1. Introduction
Acute respiratory distress syndrome (ARDS) is a
life-threatening respiratory disorder with high mortality rates,
affecting ~10% of intensive care unit admissions worldwide
(1-4).
Despite advances in ventilatory strategies and critical care,
mortality remains between 35-46%. ARDS arises from a complex
interplay of numerous concurrent injuries, inflammatory responses
and dysregulated coagulation pathways, affecting both the pulmonary
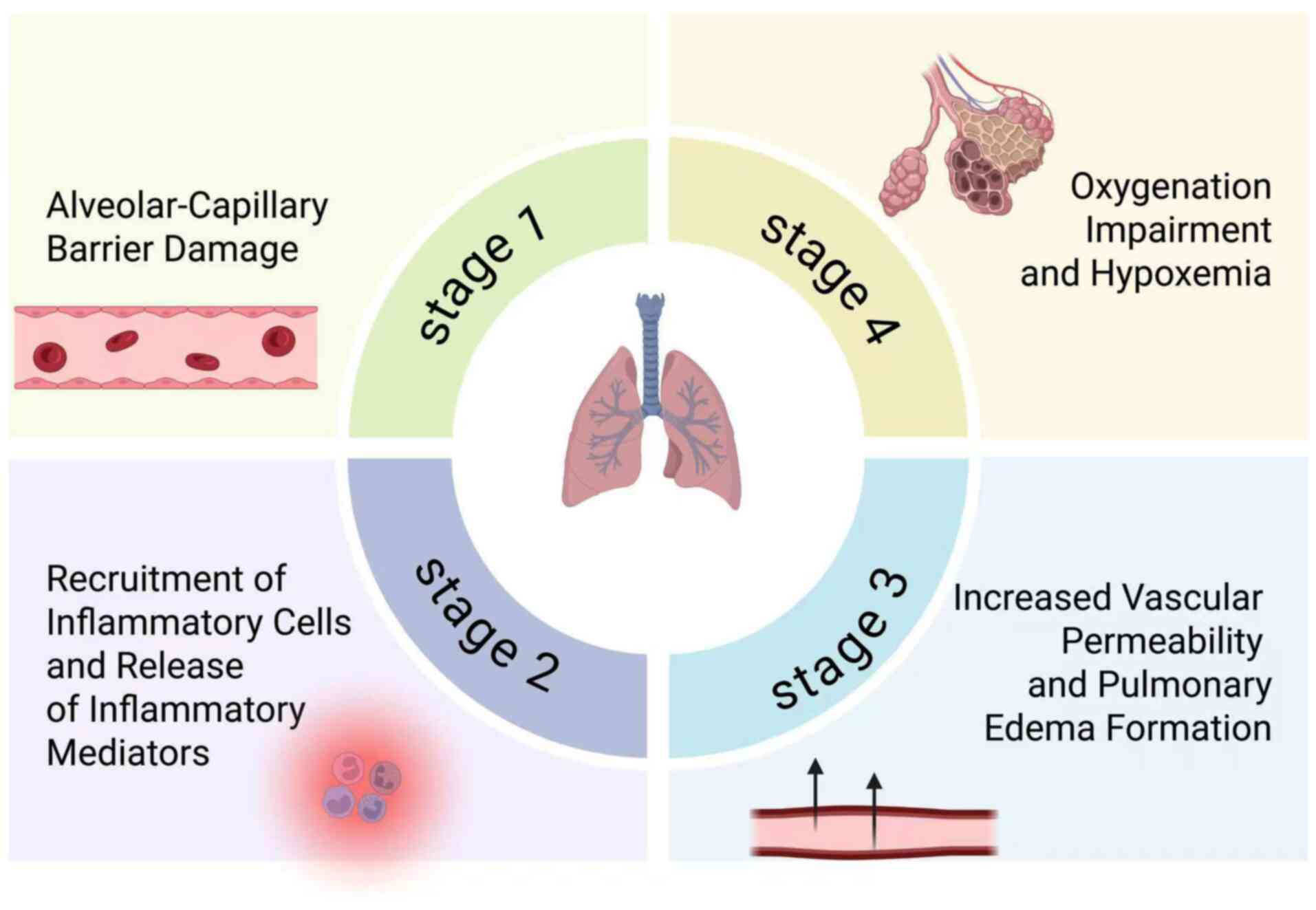
system and the whole body. As shown in Fig. 1, ARDS progression involves a
cascade of pathological events, including alveolar-capillary
barrier disruption, inflammatory cell recruitment, and increased
vascular permeability, leading to pulmonary edema and subsequent
hypoxemia. Furthermore, early fibroproliferative responses are key
determinants of lung remodeling and prognosis. A hallmark of ARDS
is the early onset of fibroproliferative changes in the lung, which
are associated with poor outcomes (5). These fibroproliferative responses are
among the earliest events in ARDS and highlight the need to
understand molecular drivers of lung remodeling and to develop
novel therapeutic strategies targeting early pathogenic events.
Autophagy, recognized by the 2016 Nobel Prize in
Physiology or Medicine awarded to Ohsumi (6), is a fundamental cellular
quality-control process responsible for degrading damaged
organelles, aggregated proteins and invading pathogens (7,8). It
exists in both non-selective and selective forms, such as
mitophagy, ferritinophagy and reticulophagy. Dysregulated autophagy
has been implicated in a number of pathological conditions,
including acute lung injury (ALI); however, its specific and
context-dependent roles in ARDS pathogenesis remain incompletely
understood.
The epithelial-mesenchymal transition (EMT) is an
additional key biological process contributing to tissue remodeling
and fibrosis. In the lung, EMT of epithelial or endothelial cells
can generate fibroblasts that deposit extracellular matrix (ECM),
contributing to fibrosis (9). In
the present study, the term EMT is used broadly to include both the
classical EMT in epithelial cells and the
endothelial-to-mesenchymal transition (EndMT) in endothelial cells.
Emerging evidence has indicated that autophagy can modulate EMT,
either promoting or restraining it, depending on the context and
type of selective autophagy involved (10-13).
However, the molecular crosstalk between autophagy and EMT in ARDS
is incompletely understood and existing studies often examine these
processes in isolation (14-16).
The present review assesses current knowledge
regarding the interplay between classical and selective autophagy
(mitophagy, ferritinophagy and reticulophagy) and EMT in ARDS,
highlighting converging and diverging findings and proposing a
hypothesis-driven framework to guide future research. Through
focusing on the intersection of these pathways, the present review
provides a framework beyond descriptive summaries and toward
mechanistic and translational perspectives, which may inform
rational targeting of autophagy pathways in future ARDS
therapies.
2. Literature search and study
selection
A comprehensive literature search was conducted to
identify studies relevant to autophagy-EMT crosstalk in ARDS and
pulmonary fibrosis. The databases PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Web of Science
(https://www.webofscience.com/) were
systematically searched for articles published in English between
January 2000 and March 2025.
The search strategy combined medical subject heading
terms and free-text keywords, including but not limited to: ‘acute
respiratory distress syndrome’, ‘acute lung injury’, ‘pulmonary
fibrosis’, ‘epithelial-mesenchymal transition’,
‘endothelial-mesenchymal transition’, ‘autophagy’,
‘macroautophagy’, ‘mitophagy’, ‘ferritinophagy’ and endoplasmic
reticulum (ER)-selective autophagy (‘ER-phagy’). Boolean operators
(‘AND’ and ‘OR’) were applied to refine the search.
Studies were included if they met one or more of the
following criteria: i) Investigated autophagy or selective
autophagy subtypes in ARDS or ALI models; ii) examined EMT or EndMT
in the context of lung injury or pulmonary fibrosis; or iii)
provided mechanistic insights into autophagy-EMT interactions
derived from other disease models (such as cancer, fibrotic or
metabolic disorders), provided that the reported mechanisms were
mechanistically linked to processes known to contribute to ARDS,
including inflammation, epithelial or endothelial injury, and
alveolar-capillary barrier dysfunction. Reviews, original
experimental studies and translational research articles were
considered.
Studies were excluded if they: i) Lacked mechanistic
relevance to autophagy or EMT; ii) were not available in full text;
or iii) were published in languages other than English. Article
screening and study selection were independently performed by two
authors. Any discrepancies were resolved through discussion until a
consensus was reached. Given the heterogeneity of experimental
models and outcomes, the present research was designed as a
narrative review and no formal meta-analysis was performed.
3. Role of autophagy in ARDS
Macroautophagy in ARDS: Mechanisms and
evidence
Macroautophagy, a lysosome-dependent degradation
pathway, serves a stage- and context-dependent role in the
pathogenesis of ARDS. During the early phase of injury, moderate
autophagy activation can limit inflammatory damage, maintain
epithelial-endothelial barrier integrity and support cellular
homeostasis. By contrast, excessive or prolonged autophagy may
induce autophagic cell death and promote fibrotic remodeling,
underscoring the need for precise temporal and quantitative
regulation (17).
Macroautophagy proceeds through initiation,
phagophore formation, autophagosome maturation and lysosomal
degradation, as shown in Fig. 2
(18), which illustrates key
regulatory pathways, including mTOR, unc-51 like autophagy
activating kinase 1 (ULK1), AMP-activated protein kinase (AMPK) and
PI3K/AKT, which integrate environmental and cellular stress signals
to modulate autophagic activity during ARDS. Under healthy
conditions, temporal regulation of these pathways limits
inflammation and prevents fibrotic remodeling (19).
In ARDS models, mTOR-centered regulation has been
extensively studied (20-22).
Rapamycin-mediated mTOR inhibition enhances autophagy and
alleviates lipopolysaccharide (LPS)-induced lung injury, whereas
pharmacological blockade of autophagy by 3-methyladenine (3-MA)
generally aggravates acute-phase pathology (21). Notably, in chronic or fibrotic
contexts, 3-MA may attenuate tissue injury by limiting maladaptive
or excessive autophagy, suggesting that the effects of autophagy
modulation are highly context-dependent, varying with disease
stage, target cell populations and timing of intervention (23).
Cell-type-specific roles further complicate this. In
macrophages, sirtuin 6 promotes M2 polarization and suppresses
inflammatory responses partially through autophagy activation
(24-27).
Dioscin enhances alveolar macrophage autophagy, mitigating
silica-induced lung injury and fibrosis (28). Mesenchymal stem cell-derived
exosomes demonstrate cargo-dependent effects, as
microRNA-377-3p-containing exosomes promote protective autophagy
(29), whereas heparanase-rich
exosomes may exacerbate fibrotic remodeling (30).
