Introduction
Hypersensitivity pneumonitis (HP) is an immunologically mediated interstitial lung disease triggered by repeated inhalation of organic antigens. It is classically considered a type III hypersensitivity reaction in which immune responses to inhaled allergens lead to inflammation in the alveoli and interstitium, manifesting across a spectrum of acute, subacute and chronic disease forms (1,2). A wide range of environmental exposures have been implicated, including bird proteins, moldy hay, thermophilic actinobacteria and certain occupational antigens (3). Genetic susceptibility, particularly associations with specific human leukocyte antigen class II alleles, may further influence disease development (2,3).
Pathogenetically, re-exposure to an inciting antigen activates sensitized T lymphocytes, especially CD4+ helper T cells, which release pro-inflammatory cytokines such as interleukin-1, interleukin-6 and tumor necrosis factor-α. These mediators recruit neutrophils and macrophages to the alveolar spaces, driving alveolitis and interstitial inflammation. Chronic antigen exposure leads to fibroblast activation and extracellular matrix deposition, culminating in pulmonary fibrosis in a subset of patients (1-3).
Clinically, HP presentation varies by exposure intensity and chronicity. Acute HP may present within hours of exposure with fever, chills, cough and dyspnea, often accompanied by auscultatory crackles. Chronic forms develop insidiously, with persistent cough, progressive exertional dyspnea, fatigue and radiographic findings of fibrosis such as reticular opacities and honeycombing, with finger clubbing and functional impairment are common in advanced disease (1-5). While acute HP often resolves with antigen avoidance, chronic HP is frequently progressive, with irreversible fibrosis and impaired quality of life (6).
Current treatment of HP relies primarily on antigen identification and avoidance of the inciting environmental antigen (trigger exposure) that is causing the immune-mediated lung inflammation, and this remains the cornerstone of management. Corticosteroids and other immunosuppressive agents are often used in patients with persistent or progressive disease, although their long-term benefit remains uncertain and adverse effects are common, particularly in older adults. In this population, prolonged corticosteroid therapy is associated with an increased risk of osteoporosis and fragility fractures, hyperglycemia and worsening diabetes, hypertension, weight gain, sarcopenia, skin fragility, cataracts, glaucoma, neuropsychiatric disturbances and heightened susceptibility to infections. In chronic HP (CHP) or fibrotic HP (FHP), outcomes are frequently poor despite immunosuppression, and numerous patients continue to progress to respiratory failure. The advent of antifibrotic therapies, such as nintedanib and pirfenidone, has introduced new therapeutic possibilities, but their efficacy in HP is supported mainly by post-hoc or small cohort analyses, and evidence remains limited compared with idiopathic pulmonary fibrosis (IPF). Thus, current treatment strategies are constrained by incomplete efficacy, variable tolerability and the lack of robust randomized data, underscoring the need for improved prognostic tools and individualized approaches (7-10).
Over the past 5 years, the conceptual framework for HP diagnosis and disease-behavior assessment has undergone marked refinement. The 2020 American Thoracic Society (ATS), Japanese Respiratory Society (JRS), and Asociación Latinoamericana de Tórax (ALAT) clinical practice guidelines introduced a probability-based, multidisciplinary diagnostic model incorporating exposure history, high-resolution computed tomography (HRCT) patterns and bronchoalveolar lavage (BAL) lymphocytosis (11). In 2022, the ATS/ERS/JRS/ALAT guideline on progressive pulmonary fibrosis (PPF) established standardized progression criteria and issued a conditional recommendation for antifibrotic therapy with nintedanib in non-IPF PPF (12). An international Delphi survey provided a consensus-based diagnostic framework for CHP, highlighting the central role of identifying a causative antigen, along with radiologic and pathologic features, in establishing diagnostic confidence (13).
Age is increasingly recognized as a notable determinant of outcomes in HP. Older patients have a higher incidence of progression to fibrosis, greater mortality risk and more frequent complications such as respiratory failure, partly due to reduced physiological reserve, immune senescence and the presence of comorbidities (14-18). Despite these observations, the specific prognostic impact of age in HP has not been systematically quantified.
To address this gap, the present study, to the best of our knowledge, provides the first meta-analysis to synthesize available evidence on the prognostic implications of advanced age in HP. By integrating results across multiple cohorts, the aim was to establish a robust estimate of the age-prognosis relationship and highlight its clinical implications for risk stratification and management in older populations.
To the best of our knowledge, the present study is the first meta-analysis to systematically and quantitatively assess the prognostic implications of advanced age in HP. While individual studies have suggested a potential association, no prior effort has synthesized the evidence across multiple cohorts to provide a robust and generalizable estimate of this correlation. The present study not only consolidates current knowledge but also highlights key gaps in literature and provides a framework for age-stratified clinical decision-making. By focusing on an underexplored prognostic factor in a disease characterized by heterogeneous presentation and outcomes, the present meta-analysis offers novel insights that can inform future research, clinical guidelines and therapeutic prioritization in older populations affected by HP.
Materials and methods
The present study was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (19). The present systematic review has been registered in the International Prospective Register of Systematic Reviews (PROSPERO) with ID number CRD420251004649.
