggering apoptosis remains subject to
debate (37), although sustained
or severe ER stress leads to cell death (38). While numerous studies using various
cell lines, mostly consistent with the present findings, have
demonstrated that metformin causes ER stress-induced apoptosis
(26-28,39-45),
there are also studies suggesting it reduces ER stress (46-48).
However this may be due to the cell lines used as well as the
metformin dose. According to studies in non-cancerous cell lines,
low-dose metformin suppresses ER stress-induced apoptosis (46-48).
For example, in a colitis model, 1 mM metformin decreases GRP78,
CHOP, caspase-12, PERK and eIF2α expression levels and apoptosis
(46). Furthermore, in pancreatic
β cells, 0.05 mM metformin suppresses palmitate-induced ER stress
induction in an AMPK-independent manner (47), and in ovarian granulosa cells, 1 mM
metformin inhibits testosterone-induced ER stress and UPR
activation by suppressing mitogen-activated protein kinase P38-α
phosphorylation (48). In studies
with cancer cell lines, metformin causes ER stress-induced
apoptosis, supporting the present findings (39-45);
in a study using prostate cancer cells, application of 5 mM
metformin increased the expression of ER stress-associated CHOP,
phosphorylated (p)-eIF2α, calreticulin, GRP78 and SR
Ca2+-ATPase 1 genes through miR-708-5p, thereby inducing
apoptosis (39). In addition, a
study of colon cancer cells demonstrated that the application of 1,
5 and 25 mM metformin induces dose-dependent increases in PERK,
p-eIF2α, ATF4 and CHOP levels (40). In colon cancer cells, 5 mM
metformin activates CHOP and inhibites cell proliferation by
causing cell cycle arrest (41)
and in thyroid cancer cells, metformin application inhibits
proliferation and induces apoptosis in a concentration- (1.25,
2.50, 5, 10 or 20 mM) and time-dependent (24-48 h) manner.
Furthermore, 5, 10 and 20 mM metformin activate ER stress by
increasing GRP78, CHOP and caspase-12 expression (42). Similarly, metformin (0-20 mM and
0-48 h) decreases the cell proliferation index and increases the
expression of GRP78, CHOP and caspase-12 in papillary thyroid
carcinoma cells (43).
Furthermore, in endometrial cancer cells, metformin increases CHOP
expression levels while decreasing GRP78 expression levels
(44). Similarly, metformin
induces UPR-mediated cell death by decreasing GRP78 expression and
upregulating IRE1α and CHOP in acute lymphoblastic leukemia cells
(45).
Studies demonstrating metformin-induced ER
stress-mediated apoptosis in BC remain limited (26-28).
In MCF-7 cells, Huang et al (26) reported that metformin (0-40 mM)
dose-dependently increases CHOP expression, inhibits proliferation,
promotes apoptosis and induces cell cycle arrest. Alizade et
al (27) found that 10 µM
metformin decreases viability and migration after 48 h,
upregulating apoptosis-related caspase-3, Bax, apoptosis regulator,
apoptosis-inducing factor mitochondria associated 1, CHOP and GRP78
expression while downregulating Bcl-2 and WEE1 G2
checkpoint kinase. In MDA-MB-231 cells, Li et al (28) demonstrated that 0.125 mg/ml
metformin under glucose deprivation enhances UPR through the
ATF4/ATF3/CHOP pathway while inhibiting anti-apoptotic Bcl-2 and
BCL-xl effects. To the best of our knowledge, no studies have
investigated the association between metformin and ER
stress-induced apoptosis in aggressive HER2-positive BC cells. In
the present study, a dose-dependent induction of endoplasmic
reticulum stress was suggested, as increases in the expression of
ER stress markers GRP78, PERK, IRE1, ATF6 and CHOP following
treatment with 5, 10 and 20 mM metformin was observed. The
upregulation of chaperone GRP78 and the three primary UPR sensors
(PERK, IRE1 and ATF6) demonstrated ER stress induction. Notable
increases in CHOP gene expression at 10 and 20 mM metformin doses
were detected, mediated by PERK and ATF6 activation, an indicator
of ER stress-induced apoptosis. This was supported by Annexin
V-FITC/PI assays showing significant increases in early and late
apoptotic cells at these concentrations. IRE1 upregulation may
promote apoptosis through JNK or caspase-12 pathways. Furthermore,
elevated IRE1, PERK and ATF6 levels may sustain the ER stress
response by maintaining GRP78 activation.