Natural compounds such as astragaloside IV have also
been investigated. In ARDS cell models, astragaloside IV inhibits
excessive autophagy, reduces oxidative stress and preserves
epithelial barrier integrity (31). While such agents are attractive for
their relative safety and multitarget activity, current evidence is
largely preclinical, with limited data regarding long-term efficacy
or clinical applicability.
Overall, the effects of macroautophagy depend on
activation intensity, timing and cellular context, with both
protective and maladaptive roles reported. Existing studies are
limited by heterogeneous models and predominantly short-term
endpoints. Future research should therefore prioritize standardized
ARDS models, longitudinal analyses and targeted modulation
strategies, particularly within immune cell populations and
stem-cell-derived vesicle systems. Key molecular targets,
experimental models and main conclusions are summarized in Table I.
 | Table IRoles and molecular mechanisms of
macroautophagy in ARDS. |
Table I
Roles and molecular mechanisms of
macroautophagy in ARDS.
| First author,
year | Drug/molecule | Autophagy
state | Main molecular
targets and pathways | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Qin et al,
2020 | Rapamycin | Activates | mTOR inhibition;
ULK1-VPS34 activation | LPS-induced ALI rat
model (ARDS-specific) | Enhances autophagy
and attenuates inflammatory lung injury | (21) |
| Wang et al,
2023 | WWOX | Activates | mTOR-ULK1 signaling
axis | LPS-induced ALI
cells and mouse models (ARDS-specific) | Promotes protective
autophagy and reduces lung inflammation | (23) |
| Wei et al,
2020 | MicroRNA-377-3p
(MSC-derived exosomes) | Activates | RPTOR (mTOR complex
component) | LPS-induced ALI
mouse model (ARDS-specific) | Enhances protective
autophagy and mitigates ARDS severity | (29) |
| Liu et al,
2023 | SIRT6 | Activates | ERK1/2 signaling
pathway | LPS-treated A549
cells and a murine ARDS model (ARDS-specific) | Suppresses
inflammation and alleviates ARDS | (27) |
| Wang et al,
2022 | SIRT6 | Activates | Macrophage M2
polarization | LPS-induced BMDMs
and mouse ARDS model (ARDS-specific) | Reduces
inflammatory injury through immunomodulatory autophagy | (24) |
| Du et al,
2019 | Dioscin | Activates | Alveolar macrophage
autophagy | CS-induced
silicosis in mouse ARDS model (pulmonary relevant) | Attenuates lung
injury and fibrosis through macrophage autophagy | (28) |
| Liu et al,
2020 | Astragaloside
IV | Inhibits | Oxidative stress
and inflammatory signaling | LPS-induced ARDS
cell model (ARDS-specific) | Prevents
maladaptive autophagy and preserves epithelial barrier
integrity | (31) |
Ferritinophagy in ARDS: Functions and
implications
Ferritinophagy, a selective autophagy pathway
mediated by nuclear receptor coactivator 4 (NCOA4), degrades
ferritin to release stored intracellular iron. While ferritin
serves as the main cytosolic iron reservoir and is key in iron
homeostasis, dysregulated ferritinophagy can lead to iron overload,
driving ferroptosis, a regulated, iron-dependent form of cell death
implicated in ARDS pathogenesis (32,33).
Moderate iron availability supports reactive oxygen
species (ROS) production, contributing to antimicrobial defense.
However, excessive iron accelerates lipid peroxidation, ferroptosis
and prolonged tissue injury (34).
Pulmonary iron accumulation has been observed in both patients with
ARDS and murine models, correlating with oxidative stress, lipid
peroxidation and fibrotic remodeling (35). Therapeutic interventions using iron
chelators such as deferoxamine can attenuate fibrosis by reducing
pulmonary iron burden, although their long-term efficacy and
cell-type-specific effects, particularly on fibroblasts and
macrophages, remain to be fully elucidated (36).
A number of regulators of ferritinophagy have been
investigated in ARDS models. Hepcidin alleviates LPS-induced ARDS
by suppressing ferroptosis through downregulation of transferrin
receptor 1 and upregulation of ferritin heavy chain (FTH), with FTH
being central to its protective effect (37). Conversely, NCOA4 actively promotes
ferritinophagy, elevating free iron, enhancing ROS generation and
triggering lipid peroxidation-mediated cell death (38). Pharmacological inhibition of NCOA4,
as demonstrated with melatonin treatment, reduces ferritinophagy in
alveolar macrophages, limits iron release and improves outcomes in
septic ARDS (39). Similarly, Yes1
associated transcriptional regulator (YAP1) suppresses
ferritinophagy, lowers intracellular free iron, reduces ROS
production and alleviates lung injury in sepsis-induced ALI models
(40).
Collectively, these studies indicate that
NCOA4-driven ferritinophagy is the association between iron
metabolism dysregulation and ferroptotic cell death, perhaps
contributing to fibrosis in ARDS. Its effects are
context-dependent, potentially varying with injury stage, pulmonary
cell type and systemic iron status. Modulation of ferritinophagy,
either through endogenous regulators (hepcidin and YAP1) or
pharmacological agents (melatonin and iron chelators), represents a
promising therapeutic approach. Future research should therefore
prioritize safer, more effective iron-targeted therapies, explore
cell-type-specific interventions and optimize pulmonary delivery
strategies (such as inhalation) to maximize local efficacy while
minimizing systemic toxicity. Key molecules, ferritinophagy states,
molecular targets, experimental models and main conclusions are
summarized in Table II.
| Table IIRoles and molecular mechanisms of
ferritinophagy in ARDS. |
Table II
Roles and molecular mechanisms of
ferritinophagy in ARDS.
| First author,
year | Drug/molecule | Ferritinophagy
state | Main molecular
targets and pathways | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Jiao et al,
2022 | Hepcidin | Inhibits | FTH and TfR1 | LPS-induced ARDS
mouse model (ARDS-specific) | Limits iron release
and ferroptosis, alleviating ARDS | (37) |
| Zhou et al,
2022 | NCOA4 | Activates | Ferritin
degradation; free iron release | Ionizing
radiation-treated intestinal epithelial cells | Excessive
ferritinophagy induces iron-dependent cell death | (38) |
| Xu et al,
2024 | Melatonin | Inhibits | NCOA4 and ferritin
axis | Septic ARDS mouse
model and alveolar macrophages (ARDS-specific) | Reduces iron
overload and ferroptosis, improving septic ARDS outcomes | (39) |
| Zhang et al,
2022 | YAP1 | Inhibits | Ferritin stability;
intracellular free iron | Sepsis-induced ALI
mouse model (pulmonary relevant) | Suppresses ROS
generation and attenuates lung injury | (40) |
Mitophagy in ARDS: Mitochondrial
quality control
Mitophagy, the selective autophagic removal of
damaged or excess mitochondria, is a key quality control mechanism
that preserves mitochondrial function and cellular homeostasis in
lung tissue. Proper regulation of mitophagy is key in maintaining
alveolar epithelial and immune cell function, whereas dysregulated
mitophagy contributes to mitochondrial dysfunction, excessive ROS
production and inflammatory injury, collectively exacerbating ARDS
pathogenesis.
Mechanistically, the PTEN-induced kinase 1
(PINK1)/parkin RBR E3 ubiquitin protein ligase (Parkin) signaling
pathway serves as the central axis orchestrating mitophagy in
response to mitochondrial damage. In ARDS models, polydatin, a
natural polyphenol, activates Parkin-dependent mitophagy,
preventing LPS-induced mitochondrial apoptosis and attenuating lung
injury (41). Similarly, sestrin 2
enhances mitophagy in alveolar macrophages through the PINK1/Parkin
pathway, offering protection against LPS-induced ALI and ARDS
(42).
Beyond canonical regulators, additional modulators
have been identified. In cecal ligation and puncture sepsis models,
resveratrol restores mitochondrial function by modulating
phospholipid scramblase 3, thereby reducing alveolar injury
(43). The transcription factor
RUNX family transcription factor 1 promotes mitophagy through
upregulation of adaptor proteins p62 and BCL2 interacting protein 3
like, preserving mitochondrial integrity and limiting epithelial
cell injury and inflammation (44). Resveratrol additionally exerts dual
benefits by activating PINK1/Parkin-mediated mitophagy while
concurrently suppressing NLR family pyrin domain containing 3
inflammasome activation, further facilitating lung tissue recovery
(45).
These findings underscore mitophagy as a
context-dependent regulator of mitochondrial quality and
inflammatory responses in ARDS. The effects may vary with disease
stage, cell type and injury severity, highlighting the need to
define temporal and cell-type-specific dynamics. Translationally,
targeted mitophagy modulation, potentially combined with
anti-inflammatory or antifibrotic strategies, represents a
promising therapeutic approach. Future studies should therefore
focus on mechanistic characterization, optimal timing of
intervention and combination therapies to maximize clinical
benefit. Key molecular regulators, experimental models and main
outcomes of mitophagy in ARDS are summarized in Table III.
| Table IIIRoles and molecular mechanisms of
mitophagy in ARDS. |
Table III
Roles and molecular mechanisms of
mitophagy in ARDS.
| First author,
year | Drug/molecule | Mitophagy
state | Main molecular
targets and pathways | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Li et al,
2019 | Polydatin | Activates
mitophagy | Parkin-mediated
mitochondrial clearance | LPS-induced ARDS
mouse model | Protects against
mitochondrial apoptosis and lung injury | (41) |
| Wu et al,
2021 | Sestrin 2 | Activates
mitophagy | PINK1/Parkin
pathway | LPS-induced mouse
ALI model | Preserves
mitochondrial homeostasis and attenuates ARDS | (42) |
| Wang et al,
2021 | Resveratrol | Activates
mitophagy |
PLSCR-3-mitochondrial signaling | CLP-induced septic
ARDS mouse model | Restores
mitochondrial function and mitigates lung injury | (43) |
| Tang et al,
2023 | RUNX1 | Activates
mitophagy | p62 and BNIP3L
upregulation | LPS-induced ALI
mouse model | Limits epithelial
injury and inflammatory responses | (44) |
| Wu et al,
2024 | Resveratrol | Activates
mitophagy | PINK1/Parkin, NLRP3
inflammasome inhibition | LPS-induced ALI
mouse model | Coordinates
mitophagy activation with inflammasome suppression | (45) |
ER-phagy in ARDS: ER homeostasis
ER-phagy is a specialized form of autophagy that
selectively degrades excess or damaged ER components to maintain ER
homeostasis. The ER is key in protein folding, calcium storage and
lipid and carbohydrate metabolism. Under stress conditions,
accumulation of misfolded proteins triggers the unfolded protein
response (UPR), an adaptive signaling pathway that increases
chaperone production and reduces protein load (46). Notably, UPR activation directly
interfaces with ER-phagy, ensuring selective clearance of
dysfunctional ER fragments and alleviating cellular stress
(12).