Search strategy
A systematic search was conducted in three main databases (Medline, Google Scholar and Cochrane), with time limits set between 1980-2025; grey literature was also included in the search. The references of the studies included were also manually and individually searched for more possible results. For the PubMed search, both free text and Medical Subject Headings (MeSH) terms were utilized. The MeSH terms employed in the search strategy included: Hypersensitivity pneumonitis [MeSH]; age factors [MeSH]; mortality [MeSH]; pulmonary fibrosis [MeSH]; respiratory failure [MeSH]; aged [MeSH]; prognosis [MeSH]; fibrosis [MeSH]; chronic disease [MeSH]; inflammation [MeSH]. Boolean operators (AND, OR) were used to combine these terms to maximize search efficiency. Specifically, the following search string was employed: ‘Hypersensitivity pneumonitis’ [MeSH] AND ‘age factors’ [MeSH] AND [‘mortality’ (MeSH) OR ‘pulmonary fibrosis’ (MeSH) OR ‘respiratory failure’(MeSH)].
Inclusion and exclusion criteria
Eligible studies for the present systematic review included populations of patients diagnosed with HP, encompassing various types such as bird fancier's lung, farmer's lung and other unidentified forms. Only studies involving patients aged ≥60 years were included, with the diagnosis confirmed through open lung biopsy, clinical medical history, HRCT findings or a combination of these diagnostic methods. The age range of the included participants spanned from 18-80 years. A comparator group was composed of patients with HP <60 years old.
The primary outcomes indicative of worse prognosis included mortality, rapid decline in forced vital capacity (FVC), rapid onset of respiratory failure and the need for long-term oxygen therapy (LTOT). The present systematic review incorporated observational studies, as well as randomized, quasi-randomized, pre-post and historical control studies. Studies that were limited to letters to the editor, editorials, comments, case reports and case series were excluded. Additionally, studies involving populations with comorbidities such as IPF, non-specific interstitial pneumonia, sarcoidosis or heart failure were excluded; non-human studies were also not considered.
Population, Intervention, Comparator, Outcome (PICO) framework
The PICO framework for this review is outlined as follows: Population, patients with HP aged ≥60 years, diagnosed using the criteria described in the inclusion section; intervention, the primary intervention is the assessment of worse prognosis through the examination of the association between older age and poorer clinical outcomes, as defined by the inclusion criteria; comparator, patients with HP aged 18-60 years were used as the comparator group; outcome, the primary outcomes are the differences in the measured correlations between older age and worse prognosis, focusing on mortality, rapid FVC decline, rapid onset of respiratory failure and the requirement for LTOT.
Outcomes
The primary outcomes indicative of worse prognosis were mortality, rapid FVC decline, rapid onset of respiratory failure and the requirement for LTOT. For each included study, the exact operational definition and measurement scale used for these endpoints was extracted.
Mortality was captured as all-cause death during follow-up and was analyzed as a binary outcome (dead/alive at the end of follow-up) or as time-to-event, depending on the original report. Rapid FVC decline was generally defined as a clinically marked fall in FVC, most commonly an absolute decline of ≥10 percentage points in % predicted FVC over 12 months or an equivalent annualized decline; when studies reported FVC as a continuous variable (ml or % predicted per year) was extracted accordingly. Rapid onset of respiratory failure was extracted as a binary variable indicating the development of chronic hypoxemic respiratory failure during follow-up, typically defined by the onset of resting hypoxemia requiring supplemental oxygen or by the occurrence of acute respiratory failure requiring hospitalization, as specified in each study. The requirement for LTOT was recorded as a yes/no variable based on the initiation of domiciliary oxygen therapy (usually ≥15 h/day) during follow-up.
PRISMA process
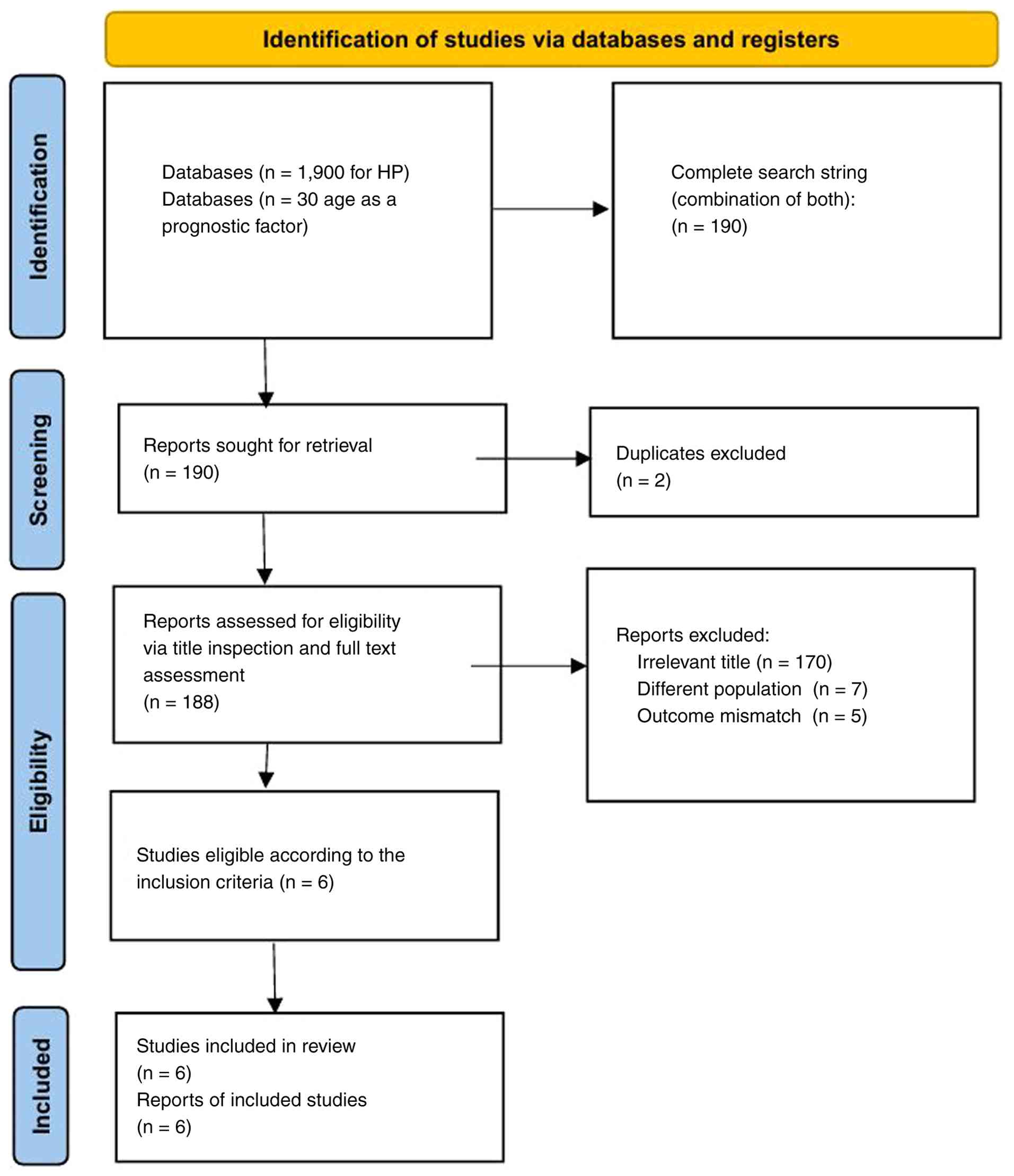