The present study provided functional and
transcriptional evidence that metformin induces ER stress and
triggers apoptosis in HER2-positive BC cells. However, certain
limitations must be acknowledged. Activation of ER stress pathways
could not be directly demonstrated at the protein level. Secondly,
the present study was not conducted in multiple HER2-positive cell
lines to strengthen the generalizability of the findings. However,
the present results in SKBR3 cells are consistent with the
literature, which reports that metformin reduces viability in a
dose-dependent manner in HER2-positive BT-474 cells (BC cell line,
ER/PR/HER2-positive). Finally, the present study employed high
metformin concentrations (49),
consistent with preclinical literature, to elucidate ER stress and
apoptosis mechanisms. However, these concentrations are markedly
higher than those of therapeutic plasma levels (10-40 µM) achieved
with standard oral dosing in diabetes treatment (50). Reasons behind the high in
vitro dose requirement may include continuous drug exposure,
the absence of serum protein binding and non-physiological
conditions of the cell culture environment, such as high glucose
and growth factor levels. These conditions may maximize cell
proliferation and survival signals, necessitating higher drug
concentrations to suppress oncogenic mechanisms (51). This represents a key limitation for
the direct clinical translation of the present results, which
require validation through in vivo animal experiments. The
effects of metformin on ER stress and apoptosis are
context-dependent. The literature reports varying and contradictory
outcomes depending on cell type, metabolic profile,
microenvironmental conditions and the dose (21,25,39,48,52,53).
Therefore, validating the present findings in other cancer models
and in vivo systems is key to define the therapeutic
potential of metformin.
In conclusion, the present findings demonstrated
that metformin treatment inhibited proliferation in a time- and
dose-dependent manner in SKBR3 cells, activated the ER stress
response to potentiate apoptotic signaling and disrupted cell
survival mechanisms. These results suggest that the ER
stress-apoptosis axis may represent a therapeutic target in
HER2-positive BC. However, further in vivo investigations
are warranted to evaluate the clinical translatability of
metformin.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
EÇ and BB conceived and designed the study. EÇ
conducted data analysis and wrote the manuscript. All authors have
read and approved the final manuscript. EÇ and BB confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Filho AM, Laversanne M, Ferlay J, Colombet
M, Piñeros M, Znaor A, Parkin DM, Soerjomataram I and Bray F: The
GLOBOCAN 2022 cancer estimates: Data sources, methods, and a
snapshot of the cancer burden worldwide. Int J Cancer.
156:1336–1346. 2025.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Amin MB, Greene FL, Edge SB, Compton CC,
Gershenwald JE, Brookland RK, Meyer L, Gress DM, Byrd DR and
Winchester DP: The Eighth Edition AJCC Cancer Staging Manual:
Continuing to build a bridge from a population-based to a more
‘personalized’ approach to cancer staging. CA Cancer J Clin.
67:93–99. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chavez-MacGregor M, Mittendorf EA, Clarke
CA, Lichtensztajn DY, Hunt KK and Giordano SH: Incorporating tumor
characteristics to the American joint committee on cancer breast
cancer staging system. Oncologist. 22:1292–1300. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yu X, Li W, Sun S and Li J: Endoplasmic
reticulum stress in cancer progression: A comprehensive review of
its role and mechanisms. Int J Med Sci. 22:4561–4585.