In ALI and ARDS, ER stress contributes to epithelial
apoptosis and inflammation, exacerbating lung injury. ER-phagy
mitigates these effects by restoring ER homeostasis, reducing
apoptosis and modulating inflammatory signaling (47). A number of ER-phagy receptors,
including family with sequence similarity 134 member B (FAM134B),
reticulon 3 long isoform (RTN3L), SEC62 preprotein translocation
regulator (SEC62) and atlastin GTPase 3 (ATL3), mediate these
protective effects and may serve as potential therapeutic targets
(48).
Emerging evidence suggests ER-phagy also influences
immune cell function (47,49-51).
In ARDS, macrophage polarization and inflammatory responses are
tightly regulated by ER-phagy, which may indirectly affect EMT and
fibrosis. Dysregulated ER-phagy could exacerbate inflammation,
impair host defense and promote maladaptive remodeling. Conversely,
therapeutic modulation of ER-phagy may enhance resolution of lung
injury and support tissue repair (47).
Although preclinical studies have highlighted the
protective role of ER-phagy, the temporal dynamics,
receptor-specific functions and cell-type specificity remain
incompletely understood (47,52-54).
Future research should therefore investigate: i) How ER-phagy
influences immune cell subsets, particularly macrophages; ii) the
interactions between ER-phagy and other selective autophagy
pathways (such as mitophagy and ferritinophagy); and iii) potential
pharmacological modulators to enhance ER-phagy-mediated
cytoprotection without inducing excessive ER degradation. Key
ER-phagy receptors, mechanisms, experimental models and main
outcomes in ARDS are summarized in Table IV.
| Table IVRoles and molecular mechanisms of
ER-phagy in ARDS. |
Table IV
Roles and molecular mechanisms of
ER-phagy in ARDS.
| First author,
year | Drug/molecule or
receptor | ER-phagy state | Main molecular
targets and pathways | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Liu et al,
2023 | ER-phagy (general
activation) | Activates | ER stress, UPR
signaling and apoptosis | ALI/ARDS animal and
cellular models (ARDS-specific) | Maintains ER
homeostasis, reduces inflammation and cell death | (47) |
| Hübner et
al, 2020 | FAM134B, RTN3L,
SEC62 and ATL3 | Activates | Selective ER
fragment recognition and degradation | Various disease
models including ALI-related studies (pulmonary relevant) | ER-phagy receptors
represent potential therapeutic targets for ARDS | (48) |
Collectively, Tables
I, II, III and IV summarize the molecular regulators,
signaling pathways and experimental evidence that associate
different autophagy subtypes with ARDS pathogenesis. Macroautophagy
(Table I) exhibits clear
context-dependent effects, exerting protective roles during the
acute inflammatory phase while becoming potentially maladaptive
when excessively or persistently activated. Ferritinophagy
(Table II) has emerged as a key
regulator of iron homeostasis, associating NCOA4-mediated ferritin
degradation with ferroptotic cell death and lung injury,
particularly in septic ARDS models. Mitophagy (Table III), primarily governed by the
PINK1/Parkin axis and its upstream modulators, preserves
mitochondrial integrity and limits inflammatory damage; however,
the therapeutic efficacy of interventions targeting the
PINK1/Parkin-mediated mitophagy pathway-that is, the effectiveness
of modulating-mitophagy to preserve mitochondrial integrity and
limit inflammatory damage likely depends on precise temporal and
cell-type-specific regulation. ER-phagy (Table IV), through UPR-associated
pathways and selective ER receptors, contributes to the maintenance
of ER homeostasis, immune regulation and stress adaptation in
ALI/ARDS.
Collectively, this evidence highlights both the
shared and distinct mechanisms by which selective autophagy
pathways influence inflammation, cell survival and tissue
remodeling in ARDS, underscoring the importance of coordinated and
context-aware modulation of autophagy for therapeutic
intervention.
4. EMT in ARDS pathogenesis
EMT and core mechanisms
EMT is a dynamic and reversible biological process
in which epithelial cells progressively lose apical-basal polarity
and intercellular junctions while acquiring mesenchymal
characteristics, including enhanced migratory capacity and
increased ECM production (55).
EMT serves key roles in embryonic development, wound healing and
tissue regeneration. However, persistent or dysregulated EMT
contributes to pathological conditions such as organ fibrosis and
cancer progression (56).
Notably, EMT is not restricted to epithelial cells
of ectodermal origin. Endothelial cells can undergo a closely
related process termed the EndMT, which has been increasingly
implicated in vascular dysfunction and fibrotic remodeling. Within
the context of lung injury and ARDS, both epithelial EMT and EndMT
contribute to fibroblast accumulation and ECM deposition. For
conceptual clarity and consistency, the present review uses the
term ‘EMT’ as an umbrella concept, while explicitly specifying
EndMT where endothelial-derived transitions are discussed.
At the molecular level, EMT is characterized by the
coordinated downregulation of epithelial markers, such as
E-cadherin and zonula occludens 1 and upregulation of mesenchymal
markers, including α-smooth muscle actin, vimentin and fibronectin
(55-57).
This phenotypic shift disrupts adherens and tight junctions, alters
cytoskeletal organization and compromises epithelial barrier
integrity, features highly relevant to ARDS pathophysiology
(58-60).
Based on biological context, EMT is commonly
classified into three subtypes (Fig.
3): i) Type I EMT during embryogenesis; ii) type II EMT
associated with tissue repair and organ fibrosis; and iii) type III
EMT involved in cancer invasion and metastasis (61). In ARDS and other fibrotic lung
diseases, EMT most closely resembles type II EMT and is driven by
profibrotic and inflammatory signaling pathways, including TGF-β,
Sonic Hedgehog WNT/β-catenin and Notch pathways (62). Fig.
3 highlights how epithelial cells transition to mesenchymal
phenotypes, contributing to fibroproliferative remodeling. These
pathways converge on EMT-associated transcription factors such as
zinc-finger transcription factor SNAI1 (Snail), Snail family
transcriptional repressor 2 (SLUG) and zinc finger E-box-binding
homeobox family members to initiate and sustain mesenchymal
reprogramming (63). Aberrant or
prolonged activation of these signaling networks promotes
pathological fibrosis, highlighting EMT as a potential therapeutic
target in fibrotic lung disease.
EMT contribution to ARDS
progression
EMT has emerged as an important contributor to
pulmonary fibrotic remodeling in a subset of ARDS survivors. In the
injured lung, persistent epithelial damage, oxidative stress and
profibrotic mediators, most prominently TGF-β1, foster a
microenvironment that favors partial or sustained EMT activation.
While EMT is well characterized in cancer biology, its extent,
timing and functional relevance in ARDS-associated fibrosis remain
incompletely defined (64-67).
Clinically, post-ARDS pulmonary fibrosis is
associated with impaired lung compliance, prolonged ventilator
dependence and increased long-term mortality. Histopathological
analyses of fibrotic lung tissue from ARDS models and patients has
revealed that epithelial cells express mesenchymal markers,
supporting the involvement of EMT-related programs in fibrogenesis
(5,16). Rather than representing a complete
phenotypic conversion, EMT in ARDS is increasingly regarded as a
partial or hybrid state, in which epithelial cells acquire
mesenchymal features that promote fibroblast activation, ECM
deposition and disruption of alveolar architecture (68-71).
Experimental intervention studies provide
mechanistic evidence associating EMT with fibrotic outcomes in ARDS
(65,72-74).
In an LPS-induced ARDS model, treatment with the histone
methyltransferase inhibitor 3-deazaneplanocin A was found to
attenuate lung injury and fibrosis by suppressing EMT through
inhibition of the TGF-β1/Smad signaling pathway (75). Similarly, pirfenidone, a clinically
approved antifibrotic agent, reduces fibrotic remodeling by
inhibiting EndMT, highlighting the contribution of mesenchymal
transition programs beyond epithelial cells in ARDS (76). Resveratrol has also been shown to
suppress EMT-related marker expression by alleviating oxidative
stress and downregulating TGF-β1 signaling, further supporting EMT
as a modifiable process in experimental ARDS (77).
At the molecular level, TGF-β1 acts as a central
driver of EMT by inducing phosphorylation of Smad2 and Smad3, which
translocate to the nucleus and activate transcriptional programs
favoring mesenchymal differentiation. By contrast, Smad7 functions
as an endogenous inhibitory regulator that restrains excessive
TGF-β signaling and limits fibrotic progression (78). The balance between these signaling
components perhaps determines whether EMT contributes to adaptive
repair or maladaptive fibrosis in ARDS. Despite growing
experimental evidence, knowledge gaps still persist (65,73,74,79).
The temporal dynamics of EMT activation during the acute, resolving
and fibrotic phases of ARDS remain unclear, as does the relative
contribution of different cell types, including alveolar epithelial
cells, endothelial cells and fibroblasts, to EMT-driven remodeling.
These uncertainties limit the translation of EMT-targeted
strategies into clinical practice. Overall, EMT represents a key
but context-dependent mechanism in ARDS-associated pulmonary
fibrosis. Therapeutic modulation of EMT-related signaling pathways,
particularly TGF-β-Smad signaling, holds promise but requires
precise consideration of disease stage, cellular targets and
interaction with parallel injury and repair pathways. Future
studies integrating cell-type-specific approaches and longitudinal
analyses are therefore key in clarifying the pathogenic vs.
reparative roles of EMT in ARDS.
5. Interplay between autophagy and EMT
Evidence scope and interpretative
framework
Mechanistic associations between autophagy and EMT
discussed in the following section are derived from a combination
of ARDS/ALI models and extrapolative evidence from other disease
contexts, including cancer, metabolic disorders and chronic
fibrotic diseases. Where available, findings directly obtained from
ARDS- or lung injury-relevant experimental systems are explicitly
highlighted. By contrast, mechanistic insights originating from
non-pulmonary or non-ARDS models are clearly identified as
extrapolative and are discussed in light of their potential
relevance and limitations for ARDS pathophysiology. This
integrative approach was adopted as evidence associating specific
autophagy subtypes with EMT in ARDS remains limited, yet shared
stress-response pathways, such as oxidative stress, metabolic
reprogramming, mitochondrial dysfunction, iron dysregulation and ER
stress, providing a rational basis for cautious mechanistic
inference. Throughout the following sections, emphasis is placed
upon context-dependent determinants, including cell type, stage of
injury and autophagy subtype, to avoid overgeneralization and frame
testable hypotheses for future ARDS-focused studies.