In the identification phase, a comprehensive search was conducted across databases, retrieving a total of 190 records. These records were derived from two search components: 1,900 records related to HP and 30 records considering age as a prognostic factor. After removing two duplicate records, 188 unique records remained for screening.
During the screening phase, all 188 records were evaluated based on their titles and abstracts. Of these, 170 were excluded due to irrelevant titles, seven were removed for involving a different population and five were excluded due to an outcome mismatch. This resulted in six studies progressing to full-text eligibility assessment.
In the eligibility phase, these six studies were thoroughly evaluated against the inclusion criteria. All six studies met the criteria and were deemed eligible for inclusion in the systematic review. Finally, in the inclusion phase, all six eligible studies were formally included in the systematic review. The flow diagram of the search strategy can be seen in Fig. 1.
Data extraction
Data extraction was conducted by two independent reviewers using a data extraction sheet that contained all the crucial information of each included study (study characteristics, author, year, location, sample size, population demographics, intervention details, outcome measures, results effect sizes, confidence intervals). Any discrepancies were resolved through constructive dialogue. The information included in the sheets were computed in an excel database form allowing easy retrieval and analysis during the synthesis phase of the present study.
Quality assessment-risk of bias
To measure and evaluate the quality of the included studies the Newcastle-Ottawa Scale (NOS) was used (20) for non-randomized observational studies. The NOS tool is a checklist designed to assess the quality of case-control studies. Key components of the tool evaluate whether cases are representative of the population, whether controls are drawn from the same population as cases, whether the exposure is well determined and whether the study appropriately accounts for confounding variables and presents outcomes in a consistent way. The tool typically includes a series of questions that are answered as ‘yes’, ‘no’, or ‘unclear’. Each response contributes to an overall quality score, helping researchers and practitioners gauge the strength of the evidence presented. The overall score of each study can be noted in Table I.
 |
Table I
Basic characteristics of the included studies with a population or sub-population of healthy individuals.
|
Table I
Basic characteristics of the included studies with a population or sub-population of healthy individuals.
| Author, year |
Study population |
Female sex, % |
Age, years |
Main comorbidities |
Underlying disease |
Outcome |
Country |
(Refs.) |
| Fernández Pérez et al, 2018 |
851 |
58 |
52 |
Not reported |
Hypersensitivity pneumonitis |
Death |
USA |
(6) |
| Jacob et al, 2018 |
233 |
60 |
62 |
Not reported |
Hypersensitivity pneumonitis |
FVC decline-Death |
USA |
(14) |
| Lima et al, 2009 |
103 |
62 |
56 |
Not reported |
Subacute and chronic hypersensitivity pneumonitis |
Death-respiratory failure |
Mexico |
(15) |
| Pérez-Padilla et al, 1993 |
125 |
95 |
41 (±14) |
Not reported |
Chronic pigeon breeder's lung (chronic HP due to domestic pigeon exposure) |
Death |
Brazil |
(16) |
| Ojanguren et al, 2019 |
160 |
58.1 |
60.9 (±12.9) |
Not reported |
Chronic hypersensitivity pneumonitis |
Death |
Spain |
(17) |
| Vourlekis et al, 2021 |
46 |
48.6 |
Fibrotic: 61 (±13); non-fibrotic: 52 (±13) |
Not reported |
Subacute/chronic hypersensitivity pneumonitis (biopsy-proven; fibrotic and non-fibrotic groups) |
Death |
USA |
(18) |
Statistical analysis
Statistical analysis was conducted using the Meta-Mar tool (https://www.meta-mar.com/) for meta-analysis (GT7.1). The primary effect measure was the hazard ratio (HR) with corresponding 95% confidence intervals (CIs), as reported in the included studies. All eligible studies assessed the association between older age and worse prognosis using multivariable regression models and provided adjusted HR estimates. When multiple models were available, the most fully adjusted HRs were extracted for quantitative synthesis.