2025.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ghemrawi R, Kremesh S, Mousa WK and Khair
M: The Role of ER stress and the unfolded protein response in
cancer. Cancer Genomics Proteomics. 22:363–381. 2025.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Çakmak E and Bilgici B: The relationship
between cancer and endoplasmic reticulum stress-induced apoptosis.
Kırıkkale Üni Tıp Derg. 27:111–119. 2025.
|
|
8
|
Ding L, Yan J, Zhu J, Zhong H, Lu Q, Wang
Z, Huang C and Ye Q: Ligand-independent activation of estrogen
receptor alpha by XBP-1. Nucleic Acids Res. 31:5266–5274.
2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Scriven P, Coulson S, Haines R,
Balasubramanian S, Cross S and Wyld L: Activation and clinical
significance of the unfolded protein response in breast cancer. Br
J Cancer. 101:1692–1698. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kiang JG, Gist ID and Tsokos GC: 17
beta-estradiol-induced increases in glucose-regulated protein 78kD
and 94kD protect human breast cancer T47-D cells from thermal
injury. Chin J Physiol. 40:213–219. 1997.PubMed/NCBI
|
|
11
|
Chen X, Iliopoulos D, Zhang Q, Tang Q,
Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et
al: Xbp1 promotes triple-negative breast cancer by controlling the
hif1alpha pathway. Nature. 508:103–107. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhao N, Cao J, Xu L, Tang Q, Dobrolecki
LE, Lv X, Talukdar M, Lu Y, Wang X, Hu DZ, et al: Pharmacological
targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven
breast cancer. J Clin Invest. 128:1283–1299. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Cano-González A, Mauro-Lizcano M,
Iglesias-Serret D, Gil J and López-Rivas A: Involvement of both
caspase-8 and Noxa-activated pathways in endoplasmic reticulum
stress-induced apoptosis in triple-negative breast tumor cells.
Cell Death Dis. 9(134)2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhou W, Fang H, Wu Q, Wang X, Liu R, Li F,
Xiao J, Yuan L, Zhou Z, Ma J, et al: Ilamycin E, a natural product
of marine actinomycete, inhibits triple-negative breast cancer
partially through ER stress-CHOP-Bcl-2. Int J Biol Sci.
15:1723–1732. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Dávila-González D, Choi DS, Rosato RR,
Granados-Principal SM, Kuhn JG, Li WF, Qian W, Chen W, Kozielski
AJ, Wong H, et al: Pharmacological inhibition of NOS Activates
ASK1/JNK pathway augmenting docetaxel-mediated apoptosis in
triple-negative breast cancer. Clin Cancer Res. 24:1152–1162.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Martín-Pérez R, Palacios C, Yerbes R,
Cano-González A, Iglesias-Serret D, Gil J, Reginato MJ and
López-Rivas A: Activated ERBB2/HER2 licenses sensitivity to
apoptosis upon endoplasmic reticulum stress through a
PERK-dependent pathway. Cancer Res. 74:1766–1777. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Benner SE and Hong WK: Clinical
chemoprevention: Developing a cancer prevention strategy. J Natl
Cancer Inst. 85:1446–1447. 1993.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Campbell KJ and Tait SWG: Targeting BCL-2
regulated apoptosis in cancer. Open Biol. 8(180002)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chen X and Cubillos-Ruiz JR: Endoplasmic
reticulum stress signals in the tumour and its microenvironment.
Nat Rev Cancer. 21:71–88. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Nathan DM, Buse JB, Davidson MB,
Ferrannini E, Holman RR, Sherwin R and Zinman B: American Diabetes
Association; European Association for Study of Diabetes. Medical
management of hyperglycemia in type 2 diabetes: A consensus
algorithm for the initiation and adjustment of therapy: A consensus
statement of the American diabetes association and the European
association for the study of diabetes. Diabetes Care. 32:193–203.