Macroautophagy and EMT:
Context-dependent regulatory roles
Macroautophagy is a key cellular process that
influences EMT by regulating energy homeostasis, redox balance and
selective protein turnover, all of which are important in
EMT-associated phenotypic plasticity. Accumulating evidence has
indicated that macroautophagy does not exert a uniform effect on
EMT; instead, its impact is highly context-dependent, varying with
cell type, metabolic state and disease stage (65,73,74,79).
Early mechanistic insights from liver-specific
autophagy-deficient mice (Albumin-Cre; autophagy-related Gene
7fl/fl) demonstrated that autophagy impairment is
associated with downregulation of epithelial markers and
upregulation of mesenchymal markers, suggesting that autophagy
deficiency can facilitate EMT progression (80). One well-characterized mechanism
underlying this effect is the selective autophagic degradation of
the EMT-inducing transcription factor Snail through a
p62/sequestosome 1 (SQSTM1)-dependent pathway (80-83).
By limiting Snail accumulation, basal autophagy acts as a
restraining force on EMT initiation.
In addition to selective protein degradation, core
autophagy-related proteins such as LC3 and beclin-1 influence EMT
by modulating cytoskeletal organization and the balance of
epithelial and mesenchymal adhesion molecules, including E-cadherin
and N-cadherin (84). These
structural and signaling effects underscore a bidirectional
relationship in which EMT-associated cytoskeletal remodeling and
metabolic reprogramming can, in turn, feedback to regulate
autophagic flux.
Notably, ARDS-relevant studies provide evidence that
macroautophagy can suppress EMT under inflammatory and hypoxic
conditions (64,83,85,86).
In LPS-induced ARDS models, pharmacological activation of autophagy
by inositol inhibits the hypoxia-inducible factor 1 α (HIF-1α)/SLUG
signaling axis, leading to reduced EMT marker expression and
attenuation of pulmonary fibrosis (85). These findings support a protective
role for macroautophagy in limiting maladaptive EMT during lung
injury and repair.
Conversely, evidence from non-pulmonary disease
models highlights the pro-EMT role of autophagy under specific
metabolic conditions (82,87-89).
In cancer cells, autophagy-derived acetyl-CoA promotes acetylation
and stabilization of Snail, thereby enhancing EMT through
upregulation of mesenchymal markers such as vimentin and repression
of epithelial markers including E-cadherin (87). This metabolic-epigenetic mechanism
illustrates how sustained or excessive autophagy may facilitate EMT
by fueling transcriptional programs that favor mesenchymal
differentiation.
Collectively, these conflicting findings can be
reconciled by a context-dependent model. In the early or acute
phase of tissue injury, moderate autophagy may exert protective
effects by degrading EMT drivers, limiting oxidative stress and
preserving epithelial identity. By contrast, during prolonged
stress, chronic inflammation or altered metabolic states, autophagy
may support EMT progression by providing biosynthetic substrates
and epigenetic regulators that reinforce mesenchymal programs. In
addition, cell-type specificity, such as differences between
epithelial cells, fibroblasts and immune cells, likely further
determines the direction and magnitude of autophagy-EMT
interactions.
From an ARDS perspective, these insights suggest
that therapeutic modulation of macroautophagy must consider timing,
intensity and cellular targets. Non-selective activation or
inhibition of autophagy may yield divergent outcomes depending on
disease stage and microenvironment. Therefore, future studies
integrating temporal analysis, cell-specific genetic models and
multi-omics approaches are required to delineate when
macroautophagy restrains EMT and when it inadvertently promotes
fibrotic remodeling. Such findings will be key in translating
autophagy-EMT crosstalk into rational therapeutic strategies for
ARDS. The available evidence supporting context-dependent roles of
macroautophagy in EMT regulation, including ARDS/ALI-relevant and
extrapolative studies, is summarized in Table V.
| Table VRelationship between macroautophagy
and EMT. |
Table V
Relationship between macroautophagy
and EMT.
| First author,
year | Drug/molecule | Autophagy
state | Molecular
targets | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Grassi et
al, 2015 | Basal
autophagy | Activates | p62/SQSTM1 | Liver-specific
autophagy-deficient mice (Alb-Cre; ATG7fl/fl) | Deficiency promotes
EMT | (80) |
| Colella et
al, 2019 | LC3 and
beclin-1 | Activates | Snail, N-cadherin
and E-cadherin | In vitro
alveolar epithelial cells (pulmonary relevant) | Modulates EMT | (84) |
| Liang et al,
2022 | Inositol | Activates | HIF-1α/SLUG
signaling pathway | LPS-induced
alveolar epithelial cells and LPS-induced ARDS mouse model
(ARDS-specific) | Inhibits EMT and
alleviates pulmonary fibrosis | (85) |
| Han et al,
2022 | Autophagy-derived
acetyl-CoA | Activates | Snail, vimentin and
E-cadherin | KL cancer
cells | Promotes EMT
through Snail acetylation | (87) |
Mitophagy and EMT: Context-dependent
crosstalk and mechanisms insights
Mitophagy, the selective autophagic elimination of
damaged or dysfunctional mitochondria, has been increasingly
recognized as a regulator of EMT through its control of
mitochondrial quality, ROS generation and metabolic signaling
(90-93).
However, similar to macroautophagy, the impact of mitophagy on EMT
is highly context-dependent and varies across cell types and
pathological conditions.
Evidence from non-pulmonary disease models has
illustrated that suppression of mitophagy can facilitate EMT. In
endothelial cells infected with Kaposi's sarcoma-associated
herpesvirus, activation of the mTOR pathway and its downstream
effectors 4E binding protein 1 and ULK1 inhibits mitophagy, leading
to mitochondrial dysfunction and induction of EMT programs
(94). Similarly, in retinal
pigment epithelial cells, oxidative stress-induced impairment of
mitophagy has resulted in mitochondrial damage and elevated ROS
production, which activated EMT signaling pathways and exacerbated
epithelial dysfunction in age-related macular degeneration models
(95). These studies support a
model in which insufficient mitophagic clearance promotes EMT by
amplifying mitochondrial stress signals.
By contrast, pulmonary-relevant models have
suggested that excessive or sustained mitophagy may also contribute
to EMT-associated pathology. In mice chronically exposed to
particulate matter 2.5, enhanced mitophagy, reflected by increased
Parkin, SQSTM1/p62 and light chain (LC)3B-II/LC3B-I ratios,
coincided with elevated TGF-β1 expression and upregulation of
mesenchymal markers, thereby promoting pulmonary inflammation and
fibrotic remodeling through EMT activation (10). Although not classical ARDS models,
these findings are relevant to lung injury and fibrosis and suggest
that prolonged mitophagy activation under persistent environmental
stress may support EMT-driven pathological remodeling.
Collectively, these seemingly contradictory observations can be
interpreted as biphasic, context-dependent models of mitophagy-EMT
crosstalk. During acute injury or transient stress, mitophagy is
likely protective by preserving mitochondrial integrity, limiting
ROS accumulation and preventing EMT initiation. Conversely, during
chronic injury, sustained inflammation or repeated environmental
insults and excessive or dysregulated mitophagy may facilitate EMT
by reinforcing profibrotic signaling pathways such as TGF-β1,
metabolic reprogramming and persistent cellular stress responses.
The direction of this effect is further shaped by cell type
(epithelial vs. endothelial), mitochondrial reserve capacity and
disease stage. From an ARDS perspective, direct evidence to
associate mitophagy with EMT remains limited and much of the
current understanding is extrapolated from other pulmonary or
non-pulmonary disease models (72,90,96,97).
This highlights an important knowledge gap and underscores the need
for ARDS-specific studies that interrogate mitophagy-EMT
interactions in alveolar epithelial cells, endothelial cells and
immune cells across different phases of lung injury and repair.
Future investigations should therefore focus on
defining the spatiotemporal dynamics of mitophagy during EMT
transitions in ARDS-relevant models. Integration of cell-specific
genetic approaches, live-cell imaging of mitochondrial turnover and
single-cell transcriptomic and metabolomic analyses will be key in
clarifying when mitophagy restrains EMT and when it contributes to
fibrotic progression. Such insights will be important in the
rational design of mitophagy-targeted interventions aimed at
limiting EMT-driven lung fibrosis in ARDS. Current evidence
regarding mitophagy and EMT regulation, derived from ARDS-relevant
models and extrapolative disease settings, is summarized in
Table VI.
| Table VIThe relationship between mitophagy
and EMT. |
Table VI
The relationship between mitophagy
and EMT.
| First author,
year | Drug/molecule | Mitophagy
state | Molecular
targets | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Santarelli et
al, 2020 | KSHV | Inhibits | mTOR, 4EBP1 and
ULK1 | HUVEC cells and
Kaposi's sarcoma model | Promotes EMT | (94) |
| Xu et al,
2021 | PM2.5 | Activates | Parkin, SQSTM1/p62,
LC3B-II/LC3B-I and TGF-β1 | PM2.5-exposed
mice | Promotes EMT and
pulmonary fibrosis | (10) |
| Hyttinen et
al, 2018 | Oxidative
stress | Inhibits | Mitochondria | RPE cells | Promotes EMT | (95) |
Ferritinophagy and EMT:
Context-dependent regulation and translational implications
Ferritinophagy is a selective autophagic process
that degrades ferritin to regulate intracellular iron availability
and redox homeostasis. By releasing iron from ferritin complexes,
ferritinophagy expands the labile iron pool, thereby enhancing ROS
generation and lipid peroxidation. Given that oxidative stress is a
key modulator of EMT, ferritinophagy has emerged as a potential
upstream regulator of EMT through iron- and ROS-dependent signaling
pathways (98).