Pooled HRs were calculated using a random-effects model to account for potential clinical and methodological heterogeneity among studies. Statistical heterogeneity was evaluated using Cochran's Q test and quantified with the I² statistic, with I²>50% considered indicative of substantial heterogeneity.
Publication bias was assessed through visual inspection of funnel plots and formally evaluated using Begg's rank correlation test and Egger's linear regression test. P<0.05 was considered suggestive of potential small-study effects.
Sensitivity analyses were performed to examine the stability and robustness of the pooled estimates. All statistical tests were two-sided, and P<0.05 was considered to indicate a statistically significant difference.
Results
Overview of the included studies
The present meta-analysis synthesized data from six observational studies (6,14-18) evaluating the correlation between older age and worse prognosis in patients with HP. A total of 1,518 patients were included across these studies.
Description of the included studies
Across cohorts, advanced age emerged as an independent predictor of adverse outcomes, with most studies confirming reduced survival and increased progression risk among older individuals.
In a large retrospective cohort study using U.S. health insurance claims data from >150 million individuals, Fernández Pérez et al (6) investigated the incidence, prevalence and survival outcomes of patients with HP, including FHP and CHP. The prevalence of HP increased with age, reaching 11.2 cases per 100,000 in individuals aged ≥65. Mortality analysis showed that patients aged ≥65 had the highest all-cause mortality rate at 115.9 deaths per 1,000 person-years, compared with 37.5 in patients aged 55-64 and 16.0 in patients aged 45-54. Notably, patients with FHP died markedly faster than non-fibrotic cases, and survival at 7 years was only 58% for patients FHP vs. 71% for HP overall. These findings support older age, particularly in the presence of fibrosis, as a strong negative prognostic indicator in HP.
Jacob et al (14) conducted a retrospective analysis of 233 patients with HP to evaluate the prevalence and prognostic implications of pleuroparenchymal fibroelastosis (PPFE) and emphysema using HRCT. Among the cohort, 23% exhibited marked PPFE, which was notably associated with impaired lung function and higher mortality. Patients with marked PPFE had markedly lower mean FVC (FVC: 57.5% vs. 73.4% predicted) and diffusing capacity for carbon monoxide (DLCO: 34.9% vs. 43.7% predicted) compared with those without or with only minor PPFE. In multivariable Cox regression, marked PPFE independently predicted mortality with a HR of 1.94, even after adjusting for age, sex, interstitial lung disease extent, emphysema and smoking status.
Lima et al (15) conducted a retrospective cohort study of 103 patients with biopsy-proven subacute and CHP in Brazil, aiming to determine the prognostic significance of clinical, radiologic and histopathological features. The median age of participants was 56 years, and 74% had HRCT evidence of fibrosis, qualifying them as CHP cases. By univariate Cox regression, older age was a notable predictor of mortality (HR=1.05 per year increase). In multivariate analysis, older age remained independently associated with mortality (HR=1.10), along with oxygen desaturation during exercise and absence of mosaic attenuation on HRCT.
Pérez-Padilla et al (16) performed a prospective analysis of 125 patients from Mexico with interstitial lung disease, including 78 with chronic pigeon breeder's lung (CPBL) and 47 with usual interstitial pneumonia (UIP), with or without bird exposure. The study aimed to compare mortality outcomes and prognostic indicators among these subgroups. Older age was found to be a notable predictor of mortality, with patients aged >44 years having a HR of 2.5 compared with younger patients. Median survival for patients with CPBL was >84 months, whereas it was only 31 months for those with UIP. The 5-year survival probability was 71% for CPBL and 23% for UIP (including bird-exposed and non-exposed), highlighting the improved prognosis of CPBL despite marked mortality. Multivariable Cox regression confirmed that, along with fibrosis and radiologic honeycombing, increasing age remained independently associated with reduced survival (RR=1.5 per decade).
Ojanguren et al (17) conducted a longitudinal study of 160 patients diagnosed with CHP between 2004-2013, aiming to identify clinical, radiological and histopathological predictors of survival. After a median follow-up of 7.0 years, 73 patients had died or undergone lung transplantation. Older age at diagnosis was an independent predictor of mortality in multivariate Cox regression analysis (HR=1.038 per year). Other independent predictors included low lymphocyte percentage in BAL (HR=0.973 per percent increase), reduced DLCO (% predicted; HR=0.956), presence of honeycombing on HRCT (HR=1.745) and an UIP histopathologic pattern (HR=2.260).
Vourlekis et al (18) conducted a retrospective cohort study involving 72 patients with biopsy-confirmed HP to evaluate the effect of pulmonary fibrosis on survival. A total of 46 patients (64%) had histopathological evidence of fibrosis, whereas 26 were classified as non-fibrotic. Patients in the fibrotic group were markedly older, with a mean age of 61 years compared with 52 years in the non-fibrotic group. During follow-up, mortality was markedly higher in patients with fibrosis (48%) compared with patients with no fibrosis, with respiratory deaths comprising the majority of cases (14 vs. 2). The median survival in patients with fibrosis was 7.1 years, compared with >20.9 years in the non-fibrotic group. In multivariable Cox regression analysis adjusted for age, fibrosis was the strongest predictor of mortality, with a HR of 6.01.