2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Faria J, Negalha G, Azevedo A and Martel
F: Metformin and breast cancer: Molecular targets. J Mammary Gland
Biol Neoplasia. 24:111–123. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ala M and Ala M: Metformin for
cardiovascular protection, inflammatory bowel disease,
osteoporosis, periodontitis, polycystic ovarian syndrome,
neurodegeneration, cancer, inflammation and senescence: What is
next? ACS Pharmacol Transl Sci. 4:1747–1770. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Cejuela M, Martin-Castillo B, Menendez JA
and Pernas S: Metformin and breast cancer: Where are we now? Int J
Mol Sci. 26(5407)2025.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ahmadpour F, Igder S, Eftekhari Moghadam
AR, Moradipoodeh B, Sepahdar A, Mokarram P, Fallahi J and
Mohammadzadeh G: Metformin as a potential therapeutic agent in
breast cancer: Targeting miR-125a methylation and epigenetic
regulation. Int J Mol Cell Med. 13:272–285. 2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Amaral MEA, Nery LR, Leite CE, de Azevedo
Junior WF and Campos MM: Pre-clinical effects of metformin and
aspirin on the cell lines of different breast cancer subtypes.
Invest New Drugs. 36:782–796. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Huang Y, Zhou Z, Zhang J, Hao Z, He Y, Wu
Z, Song Y, Yuan K, Zheng S, Zhao Q, et al: lncRNA MALAT1
participates in metformin inhibiting the proliferation of breast
cancer cell. J Cell Mol Med. 25:7135–7145. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Alizade A, Evyapan G, Celik IS and Ozdem
B: Metformin induces mitochondria-mediated and endoplasmic
reticulum stress-mediated apoptosis and inhibits
angiogenesis-related gene expression in breast cancer cells via
targeting VEGF-A/VEGFR2/NRP1. Croat Med J. 66:115–124.
2025.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li Y, Zhang Q, Yang J, He W, Jiang Y, Chen
Y and Wang Y: Metformin combined with glucose starvation
synergistically suppress triple-negative breast cancer by enhanced
unfolded protein response. Biochem Biophys Res Commun. 675:146–154.
2023.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kim C and Kim B: . Anti-cancer natural
products and their bioactive compounds inducing ER stress-mediated
apoptosis: A review. Nutrients. 10(1021)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Roshan Moniri M, Young A, Reinheimer K,
Rayat J, Dai LJ and Warnock GL: Dynamic assessment of cell
viability, proliferation and migration using real time cell
analyzer system (RTCA). Cytotechnology. 67:379–386. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Saini N and Yang X: Metformin as an
anti-cancer agent: Actions and mechanisms targeting cancer stem
cells. Acta Biochim Biophys Sin (Shanghai). 50:133–143.
2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen TW, Liang YN, Feng D, Tao LY, Qi K,
Zhang HY, Wang HX, Lin QS and Kong H: Metformin inhibits
proliferation and promotes apoptosis of HER2 positive breast cancer
cells by downregulating HSP90. J BUON. 18:51–56. 2013.PubMed/NCBI
|
|
34
|
Neamati D, Khedri A, Aberomand M, Hemmati
AA, Mohammadzadeh M, Akbari Baghbani K and Mohammadzadeh G:
Metformin synergistically increases the anticancer effects of
lapatinib through induction of apoptosis and modulation of Akt/AMPK
pathway in SK-BR3 breast cancer cell line. Iran J Basic Med Sci.
24:1529–1537. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu Y, Xu T, Xiong Y and Huang J: Metformin
inhibits proliferation and promotes apoptosis of HER-2 positive
breast cancer cells possibly through the Hippo-YAP pathway. Nan
Fang Yi Ke Da Xue Xue Bao. 42:740–746. 2022.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
36
|
Liu B, Fan Z, Edgerton SM, Yang X, Lind SE
and Thor AD: Potent anti-proliferative effects of metformin on
trastuzumab-resistant breast cancer cells via inhibition of
erbB2/IGF-1 receptor interactions. Cell Cycle. 10:2959–2966.