The majority of mechanistic insights into
ferritinophagy-EMT crosstalk are currently derived from cancer
models rather than pulmonary or ARDS-specific systems. In colon
carcinoma CT26 cells, the iron chelator
2,2'-dipyridone-2-thioacetate (DpdtpA) activates NCOA4-dependent
ferritinophagy, leading to increased intracellular ROS production
and robust suppression of EMT marker expression (99). Similar observations have been
reported in a gastric cancer model, whereby dipyridylhydrazone
dithiocarbamate-induced ferritinophagy elevated ROS levels,
activated p53 signaling and inhibited EMT progression in MGC-803
cells (100). In addition, DpdtbA
enhances ferritinophagic flux and concomitantly activates the
prolyl hydroxylase domain-containing protein 2/HIF-1α axis together
with p53, collectively restraining EMT in gastric carcinoma
(11). These studies suggest that,
under certain conditions, ferritinophagy-driven oxidative stress
can function to alleviate EMT by engaging tumor suppressor pathways
in highly proliferative or metabolically active cells. Beyond
direct ROS signaling, ferritinophagy also intersects with EMT
through ferroptosis-related mechanisms. For instance, D-camphor
enhances cisplatin sensitivity by linking NCOA4-mediated
ferritinophagy with ferroptosis and EMT suppression in cancer cells
(101). However, extrapolation of
these findings to ARDS must be approached with caution. For
example, in a ventilator-induced lung injury, AMPK/ULK1-dependent
NCOA4-mediated ferritinophagy was shown to drive ferroptosis and
lung tissue damage, with inhibition of ferritinophagy attenuating
iron overload, lipid peroxidation and pulmonary injury markers
(102). Similarly, in a model of
septic ARDS, melatonin was reported to ameliorate alveolar
macrophage ferroptosis by inhibiting NCOA4-dependent
ferritinophagy, leading to reduced iron-mediated ROS accumulation
and improved lung histopathology (39). Collectively, in ARDS-relevant
models, excessive or dysregulated ferritinophagy can exacerbate
iron overload, ferroptotic cell death and inflammatory injury,
which may indirectly facilitate EMT and fibrotic signaling through
the iron-ROS-TGF-β axis. Therefore, a unifying framework could be
proposed, whereby transient or moderate ferritinophagy may suppress
EMT through ROS-mediated activation of p53 and related
stress-response pathways, whereas sustained or excessive
ferritinophagy in inflamed tissues may aggravate epithelial damage,
reinforce profibrotic signaling and ultimately favor EMT-driven
remodeling. Cell type-specific responses (epithelial cells vs.
macrophages or fibroblasts) and differences between acute vs.
chronic injury states are likely key determinants of these
divergent outcomes.
From a translational perspective, targeting
ferritinophagy is a two-sided therapeutic strategy. While inducing
ferritinophagy may be beneficial for limiting EMT in cancer,
inhibiting excessive ferritinophagy could be more appropriate in
ARDS to prevent ferroptosis, epithelial barrier disruption and
secondary fibrotic remodeling. Future ARDS-focused studies should
therefore integrate iron metabolism profiling, EMT marker analysis
and ferroptosis assessment in cell type-specific models to further
determine these associations. Evidence implicating ferritinophagy
in EMT regulation, primarily derived from extrapolative models with
emerging relevance to ARDS, is summarized in Table VII.
| Table VIIRelationship between ferritinophagy
and EMT. |
Table VII
Relationship between ferritinophagy
and EMT.
| First author,
year | Drug/molecule | Ferritinophagy
state | Molecular
targets | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Sun et al,
2019 | DpdtpA | Activates | NCOA4/ferritin | CT26 colon
carcinoma cells | Inhibits EMT | (99) |
| Feng et al,
2020 | DpdtC | Activates
ferritinophagy | ROS/p53 pathway and
ferritin | MGC-803 gastric
cancer cells | Suppresses EMT | (100) |
| Guan et al,
2021 | DpdtbA | Activates | Ferritin, p53, PHD2
and HIF-1α | SGC-7901 and
MGC-803 gastric cancer cells | Inhibits EMT | (11) |
| Li et al,
2022 | D-Camphor | Activates | NCOA4 and
EMT-related signaling | H460/CDDP xenograft
tumor model | Inhibits EMT | (101) |
| Ou et al,
2024 | Mechanical
ventilation (VILI) | Activates | AMPK/ULK1-NCOA4
pathway; ferroptosis | Ventilator-induced
lung injury mouse model | Promotes
ferroptosis and lung tissue injury, potentially facilitating
EMT-associated remodeling | (102) |
| Xu et al,
2024 | Melatonin | Inhibits | NCOA4-dependent
ferritin degradation; iron-ROS axis | Septic ARDS mouse
model; alveolar macrophages | Alleviates
ferroptosis and lung injury, indirectly limiting EMT-associated
damage | (39) |
ER-phagy and EMT: Context-dependent
regulation and therapeutic potential
As a central organelle, the ER is responsible for
protein folding, processing, calcium homeostasis and metabolic
regulation. Disruptions in ER function can lead to the accumulation
of misfolded proteins, causing ER stress that perturbs redox
balance, energy metabolism, inflammation, differentiation and cell
survival. To restore homeostasis, cells deploy two primary quality
control systems: i) The ubiquitin-proteasome pathway; and ii)
selective autophagic clearance of the ER, termed ER-phagy (103). ER stress concurrently activates
the UPR and ER-phagy and while the UPR primarily aims to reduce
protein load and reestablish folding capacity, ER-phagy selectively
degrades damaged or excess ER fragments through autophagosomes,
facilitating ER recovery and preserving cellular homeostasis
(104).
UPR activation is well regarded to promote EMT in
cancer and fibrotic contexts through pathways including X-box
binding protein 1 (XBP1), activating transcription factor 6 and
eukaryotic translation initiation factor 2 α kinase 3, which
converge on transcriptional programs that enhance mesenchymal
marker expression and suppress epithelial traits (105-108).
By contrast, the role of ER-phagy in modulating EMT has been
emerging and appears largely protective. For example, in diabetic
nephropathy, ER stress-induced ferroptosis through the XBP1-E3
ubiquitin-protein ligase Hrd-nuclear factor erythroid 2-related
factor 2 axis drives EMT and tissue fibrosis (12). Conversely, activation of the
ER-phagy receptor FAM134B in lung epithelial cells enhances
selective ER clearance, reduces apoptosis, mitigates tissue injury
and limits collagen deposition, collectively suppressing EMT and
fibrotic remodeling in preclinical models (109). In LPS-induced ALI models,
inhibition of ER stress with 4-phenylbutyric acid (4-PBA)
attenuated pulmonary inflammation, lipid peroxidation and
ferroptosis, suggesting that modulation of ER stress and related
autophagic responses contributes to lung protection in ALI/ARDS
contexts (110). Similarly, in
hyperoxia-induced ALI, 4-PBA-mediated suppression of ER stress,
alleviated pulmonary edema, reduced inflammatory responses and
preserved barrier integrity, further implicating ER homeostasis as
a determinant of injury severity in lung injury models (111). Additional ER-phagy receptors,
such as RTN3L, SEC62 and ATL3, have been implicated in ER
homeostasis and immune regulation, suggesting additional avenues
for modulating EMT indirectly through ER quality control.
Notably, these observations indicate a
context-dependent interplay between ER stress, UPR and ER-phagy,
whereby, while persistent or excessive ER stress promotes EMT and
tissue remodeling, ER-phagy functions as a protective mechanism
that alleviates stress, preserves epithelial integrity and limits
EMT progression. However, the precise molecular pathways that
associate ER-phagy with EMT in ARDS or ALI remain incompletely
defined and the majority of current evidence is extrapolated from
cancer or renal disease models. Therefore, ARDS-specific
investigations are required to further elucidate how ER-phagy
modulates EMT in alveolar epithelial cells, endothelial cells and
immune cell populations under inflammatory or fibrotic conditions.
From a translational perspective, targeting ER-phagy offers a
promising strategy to mitigate EMT-associated fibrosis and tissue
remodeling. Potential approaches include pharmacological
enhancement of ER-phagy receptors, modulation of UPR signaling to
prevent maladaptive EMT and cell type-specific interventions that
preserve ER homeostasis while minimizing systemic effects (112-114).
Future studies should therefore integrate ER stress profiling, EMT
marker assessment and functional outcomes in ARDS-relevant
preclinical models to validate these strategies. Available studies
examining the roles of ER-phagy and ER stress in EMT regulation,
including extrapolative and limited ARDS-relevant evidence, are
summarized in Table VIII.
| Table VIIIRelationship between ER-phagy/ER
stress and EMT. |
Table VIII
Relationship between ER-phagy/ER
stress and EMT.
| First author,
year | Drug/molecule | ER-phagy state | Molecular
targets | Experimental
model | Main
conclusion | (Refs.) |
|---|
| Guo et al,
2024 | FAM134B | Activates | ER stress, FAM134B,
apoptosis and collagen deposition | RLE-6TN alveolar
epithelial cells; rat model | Inhibits EMT and
fibrosis | (109) |
| Liu et al,
2023 | XBP1-HRD1-Nrf2 | Activates | XBP1, HRD1 and
Nrf2 |
Streptozotocin-induced DN mice and HK-2
cells | Promotes EMT | (12) |
| Wang et al,
2024 | 4-PBA | Inhibits | ER stress, lipid
peroxidation and ferroptosis | LPS-induced acute
lung injury mouse model | Attenuates lung
injury and limits EMT-associated remodeling | (110) |
| Pao et al,
2021 | 4-PBA | Inhibits | ER stress,
inflammation and barrier integrity | Hyperoxia-induced
acute lung injury mouse model | Alleviates
pulmonary injury and preserves epithelial integrity | (111) |
The molecular mechanisms and functional outcomes of
macroautophagy, mitophagy, ferritinophagy and ER-phagy in
regulating EMT are summarized in Tables V, VI, VII and VIII. This evidence highlights that the
effects of autophagy on EMT are highly context-dependent, varying
by autophagy subtype, cellular environment and disease stage. For
example, macroautophagy can either suppress or promote EMT
depending on metabolic and epigenetic cues, whereas mitophagy shows
bidirectional effects influenced by mitochondrial stress and ROS
levels. Ferritinophagy predominantly inhibits EMT through
ROS-mediated signaling and ferroptotic pathways, while ER-phagy
typically mitigates EMT by alleviating ER stress and collagen
deposition. Collectively, these findings support a model in which
the interplay between autophagy subtype, temporal stage of injury
and specific cell type determines whether EMT is restrained or
facilitated, providing a framework for targeted therapeutic
strategies in ARDS-associated fibrosis.
6. Discussion
Autophagy, both macroautophagy and selective
subtypes including mitophagy, ferritinophagy and ER-phagy, serves a
key role in regulating EMT and pulmonary fibrosis in ARDS.