Table II provides the key characteristics of the included studies, including study year, geographic location, population size and outcome definitions (mortality, FVC decline, respiratory failure, need for LTOT).
|
Table II
Quality assessment of included studies.
|
Table II
Quality assessment of included studies.
| Author, year |
Ottawa scale |
(Refs.) |
| Fernández Pérez et al, 2018 |
Selection: 4; comparability: 1; outcome: 3 (total 8/9) |
(6) |
| Jacob et al, 2018 |
Selection: 4; comparability: 2; outcome: 2 (total 8/9) |
(14) |
| Lima et al, 2009 |
Selection: 3; comparability: 1; outcome: 3 (total 7/9) |
(15) |
| Pérez-Padilla et al, 1993 |
Selection: 3; comparability: 1; outcome: 3 (total 7/9) |
(16) |
| Ojanguren et al, 2019 |
Selection: 3; comparability: 2; outcome: 3 (total 8/9) |
(17) |
| Vourlekis et al, 2021 |
Selection: 3; comparability: 2; outcome: 3 (total 8/9) |
(18) |
Association between age (per 1-year increase) and all-cause mortality: random-effects meta-analysis
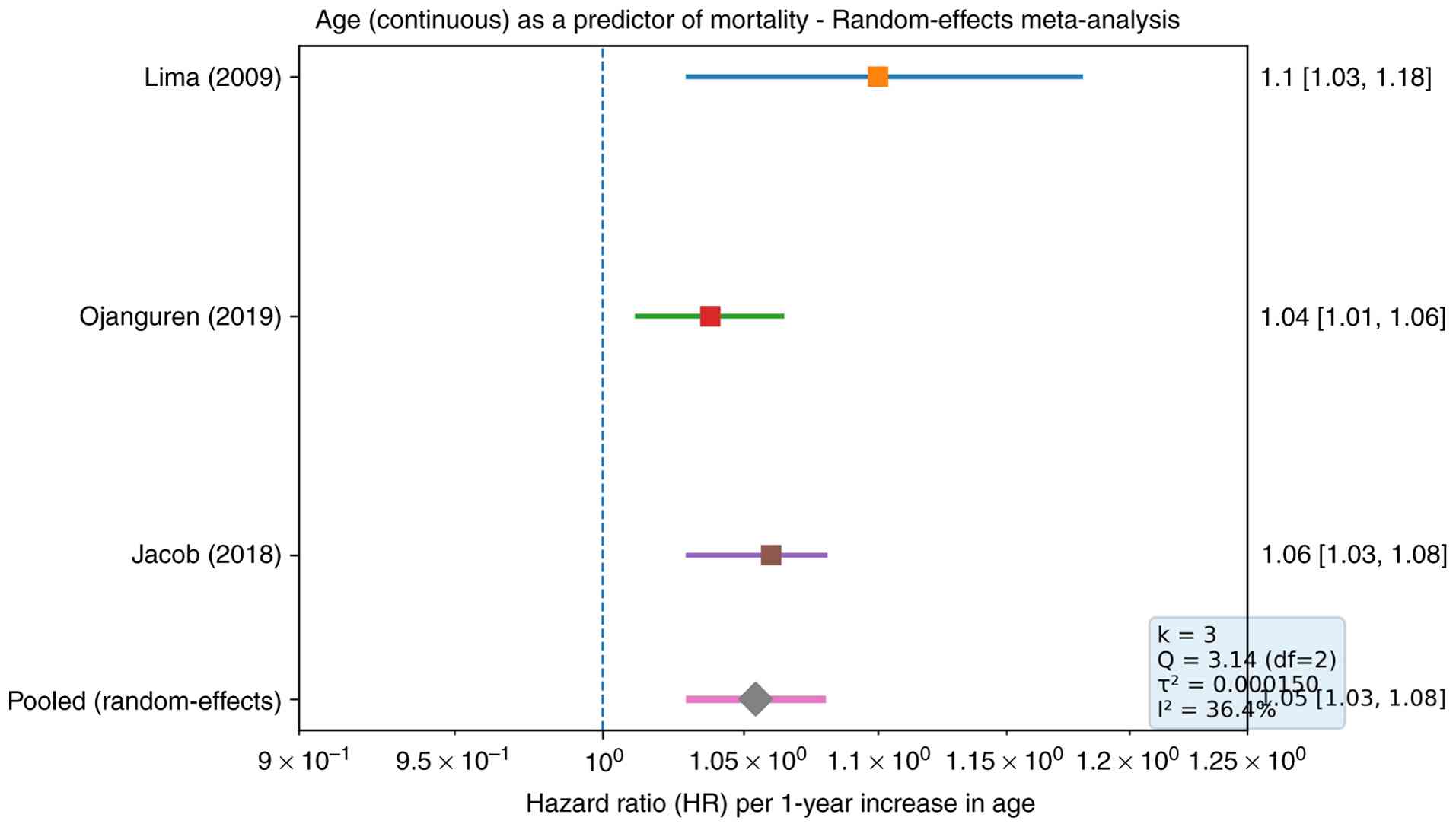
Only three of the six eligible studies reported age effects in a directly comparable form (continuous age, per 1-year increase) with extractable adjusted HRs. In Lima et al (15), older age was associated with increased mortality (adjusted HR 1.10, 95% CI 1.03-1.18). Similarly, Ojanguren et al (17) reported a notable association between age and the risk of death/transplantation (adjusted HR 1.038, 95% CI 1.012-1.064) and Jacob et al (14) found that increasing age independently predicted all-cause mortality (adjusted HR 1.06, 95% CI 1.03-1.08). Pooling these estimates using a random-effects DerSimonian-Laird model (k=3) yielded a pooled HR of 1.054 per 1-year increase in age (95% CI 1.030-1.079), indicating that each additional year of age was associated with an average 5.4% increase in the hazard of death; the pooled effect was statistically significant (P<0.00001). The between-study heterogeneity was moderate (Q=3.14; df=2; τ²=0.000150; I²=36.4%) (Fig. 2).