2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhang W, Shi Y, Oyang L, Cui S, Li S, Li
J, Liu L, Li Y, Peng M, Tan S, et al: Endoplasmic reticulum
stress-a key guardian in cancer. Cell Death Discov.
10(343)2024.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yang J, Wei J, Wu Y, Wang Z, Guo Y, Lee P
and Li X: Metformin induces ER stress-dependent apoptosis through
miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis.
4(e158)2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lee DE, Lee GY, Lee HM, Choi SY, Lee SJ
and Kwon OS: Synergistic apoptosis by combination of metformin and
an O-GlcNAcylation inhibitor in colon cancer cells. Cancer Cell
Int. 23(108)2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lee DE, Lee HM, Jun Y, Choi SY, Lee SJ and
Kwon OS: Metformin induces apoptosis in TRAIL-resistant colorectal
cancer cells. Biochim Biophys Acta Mol Cell Res.
1872(119873)2025.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ye J, Qi L, Chen K, Li R, Song S, Zhou C
and Zhai W: Metformin induces TPC-1 cell apoptosis through
endoplasmic reticulum stress-associated pathways in vitro and in
vivo. Int J Oncol. 55:331–339. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Dong LR, Li M, Li S, Liu AD, Xiong YJ,
Tang H and Song XD: Metformin induced apoptosis of papillary
thyroid carcinoma BCPAP cells. Lin Chuang Er Bi Yan Hou Tou Jing
Wai Ke Za Zhi. 31:937–940. 2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
44
|
Conza D, Mirra P, Calì G, Insabato L,
Fiory F, Beguinot F and Ulianich L: Metformin dysregulates the
unfolded protein response and the WNT/β-Catenin pathway in
endometrial cancer cells through an AMPK-Independent mechanism.
Cells. 10(1067)2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Leclerc GM, Leclerc GJ, Kuznetsov JN,
DeSalvo J and Barredo JC: Metformin induces apoptosis through
AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts.
PLoS One. 8(e74420)2013.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wang J, Chen C, Ren Y, Zhou X and Yu S:
Metformin alleviates intestinal epithelial barrier damage by
inhibiting endoplasmic reticulum stress-induced cell apoptosis in
colitis cell model. Zhejiang Da Xue Xue Bao Yi Xue Ban. 50:627–632.
2021.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
47
|
Kim HI, Lee JS, Kwak BK, Hwang WM, Kim MJ,
Kim YB, Chung SS and Park KS: Metformin ameliorates lipotoxic
β-Cell dysfunction through a concentration-dependent dual mechanism
of action. Diabetes Metab J. 43:854–866. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jin J, Ma Y, Tong X, Yang W, Dai Y, Pan Y,
Ren P, Liu L, Fan HY, Zhang Y and Zhang S: Metformin inhibits
testosterone-induced endoplasmic reticulum stress in ovarian
granulosa cells via inactivation of p38 MAPK. Hum Reprod.
35:1145–1158. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Keshandehghan A, Nikkhah S, Tahermansouri
H, Heidari-Keshel S and Gardaneh M: Co-Treatment with sulforaphane
and nano-metformin molecules accelerates apoptosis in HER2+ breast
cancer cells by inhibiting key molecules. Nutr Cancer. 72:835–848.
2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Stambolic V, Woodgett JR, Fantus IG,
Pritchard KI and Goodwin PJ: Utility of metformin in breast cancer
treatment, is neoangiogenesis a risk factor? Breast Cancer Res
Treat. 114:387–389. 2009.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Dowling RJ, Niraula S, Stambolic V and
Goodwin PJ: Metformin in cancer: Translational challenges. J Mol
Endocrinol. 48:R31–R43. 2012.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Morales DR and Morris AD: Metformin in
cancer treatment and prevention. Annu Rev Med. 66:17–29.
2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Quentin T, Steinmetz M, Poppe A and Thoms
S: Metformin differentially activates ER stress signaling pathways
without inducing apoptosis. Dis Model Mech. 5:259–269.
2012.PubMed/NCBI View Article : Google Scholar
|