Macroautophagy maintains cellular homeostasis and protein turnover,
modulating EMT through mechanisms such as p62-mediated Snail
degradation, autophagy-derived acetyl-CoA and regulation of
adhesion molecules. Mitophagy preserves mitochondrial quality,
influencing EMT through ROS production, metabolic reprogramming and
TGF-β1 signaling. Ferritinophagy regulates intracellular iron and
redox balance, with ROS-dependent modulation of EMT, while ER-phagy
maintains ER homeostasis and alleviates stress-induced EMT. The
effects of these autophagy pathways are highly context- and cell
type-dependent, with outcomes influenced by injury stage, oxidative
stress and local microenvironmental cues. Given the limited
availability of ARDS-specific mechanistic studies, the present
review incorporates evidence from other disease models, which
should be interpreted with caution and validated in pulmonary
systems. ARDS-specific mechanistic studies remain limited, yet
direct evidence in ARDS/ALI models is strongest for macroautophagy
(such as LPS-induced epithelial injury and fibrosis) and ER-phagy
(such as FAM134B-mediated protection in lung epithelial cells).
Mitophagy and ferritinophagy mechanisms are largely extrapolated
from non-pulmonary models (such as cancer, retinal degeneration and
nephropathy) but provide important mechanistic insights. This
distinction is important in interpretation and translational
planning, as extrapolated findings may not fully recapitulate
pulmonary microenvironment or cell-type interactions in ARDS.
Despite growing mechanistic insights, a number of
knowledge gaps remain. The majority of studies rely on single-cell
or in vitro systems, limiting the understanding of
multicellular interactions and spatiotemporal dynamics in the
injured lung. The temporal regulation of autophagy-EMT interplay
during ARDS, from the acute injury phase to fibrosis resolution,
remains poorly characterized. Furthermore, the contribution of
endothelial cells, alveolar macrophages and fibroblasts in shaping
EMT responses through selective autophagy is incompletely
understood. Conflicting evidence, such as bidirectional effects of
macroautophagy and mitophagy on EMT, underscores the need for
context-specific and cell-specific analyses. Furthermore, existing
animal models inadequately capture human ARDS heterogeneity,
challenging translational relevance (115-118).
To address these limitations, integrative, systems-level approaches
are required. Multi-omics strategies (transcriptomics, proteomics
and metabolomics), combined with live-cell imaging, may map
autophagy-EMT dynamics in relevant cell types and disease phases.
Well-characterized patient-derived samples, stratified by ARDS
stage and etiology, are key in validating preclinical mechanisms
and identifying biomarkers for clinical translation. Tables I, II, III, IV, V,
VI, VII and VIII summarize autophagy subtypes,
molecular targets, experimental models and outcomes, providing a
comprehensive reference for dissecting autophagy-EMT crosstalk in
ARDS. The present study proposes a phase- and cell type-dependent
model of autophagy-EMT regulation in ARDS: i) Early injury phase:
Protective macroautophagy and ER-phagy in epithelial and
endothelial cells limit EMT and fibrosis; ii) persistent or
maladaptive phase: Excessive mitophagy or ferritinophagy in
alveolar macrophages or epithelial cells may promote ROS
accumulation and mesenchymal transition, exacerbating fibrosis; and
iii) cell type specificity: Epithelial cells, endothelial cells and
immune cells display distinct autophagy-EMT responses, suggesting
targeted interventions could optimize therapeutic outcomes.
With regard to translational guidance, selective
modulation of autophagy subtypes offers potential strategies to
restrain EMT and fibrosis in ARDS: i) Macroautophagy: Moderate
activation may preserve epithelial identity and limit EMT, with
excessive activation in late injury perhaps enhancing pro-fibrotic
signaling; ii) mitophagy: Fine-tuning in macrophages and epithelial
cells may prevent ROS-driven EMT without impairing mitochondrial
quality control; iii) ferritinophagy: Inhibition of excessive
ferritinophagy could prevent ferroptosis and epithelial barrier
disruption, whereas transient activation may have protective
effects in other contexts; iv) ER-phagy: Pharmacological
enhancement of ER-phagy receptors (such as FAM134B) can relieve ER
stress, reduce apoptosis and mitigate EMT progression; and v)
practical considerations: Timing, intensity and cell type
specificity are key and off-target effects and systemic autophagy
modulation must be carefully monitored. Biomarkers, such as LC3-II,
NCOA4, ROS levels and EMT markers, are able to guide intervention
assessment.
With regard to research priorities, in order to
advance ARDS-targeted therapies, future studies should aim to focus
on the following: i) ARDS-specific validation of selective
autophagy-EMT interactions in epithelial, endothelial and immune
cells; ii) temporal mapping of autophagy-EMT dynamics across acute
injury, repair and fibrotic phases; iii) integration of
multi-omics, live-cell imaging and spatial transcriptomics to
capture cell-specific autophagy responses; iv) identification of
reliable, cell-type-specific biomarkers for clinical translation;
and v) testing targeted interventions in well-characterized ARDS
preclinical models and patient-derived samples.
In conclusion, autophagy-EMT crosstalk constitutes a
key determinant of ARDS progression and fibrosis. The present
review summarized preclinical evidence across macroautophagy,
selective autophagy subtypes and EMT mechanisms, highlighting
context-, phase- and cell-specific effects. Understanding these
regulatory networks will inform the rational design of
autophagy-targeted therapies to mitigate pulmonary fibrosis,
preserve lung function and improve outcomes in patients with ARDS.
Focused translational research is needed to bridge mechanistic
insights and clinical application.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by The Joint Project of
Guangzhou City School (Institute) Joint Funding Project (grant no.
2023A03J0528) and The Natural Science Foundation of Guangdong
Province (grant no. 2020A1515010816).
Availability of data and materials
Not applicable.
Authors' contributions
YZ wrote, reviewed and edited the original draft of
the manuscript and contributed towards visualization. HH wrote,
reviewed and edited the manuscript and conducted the literature
search and screening. CD and QG were involved in visualization. JT
and YY conducted the literature search and screening. ZG
contributed towards the conceptualization and supervision of the
present study. RZ contributed towards conceptualization,
supervision and project administration. All authors read and
approved the final version of the manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Villar J, Szakmany T, Grasselli G and
Camporota L: Redefining ARDS: A paradigm shift. Crit Care.
27(416)2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bos LDJ and Ware LB: Acute respiratory
distress syndrome: Causes, pathophysiology, and phenotypes. Lancet.
400:1145–1156. 2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bellani G, Laffey JG, Pham T, Fan E,
Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley
DF, et al: Epidemiology, patterns of care, and mortality for
patients with acute respiratory distress syndrome in intensive care
units in 50 countries. JAMA. 315:788–800. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fan E, Brodie D and Slutsky AS: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Burnham EL, Janssen WJ, Riches DW, Moss M
and Downey GP: The fibroproliferative response in acute respiratory
distress syndrome: Mechanisms and clinical significance. Eur Respir
J. 43:276–285. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Committee NP: Scientific background on the
2016 nobel prize in physiology or medicine: Discoveries of
mechanisms for autophagy, 2016.
|
|
7
|
Galluzzi L, Bravo-San Pedro JM, Levine B,
Green DR and Kroemer G: Pharmacological modulation of autophagy:
Therapeutic potential and persisting obstacles. Nat Rev Drug
Discov. 16:487–511. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Vargas JNS, Hamasaki M, Kawabata T, Youle
RJ and Yoshimori T: The mechanisms and roles of selective autophagy
in mammals. Nat Rev Mol Cell Biol. 24:167–185. 2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hashimoto N, Phan SH, Imaizumi K, Matsuo
M, Nakashima H, Kawabe T, Shimokata K and Hasegawa Y:
Endothelial-mesenchymal transition in bleomycin-induced pulmonary
fibrosis. Am J Respir Cell Mol Biol. 43:161–172. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Xu M, Wang X, Xu L, Zhang H, Li C, Liu Q,
Chen Y, Chung KF, Adcock IM and Li F: Chronic lung inflammation and
pulmonary fibrosis after multiple intranasal instillation of
PM2.5 in mice. Environ Toxicol. 36:1434–1446.
2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Guan D and Li C, Li Y, Li Y, Wang G, Gao F
and Li C: The DpdtbA induced EMT inhibition in gastric cancer cell
lines was through ferritinophagy-mediated activation of p53 and
PHD2/hif-1α pathway. J Inorg Biochem. 218(111413)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Liu Z, Nan P, Gong Y, Tian L, Zheng Y and
Wu Z: Endoplasmic reticulum stress-triggered ferroptosis via the
XBP1-Hrd1-Nrf2 pathway induces EMT progression in diabetic
nephropathy. Biomed Pharmacother. 164(114897)2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Piera-Velazquez S and Jimenez SA:
Endothelial to mesenchymal transition: Role in physiology and in
the pathogenesis of human diseases. Physiol Rev. 99:1281–1324.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Phan THG, Paliogiannis P, Nasrallah GK,
Giordo R, Eid AH, Fois AG, Zinellu A, Mangoni AA and Pintus G:
Emerging cellular and molecular determinants of idiopathic
pulmonary fibrosis. Cell Mol Life Sci. 78:2031–2057.
2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sakuma Y: Epithelial-to-mesenchymal
transition and its role in EGFR-mutant lung adenocarcinoma and
idiopathic pulmonary fibrosis. Pathol Int. 67:379–388.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kim KK, Kugler MC, Wolters PJ, Robillard
L, Galvez MG, Brumwell AN, Sheppard D and Chapman HA: Alveolar
epithelial cell mesenchymal transition develops in vivo during
pulmonary fibrosis and is regulated by the extracellular matrix.
Proc Natl Acad Sci USA. 103:13180–13185. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Shen Y, He Y, Pan Y, Liu L, Liu Y and Jia
J: Role and mechanisms of autophagy, ferroptosis, and pyroptosis in
sepsis-induced acute lung injury. Front Pharmacol.
15(1415145)2024.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yu L, Chen Y and Tooze SA: Autophagy
pathway: Cellular and molecular mechanisms. Autophagy. 14:207–215.
2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32.
2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jia X, Cao B, An Y, Zhang X and Wang C:
Rapamycin ameliorates lipopolysaccharide-induced acute lung injury
by inhibiting IL-1β and IL-18 production. Int Immunopharmacol.
67:211–219. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Qin L, Li M, Tan HL, Yang HX, Li SD, Luan
ZX, Chen YF and Yang MH: Mechanistic target of rapamycin-mediated
autophagy is involved in the alleviation of
lipopolysaccharide-induced acute lung injury in rats. Int
Immunopharmacol. 78(105790)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hu Y, Lou J, Mao YY, Lai TW, Liu LY, Zhu
C, Zhang C, Liu J, Li YY, Zhang F, et al: Activation of MTOR in
pulmonary epithelium promotes LPS-induced acute lung injury.