Publication bias
Publication bias was assessed for the meta-analysis. Visual inspection of the funnel plot did not suggest marked asymmetry; however, interpretation is limited by the small number of included studies. Formal statistical evaluation using Begg's rank correlation test showed no evidence of publication bias (P>0.99) (Fig. S1). Egger's regression asymmetry test likewise did not indicate statistically significant small-study effects (P=0.065) (Fig. S2). Nevertheless, given that fewer than 10 studies were available, both graphical and statistical assessments of publication bias are underpowered, and these results should be interpreted with caution.
Discussion
Prognosis in HP is markedly variable, influenced by factors such as antigen exposure, disease phenotype (acute, subacute or chronic) and patient-specific characteristics. Previous studies have identified age >65 as a notable predictor of worse prognosis in HP, highlighting the critical role of age in disease management and outcome prediction (6,14-18).
To the best of our knowledge, the present meta-analysis is the first to systematically synthesize data from multiple studies on the relationship between age and prognosis in HP, consistently demonstrating that patients aged ≥65 experience poorer outcomes compared with younger individuals. Key findings from previous studies include higher mortality rates, increased risk of disease progression to pulmonary fibrosis and a greater likelihood of respiratory failure in older patients. These results align with the broader understanding of aging as a risk factor for worse outcomes in chronic lung diseases, such as IPF and chronic obstructive pulmonary disease (21).
The association between advanced age and worse prognosis in HP can be attributed to several interconnected factors. Aging is associated with structural and functional changes in the lungs, including reduced elastic recoil, decreased alveolar surface area and impaired mucociliary clearance (22). These changes predispose older individuals to more severe lung injury and diminished capacity for repair following antigen exposure. The aging immune system also undergoes marked alterations, including reduced T-cell diversity, impaired antigen presentation and chronic low-grade inflammation. In HP, these immune changes may exacerbate the dysregulated response to inhaled antigens, leading to more severe and persistent inflammation (23).
Fig. 3 illustrates the immune response leading to inflammation and fibrosis in older individuals with HP.
Older patients with HP frequently have comorbidities such as cardiovascular disease, diabetes and chronic kidney disease, complicating disease management and contributing to worse outcomes. Age-related frailty and diminished physiological reserve may limit tolerance to aggressive therapies (22,23). Additionally, HP in older adults is often misdiagnosed or diagnosed at a later stage due to nonspecific symptoms (such as dyspnea and cough) that overlap with other age-related conditions. Delayed diagnosis can result in advanced disease at presentation, reducing the likelihood of favorable outcomes.
Several studies in the existing literature have underscored the role of advanced age as a prognostic determinant in HP, although most have not performed a focused quantitative synthesis on this variable. A systematic review and meta-analysis published in 2022, which included 21 studies and 3,077 patients with HP, identified increasing age as a notable predictor of mortality, with a pooled OR of 1.036 per year increase in age (95% CI, 1.025-1.046) in multivariate analysis (24). Similarly, a multicenter retrospective study analyzing 292 patients with CHP demonstrated that older age was associated with both disease progression and mortality, further supporting the necessity for age-adapted diagnostic and management strategies (25).
New multicenter cohorts corroborate older age as an independent predictor of progression in FHP and tie adverse outcomes to fibroblastic foci on biopsy, lower FVC and lower BAL lymphocytosis, whereas quantitative CT fibrosis scoring adds prognostic granularity. These findings align with our meta-analytic signal and support incorporating age into risk tools along with radiologic, histologic, and laboratory markers (14,26-29).
Additionally, a narrative review on prognostic factors in CHP highlighted older age, severe pulmonary function decline and established fibrosis as the primary cause of reduced survival (27). While these studies affirm the prognostic relevance of age, none of them offered a dedicated meta-analytic synthesis specifically quantifying the correlation between advanced age and HP outcomes. The present study addresses this gap by offering a pooled correlation estimate and evaluating heterogeneity, thus enhancing the understanding of age as a prognostic marker and contributing to evidence-based risk stratification in HP.
The identification of age >65 as a predictor of worse prognosis in HP carries notable clinical implications, and by incorporating age into prognostic models can enhance risk stratification and guide treatment decisions (28,29). Older patients may benefit from closer monitoring, early intervention and multidisciplinary care to address comorbidities and optimize outcomes (30). However, older patients with HP may have reduced tolerance for immunosuppressive therapies, such as corticosteroids, due to increased risks of adverse effects (such as osteoporosis and infections). In these cases, alternative strategies, including antigen avoidance and antifibrotic agents, should be considered.
In CHP/FHP with progression, nintedanib consistently reduces the rate of FVC decline in subgroup analyses, concordant with the PPF guideline. Evidence for pirfenidone remains low certainty but suggests benefit in selected progressive phenotypes; ongoing and real-world studies continue to refine patient selection. Where no inciting antigen is identified, data caution against routine immunosuppression (8-11). Management of HP in older adults should focus on patient-centered care, considering individual goals, functional status and quality of life. Shared decision-making is essential to balance the potential benefits and risks of treatment (9,31).