Autophagy. 12:2286–2299. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang C, Yang Y, Zhou C, Mei X, Liu J, Luo
K, Zhou J, Qin C and Zeng Z: WWOX activates autophagy to alleviate
lipopolysaccharide-induced acute lung injury by regulating mTOR.
Int Immunopharmacol. 115(109671)2023.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang QL, Yang L, Liu ZL, Peng Y, Gao M,
Deng LT, Liu X and Xing W: Sirtuin 6 regulates macrophage
polarization to alleviate sepsis-induced acute respiratory distress
syndrome via dual mechanisms dependent on and independent of
autophagy. Cytotherapy. 24:149–160. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
He J, Yu C, Shen Y, Huang J, Zhou Y, Gu J,
Cao Y and Zheng Q: Sirtuin 6 ameliorates bleomycin-induced
pulmonary fibrosis via activation of lipid catabolism. J Cell
Physiol. 239(e31027)2024.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li X, Li Y, Hao Q, Jin J and Wang Y:
Metabolic mechanisms orchestrated by Sirtuin family to modulate
inflammatory responses. Front Immunol. 15(1448535)2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu H, Wang S, Gong L, Shen Y, Xu F, Wang
Y, Hu L and Zhu L: SIRT6 ameliorates LPS-induced apoptosis and
tight junction injury in ARDS through the ERK1/2 pathway and
autophagy. Int J Med Sci. 20:581–594. 2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Du S, Li C, Lu Y, Lei X, Zhang Y, Li S,
Liu F, Chen Y, Weng D and Chen J: Dioscin alleviates crystalline
Silica-induced pulmonary inflammation and fibrosis through
promoting alveolar macrophage autophagy. Theranostics. 9:1878–1892.
2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wei X, Yi X, Lv H, Sui X, Lu P, Li L, An
Y, Yang Y, Yi H and Chen G: MicroRNA-377-3p released by mesenchymal
stem cell exosomes ameliorates lipopolysaccharide-induced acute
lung injury by targeting RPTOR to induce autophagy. Cell Death Dis.
11(657)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Feng F, Wang LJ, Li JC, Chen TT and Liu L:
Role of heparanase in ARDS through autophagy and exosome pathway
(review). Front Pharmacol. 14(1200782)2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Liu B, Zhao H, Wang Y, Zhang H and Ma Y:
Astragaloside IV attenuates Lipopolysaccharides-induced pulmonary
epithelial cell injury through inhibiting autophagy. Pharmacology.
105:90–101. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kotla NK, Dutta P, Parimi S and Das NK:
The role of ferritin in health and disease: Recent advances and
understandings. Metabolites. 12(609)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wen Y, Liu Y, Liu W, Liu W, Dong J, Liu Q,
Yu Z, Ren H and Hao H: Ferroptosis: A potential target for acute
lung injury. Inflamm Res. 73:1615–1629. 2024.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang X, Zhou J, Holbein BE and Lehmann C:
Iron chelation as a potential therapeutic approach in acute lung
injury. Life (Basel). 13(1659)2023.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Maus M, López-Polo V, Mateo L, Lafarga M,
Aguilera M, De Lama E, Meyer K, Sola A, Lopez-Martinez C,
López-Alonso I, et al: Iron accumulation drives fibrosis,
senescence and the senescence-associated secretory phenotype. Nat
Metab. 5:2111–2130. 2023.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhu Y, Chang J, Tan K, Huang SK, Liu X,
Wang X, Cao M, Zhang H, Li S, Duan X, et al: Clioquinol attenuates
pulmonary fibrosis through inactivation of fibroblasts via iron
chelation. Am J Respir Cell Mol Biol. 65:189–200. 2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jiao Y, Yong C, Zhang R, Qi D and Wang D:
Hepcidin alleviates LPS-Induced ARDS by regulating the
Ferritin-mediated suppression of ferroptosis. Shock. 57:274–281.
2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhou H, Zhou YL, Mao JA, Tang LF, Xu J,
Wang ZX, He Y and Li M: NCOA4-mediated ferritinophagy is involved
in ionizing radiation-induced ferroptosis of intestinal epithelial
cells. Redox Biol. 55(102413)2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Xu W, Wu Y, Wang S, Hu S, Wang Y, Zhou W,
Chen Y, Li Q, Zhu L, Yang H and Lv X: Melatonin alleviates septic
ARDS by inhibiting NCOA4-mediated ferritinophagy in alveolar
macrophages. Cell Death Discov. 10(253)2024.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhang J, Zheng Y, Wang Y, Wang J, Sang A,
Song X and Li X: YAP1 alleviates sepsis-induced acute lung injury
via inhibiting ferritinophagy-mediated ferroptosis. Front Immunol.
13(884362)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Li T, Liu Y, Xu W, Dai X, Liu R, Gao Y,
Chen Z and Li Y: Polydatin mediates Parkin-dependent mitophagy and
protects against mitochondria-dependent apoptosis in acute
respiratory distress syndrome. Lab Invest. 99:819–829.
2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wu D, Zhang H, Wu Q, Li F, Wang Y, Liu S
and Wang J: Sestrin 2 protects against LPS-induced acute lung
injury by inducing mitophagy in alveolar macrophages. Life Sci.
267(118941)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wang C, Yuan J and Du J: Resveratrol
alleviates acute lung injury through regulating PLSCR-3-mediated
mitochondrial dysfunction and mitophagy in a cecal ligation and
puncture model. Eur J Pharmacol. 913(174643)2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Tang X, Zhong L, Tian X, Zou Y, Hu S, Liu
J, Li P, Zhu M, Luo F and Wan H: RUNX1 promotes mitophagy and
alleviates pulmonary inflammation during acute lung injury. Signal
Transduct Target Ther. 8(288)2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wu D, Zhang H, Li F, Liu S, Wang Y, Zhang
Z, Wang J and Wu Q: Resveratrol alleviates acute lung injury in
mice by promoting Pink1/Parkin-related mitophagy and inhibiting
NLRP3 inflammasome activation. Biochim Biophys Acta Gen Subj.
1868(130612)2024.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fumagalli F, Noack J, Bergmann TJ,
Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Soldà
T, et al: Translocon component Sec62 acts in endoplasmic reticulum
turnover during stress recovery. Nat Cell Biol. 18:1173–1184.
2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Liu S, Fang X, Zhu R, Zhang J, Wang H, Lei
J, Wang C, Wang L and Zhan L: Role of endoplasmic reticulum
autophagy in acute lung injury. Front Immunol.
14(1152336)2023.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Hübner CA and Dikic I: ER-phagy and human
diseases. Cell Death Differ. 27:833–842. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
He X, He H, Hou Z, Wang Z, Shi Q, Zhou T,
Wu Y, Qin Y, Wang J, Cai Z, et al: ER-phagy restrains inflammatory
responses through its receptor UBAC2. EMBO J. 43:5057–5084.
2024.PubMed/NCBI View Article : Google Scholar
|
|
50
|
He L, Qian X and Cui Y: Advances in
ER-Phagy and its diseases relevance. Cells. 10(2328)2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wang L, Wang D, Zhang T, Ma Y, Tong X and
Fan H: The role of immunometabolism in macrophage polarization and
its impact on acute lung injury/acute respiratory distress
syndrome. Front Immunol. 14(1117548)2023.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Jin H, Yang Y, Zhu X, Zhou Y, Xu Y, Li J,
Qi C, Shao X, Wu J, Wu S, et al: DDRGK1-mediated ER-phagy
attenuates acute kidney injury through ER-stress and apoptosis.
Cell Death Dis. 15(63)2024.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Wolf D, Röder C, Sendtner M and
Lüningschrör P: An essential role for calnexin in ER-phagy and the
unfolded protein response. Cells. 13(1498)2024.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Yang M, Luo S, Wang X, Li C, Yang J, Zhu
X, Xiao L and Sun L: ER-Phagy: A new regulator of ER homeostasis.
Front Cell Dev Biol. 9(684526)2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Amack JD: Cellular dynamics of EMT:
Lessons from live in vivo imaging of embryonic development. Cell
Commun Signal. 19(79)2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Akhurst RJ: From shape-shifting embryonic
cells to oncology: The fascinating history of epithelial
mesenchymal transition. Semin Cancer Biol. 96:100–114.
2023.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Hartsock A and Nelson WJ: Adherens and
tight junctions: Structure, function and connections to the actin
cytoskeleton. Biochim Biophys Acta. 1778:660–669. 2008.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Giepmans BN and van Ijzendoorn SC:
Epithelial cell-cell junctions and plasma membrane domains. Biochim
Biophys Acta. 1788:820–831. 2009.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Bazzoni G and Dejana E: Endothelial
cell-to-cell junctions: Molecular organization and role in vascular
homeostasis. Physiol Rev. 84:869–901. 2004.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Marconi GD, Fonticoli L, Rajan TS,
Pierdomenico SD, Trubiani O, Pizzicannella J and Diomede F:
Epithelial-mesenchymal transition (EMT): The Type-2 EMT in wound
healing, tissue regeneration and organ fibrosis. Cells.
10(1587)2021.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Zhang J, Tian XJ and Xing J: Signal
transduction pathways of EMT induced by TGF-β, SHH, and WNT and
their crosstalks. J Clin Med. 5(41)2016.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Shu DY, Butcher E and Saint-Geniez M:
Emerging roles in Age-related macular degeneration. Int J Mol Sci.
21(4271)2020.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Hill C, Jones MG, Davies DE and Wang Y:
Epithelial-mesenchymal transition contributes to pulmonary fibrosis
via aberrant epithelial/fibroblastic cross-talk. J Lung Health Dis.
3:31–35. 2019.PubMed/NCBI
|
|
65
|
Pan T, Feng Y, Li Y, Yang Y, Zhou J and
Song Y: Exacerbation of pulmonary fibrosis following acute lung
injury via activin-A production by recruited alveolar macrophages.
J Thorac Dis. 16:7709–7728. 2024.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Li LF, Yu CC, Huang CY, Wu HP, Chu CM, Liu
PC and Liu YY: Attenuation of Ventilation-enhanced
Epithelial-mesenchymal transition through the phosphoinositide
3-Kinase-γ in a murine bleomycin-induced acute lung injury model.
Int J Mol Sci. 24(5538)2023.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Tan C, Zhou H, Xiong Q, Xian X, Liu Q,
Zhang Z, Xu J and Yao H: Cromolyn sodium reduces LPS-induced
pulmonary fibrosis by inhibiting the EMT process enhanced by
MC-derived IL-13. Respir Res. 26(3)2025.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Bocci F, Tripathi SC, Vilchez Mercedes SA,
George JT, Casabar JP, Wong PK, Hanash SM, Levine H, Onuchic JN and
Jolly MK: NRF2 activates a partial epithelial-mesenchymal
transition and is maximally present in a hybrid
epithelial/mesenchymal phenotype. Integr Biol (Camb). 11:251–263.