While the present meta-analysis provides valuable insights, several limitations should be acknowledged. The included studies varied in terms of diagnostic criteria, disease phenotypes, and outcome measures, which may have introduced heterogeneity and limited the generalizability of the findings. Age is often associated with other prognostic factors, such as smoking history, antigen exposure duration and comorbidities; although multivariate analyses were performed, residual confounding may have influenced the results. Additionally, most studies were retrospective or cross-sectional, which limits the ability to establish causal relationships or assess the long-term impact of age on disease progression. The inclusion of only six observational studies with a total of 1,518 participants may limit the generalizability and statistical power of the findings, underlining the need for further large-scale investigations.
The observed heterogeneity across studies (I2=36.4%) is substantial and warrants careful consideration; several factors may account for this variability. First, geographic settings and environmental exposures differ markedly, with studies conducted in North America, Europe and Latin America where inciting antigens and occupational risk profiles vary widely, potentially influencing both disease phenotype and prognosis. Second, diagnostic methods were not uniform across studies; while some cohorts relied on HRCT and multidisciplinary consensus, others used biopsy confirmation, introducing differences in case definition and disease severity at inclusion. Third, patient selection criteria varied, with certain studies focusing on FHP or CHP whereas others included both acute and chronic forms, leading to heterogeneity in disease trajectory and mortality risk. These methodological and population-level differences likely contribute to the between-study variation observed in our pooled estimates and highlight the need for standardized diagnostic criteria and subtype-specific analyses in future meta-analyses.
The findings of the present meta-analysis may also contribute to shaping future diagnostic and therapeutic strategies in HP. Given the consistent association between older age and worse prognosis, incorporating age into risk stratification models could enhance early identification of high-risk individuals, particularly those presenting with fibrotic features or reduced pulmonary reserve. Diagnostic algorithms may benefit from integrating age-adjusted thresholds for clinical suspicion and prioritize early HRCT imaging and BAL in older patients. On a therapeutic level, older patients may require individualized treatment plans that balance efficacy with tolerability, considering their reduced physiological reserve and higher burden of comorbidities. Moreover, the use of antifibrotic agents, currently under investigation in chronic fibrosing interstitial lung diseases, may prove beneficial in selected older patients with HP at high risk for progression. Thus, the present study not only reinforces the prognostic role of age but also highlights its relevance in guiding patient-centered diagnostic and treatment approaches.
The findings of the present meta-analysis underscore the need for further research to improve the understanding of the role of age in HP prognosis. Investigating the molecular and cellular mechanisms underlying age-related differences in HP pathogenesis could identify novel therapeutic targets. Longitudinal studies are required to evaluate the impact of age on disease progression, treatment response and survival in HP. Developing age-specific treatment algorithms and prognostic tools could improve outcomes for older patients with HP.
While Dasgupta et al (24) provided a comprehensive synthesis of multiple risk factors for mortality in HP, to the best of our knowledge, the present study is the first meta-analysis dedicated specifically to the prognostic role of advanced age. By focusing exclusively on age, converting HRs into correlation coefficients and incorporating broader outcomes such as FVC decline, respiratory failure and LTOT, the present study complements existing evidence and provides novel insights that are clinically relevant for age-adapted management strategies in HP.
In conclusion, future research should aim to clarify the underlying mechanisms of age-related differences in HP progression and explore more personalized treatment approaches for elderly patients. The findings support the hypothesis that age may be a relevant prognostic factor in HP. Clinicians should remain vigilant in the management of elderly patients with HP, and further research is needed to explore underlying mechanisms and age-specific therapeutic strategies.
Supplementary Material
Funnel plot for publication bias assessment using Egger's regression test. Association between age (per 1-year increase) and all-cause mortality in hypersensitivity pneumonitis.
Funnel plot for publication bias assessment using Begg's rank correlation test. Association between age (per 1-year increase) and all-cause mortality in hypersensitivity pneumonitis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
KD and VEG conceptualized the study. KD, VEG, TVK, VTM, AC, NT, ES, VEG and DAS made a substantial contribution to data interpretation and analysis and wrote and prepared the draft of the manuscript. KD and VEG analyzed the data and provided critical revisions. All authors contributed to manuscript revision and approved the final version of the manuscript. KD and VEG confirm the authenticity of all the raw data.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had no personal involvement in the reviewing process, or any influence in terms of adjudicating on the final decision, for this article. The other authors declare that they have no competing interests.
Use of artificial intelligence tools
During the preparation of this work, AI tool Chat GPT was used to improve the readability and language of the manuscript, and subsequently, the authors revised and edited the content produced by the AI tool as necessary, taking full responsibility for the ultimate content of the present manuscript.
References
|
1
|
Vasakova M, Morell F, Walsh S, Leslie K and Raghu G: Hypersensitivity pneumonitis: Perspectives in diagnosis and management. Am J Respir Crit Care Med. 196:680–689. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Martinez FJ and Flaherty KR: Diagnosis and treatment of fibrotic hypersensitivity pneumonia. Where we stand and where we need to go. Am J Respir Crit Care Med. 196:690–699. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang P, Jones KD, Urisman A, Elicker BM, Urbania T, Johannson KA, Assayag D, Lee J, Wolters PJ, Collard HR and Koth LL: Pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest. 152:502–509. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Raghu G, Amatto VC, Behr J and Stowasser S: Comorbidities in idiopathic pulmonary fibrosis patients: A systematic literature review. Eur Respir J. 46:1113–1130. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, et al: An official American thoracic society/European respiratory society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 188:733–748. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R and Cole AL: Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: A claims-based cohort analysis. Ann Am Thorac Soc. 15:460–469. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fernández Pérez ER, Travis WD, Lynch DA, Brown KK, Johannson KA, Selman M, Ryu JH, Wells AU, Tony Huang YC, Pereira CAC, et al: Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest. 160:e97–e156. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Adegunsoye A, Oldham JM, Fernández Pérez ER, Hamblin M, Patel N, Tener M, Bhanot D, Robinson L, Bullick S, Chen L, et al: Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res. 3:00016–2017. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, et al: Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 381:1718–1727. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, Moua T, Crestani B, Wuyts WA, Stowasser S, et al: Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. 8:453–460. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, Bargagli E, Chung JH, Collins BF, Bendstrup E, et al: Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 202:e36–e69. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, et al: Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 205:e18–e47. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Morisset J, Johannson KA, Jones KD, Wolters PJ, Collard HR, Walsh SLF and Ley B: HP Delphi Collaborators. Identification of diagnostic criteria for chronic hypersensitivity pneumonitis: An international modified delphi survey. Am J Respir Crit Care Med. 197:1036–1044. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jacob J, Odink A, Brun AL, Macaluso C, de Lauretis A, Kokosi M, Devaraj A, Desai S, Renzoni E and Wells AU: Functional associations of pleuroparenchymal fibroelastosis and emphysema with hypersensitivity pneumonitis. Respir Med. 138:95–101. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lima MS, Coletta ENAM, Ferreira RG, Jasinowodolinski D, Arakaki JSO, Rodrigues SCS, Rocha NANS and Pereira CAC: Subacute and chronic hypersensitivity pneumonitis: Histopathological patterns and survival. Respir Med. 103:508–515. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pérez-Padilla R, Salas J, Chapela R, Sánchez M, Carrillo G, Pérez R, Sansores R, Gaxiola M and Selman M: Mortality in Mexican patients with chronic pigeon breeder's lung compared with those with usual interstitial pneumonia. Am Rev Respir Dis. 148:49–53. 1993.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ojanguren I, Morell F, Ramón MA, Villar A, Romero C, Cruz MJ and Muñoz X: Long-term outcomes in chronic hypersensitivity pneumonitis. Allergy. 74:944–952. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Vourlekis JS, Schwarz MI, Cherniack RM, Curran-Everett D, Cool CD, Tuder RM, King TE Jr and Brown KK: The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med. 116:662–668. 2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan SE, et al: The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ. 372(n71)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wells GA, Shea BJ, O'Connell D, Peterson J, Welch V, Losos M and Tugwell P: The Newcastle-Ottawa Scale (NOS) for assessing the quality of non-randomized studies in meta-analyses. Kansas City (MO): American Academy of General Practice, 2000.
|
|
21
|
Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, et al: Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir Med. 1:309–317. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Song JW, Hong SB, Lim CM, Koh Y and Kim DS: Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur Respir J. 37:356–363. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N and Yoshizawa Y: Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest. 134:1265–1270. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dasgupta S, Bhattacharya A, Abhijit RD, Roy Chowdhury S and Chaudhury K: Risk factors associated with mortality in hypersensitivity pneumonitis: A meta-analysis. Expert Rev Respir Med. 16:801–811. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Trushenko NV, Suvorova OA, Pershina ES, Nekludova GV, Chikina SY, Levina IA, Chernyaev AL, Samsonova MV, Tyurin IE, Mustafina MK, et al: Predictors of progression and mortality in patients with chronic hypersensitivity pneumonitis: retrospective analysis of registry of fibrosing interstitial lung diseases. Life (Basel). 13(467)2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Cano-Jiménez E, Villar Gómez A, Velez Segovia E, Aburto Barrenechea M, Sellarés Torres J, Francesqui J, Portillo Carroz K, Solis Solis AJ, Acosta Fernández O, Llanos González AB, et al: Prognostic factors of progressive fibrotic hypersensitivity pneumonitis: A large, retrospective, multicentre, observational cohort study. ERJ Open Res. 10:00405–2023. 2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Creamer AW and Barratt SL: Prognostic factors in chronic hypersensitivity pneumonitis. Eur Respir Rev. 29(190167)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, Flynn A, Molyneaux PL, Braybrooke R, Divyateja H, et al: An epithelial biomarker signature for idiopathic pulmonary fibrosis: An analysis from the multicentre PROFILE cohort study. Lancet Respir Med. 5:946–955. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Long X, He X, Ohshimo S, Griese M, Sarria R, Guzman J, Costabel U and Bonella F: Serum YKL-40 as predictor of outcome in hypersensitivity pneumonitis. Eur Respir J. 49(1501924)2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sahin H, Brown KK, Curran-Everett D, Hale V, Cool CD, Vourlekis JS and Lynch DA: Chronic hypersensitivity pneumonitis: CT features comparison with pathologic evidence of fibrosis and survival. Radiology. 244:591–598. 2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Khan F, Stewart I, Howard L, McKeever TM, Jones S, Hearson G, Braybrooke R, Edwards C, Jenkins G and Saini G: The its not JUST idiopathic pulmonary fibrosis Study (INJUSTIS): Description of the protocol for a multicentre prospective observational cohort study identifying biomarkers of progressive fibrotic lung disease. BMJ Open Respir Res. 6(e000439)2019.PubMed/NCBI View Article : Google Scholar
|