2019.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Jolly MK, Tripathi SC, Jia D, Mooney SM,
Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben-Jacob E and Levine
H: Stability of the hybrid epithelial/mesenchymal phenotype.
Oncotarget. 7:27067–27084. 2016.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Li W, Xie Y, Chen Z, Cao D and Wang Y:
Epithelial-mesenchymal transition in pulmonary fibrosis: Molecular
mechanisms and emerging therapeutic strategies. Front Med
(Lausanne). 12(1658001)2025.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Jolly MK, Ward C, Eapen MS, Myers S,
Hallgren O, Levine H and Sohal SS: Epithelial-mesenchymal
transition, a spectrum of states: Role in lung development,
homeostasis, and disease. Dev Dyn. 247:346–358. 2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Feng J, Huang X, Peng Y, Yang W, Yang X,
Tang R, Xu Q, Gao Y, He Z, Xing S and Mei S: Pyruvate kinase M2
modulates mitochondrial dynamics and EMT in alveolar epithelial
cells during sepsis-associated pulmonary fibrosis. J Transl Med.
23(205)2025.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Kawami M, Takenaka S, Kadekaru Y, Akai M,
Konaka T, Yumoto R and Takano R: Evaluation on
epithelial-mesenchymal state and microRNAs focusing on isolated
alveolar epithelial cells from bleomycin injured rat lung.
Toxicology. 461(152903)2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Cabrera-Benítez NE, Parotto M, Post M, Han
B, Spieth PM, Cheng WE, Valladares F, Villar J, Liu M, Sato M, et
al: Mechanical stress induces lung fibrosis by
epithelial-mesenchymal transition. Crit Care Med. 40:510–517.
2012.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Bao X, Liu X, Liu N, Zhuang S, Yang Q, Ren
H, Zhao D, Bai J, Zhou X and Tang L: Inhibition of EZH2 prevents
acute respiratory distress syndrome (ARDS)-associated pulmonary
fibrosis by regulating the macrophage polarization phenotype.
Respir Res. 22(194)2021.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Zhang R, Tan Y, Yong C, Jiao Y, Tang X and
Wang D: Pirfenidone ameliorates early pulmonary fibrosis in
LPS-induced acute respiratory distress syndrome by inhibiting
endothelial-to-mesenchymal transition via the Hedgehog signaling
pathway. Int Immunopharmacol. 109(108805)2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Zhang YQ, Liu YJ, Mao YF, Dong WW, Zhu XY
and Jiang L: Resveratrol ameliorates lipopolysaccharide-induced
epithelial mesenchymal transition and pulmonary fibrosis through
suppression of oxidative stress and transforming growth factor-β1
signaling. Clin Nutr. 34:752–760. 2015.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Kanemaru R, Takahashi F, Kato M, Mitsuishi
Y, Tajima K, Ihara H, Hidayat M, Wirawan A, Koinuma Y, Hayakawa D,
et al: Dasatinib suppresses TGFβ-Mediated Epithelial-mesenchymal
transition in alveolar epithelial cells and inhibits pulmonary
fibrosis. Lung. 196:531–554. 2018.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Zhou G, Dada LA, Wu M, Kelly A, Trejo H,
Zhou Q, Varga J and Sznajder JI: Hypoxia-induced alveolar
epithelial-mesenchymal transition requires mitochondrial ROS and
hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol.
297:L1120–L1130. 2009.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Grassi G, Di Caprio G, Santangelo L, Fimia
GM, Cozzolino AM, Komatsu M, Ippolito G, Tripodi M and Alonzi T:
Autophagy regulates hepatocyte identity and
epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions
promoting Snail degradation. Cell Death Dis.
6(e1880)2015.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Zada S, Hwang JS, Ahmed M, Lai TH, Pham TM
and Kim DR: Control of the Epithelial-to-mesenchymal transition and
cancer metastasis by Autophagy-dependent SNAI1 degradation. Cells.
8(129)2019.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Liu X, Meng L, Li X, Li D, Liu Q, Chen Y,
Li X, Bu W and Sun H: Regulation of FN1 degradation by the
p62/SQSTM1-dependent autophagy-lysosome pathway in HNSCC. Int J
Oral Sci. 12(34)2020.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Hill C, Li J, Liu D, Conforti F, Brereton
CJ, Yao L, Zhou Y, Alzetani A, Chee SJ, Marshall BG, et al:
Autophagy inhibition-mediated epithelial-mesenchymal transition
augments local myofibroblast differentiation in pulmonary fibrosis.
Cell Death Dis. 10(591)2019.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Colella B, Faienza F and Di Bartolomeo S:
EMT regulation by autophagy: A new perspective in glioblastoma
biology. Cancers (Basel). 11(312)2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Liang Y, Xu Y, Lu B, Huang Y, Xu S, Xie J,
Liu M, Che D, Ma L, Tao J, et al: Inositol alleviates pulmonary
fibrosis by promoting autophagy via inhibiting the HIF-1α-SLUG axis
in acute respiratory distress syndrome. Oxid Med Cell Longev.
2022(1030238)2022.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Liu H, Du Y, Zhang Z, Lv L, Xiong W, Zhang
L, Li N, He H, Li Q and Liu Y: Autophagy contributes to
hypoxia-induced epithelial to mesenchymal transition of endometrial
epithelial cells in endometriosis. Biol Reprod. 99:968–981.
2018.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Han JH, Kim YK, Kim H, Lee J, Oh MJ, Kim
SB, Kim M, Kim KH, Yoon HJ, Lee MS, et al: Snail acetylation by
autophagy-derived acetyl-coenzyme A promotes invasion and
metastasis of KRAS-LKB1 co-mutated lung cancer cells. Cancer Commun
(Lond). 42:716–749. 2022.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Dower CM, Wills CA, Frisch SM and Wang HG:
Mechanisms and context underlying the role of autophagy in cancer
metastasis. Autophagy. 14:1110–1128. 2018.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Gugnoni M, Sancisi V, Manzotti G, Gandolfi
G and Ciarrocchi A: Autophagy and epithelial-mesenchymal
transition: An intricate interplay in cancer. Cell Death Dis.
7(e2520)2016.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Yan J, Zhao W, Wang H, Zhou L, Li X and
Wang G: Aldose reductase mediated mitophagy promotes
epithelial-mesenchymal transition of hepatocytes. Hepat Med.
17:141–159. 2025.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Datta S, Cano M, Satyanarayana G, Liu T,
Wang L, Wang J, Cheng J, Itoh K, Sharma A, Bhutto I, et al:
Mitophagy initiates retrograde mitochondrial-nuclear signaling to
guide retinal pigment cell heterogeneity. Autophagy. 19:966–983.
2023.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Xu C, Cao Y, Liu R, Liu L, Zhang W, Fang
X, Jia S, Ye J, Liu Y, Weng L, et al: Mitophagy-regulated
mitochondrial health strongly protects the heart against cardiac
dysfunction after acute myocardial infarction. J Cell Mol Med.
26:1315–1326. 2022.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Han S, Zhang M, Jeong YY, Margolis DJ and
Cai Q: The role of mitophagy in the regulation of mitochondrial
energetic status in neurons. Autophagy. 17:4182–4201.
2021.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Santarelli R, Arteni AMB, Gilardini
Montani MS, Romeo MA, Gaeta A, Gonnella R, Faggioni A and Cirone M:
KSHV dysregulates bulk macroautophagy, mitophagy and UPR to promote
endothelial to mesenchymal transition and CCL2 release, key events
in viral-driven sarcomagenesis. Int J Cancer. 147:3500–3510.
2020.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Hyttinen JMT, Viiri J, Kaarniranta K and
Błasiak J: Mitochondrial quality control in AMD: Does mitophagy
play a pivotal role? Cell Mol Life Sci. 75:2991–3008.
2018.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Li D, Shen C, Liu L, Hu J, Qin J, Dai L,
Gao L, Cheng M, Wang D, Bao R and Wang B: PKM2 regulates cigarette
smoke-induced airway inflammation and epithelial-to-mesenchymal
transition via modulating PINK1/Parkin-mediated mitophagy.
Toxicology. 477(153251)2022.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Lin Q, Zhang CF, Guo JL, Su JL, Guo ZK and
Li HY: Involvement of NEAT1/PINK1-mediated mitophagy in chronic
obstructive pulmonary disease induced by cigarette smoke or
PM2.5. Ann Transl Med. 10(277)2022.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Sun Y and Li C, Feng J, Li Y, Zhai X,
Zhang L and Li C: Ferritinophagic flux activation in CT26 cells
contributed to EMT inhibition induced by a novel iron chelator,
DpdtpA. Oxid Med Cell Longev. 2019(8753413)2019.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Feng J and Li C, Xu R, Li Y, Hou Q, Feng
R, Wang S, Zhang L and Li C: DpdtC-induced EMT inhibition in
MGC-803 cells was partly through ferritinophagy-mediated ROS/p53
pathway. Oxid Med Cell Longev. 2020(9762390)2020.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Li J, Yuan J, Li Y, Wang J, Xie Q, Ma R,
Wang J, Ren M, Lu D and Xu Z: d-Borneol enhances cisplatin
sensitivity via autophagy dependent EMT signaling and
NCOA4-mediated ferritinophagy. Phytomedicine.
106(154411)2022.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Ou H, Lin J, Ji L, Ye L, Ling M, Liao X,
Lin F, Wang Y, Luo B, Hu Z and Pan L: Ferritinophagy mediated by
the AMPK/ULK1 pathway is involved in ferroptosis subsequent to
ventilator-induced lung injury. Respir Res. 25(440)2024.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Chino H and Mizushima N: ER-phagy: Quality
control and turnover of endoplasmic reticulum. Trends Cell Biol.
30:384–398. 2020.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Li W, He P, Huang Y, Li YF, Lu J, Li M,
Kurihara H, Luo Z, Meng T, Onishi M, et al: Selective autophagy of
intracellular organelles: Recent research advances. Theranostics.
11:222–256. 2021.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Santamaría PG, Mazón MJ, Eraso P and
Portillo F: UPR: An upstream signal to EMT induction in cancer. J
Clin Med. 8(624)2019.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Zhou JF, Zhou Q, Chen C and Pan JX: A
review of the roles of endoplasmic reticulum stress in cancer cell
metastasis. Sichuan Da Xue Xue Bao Yi Xue Ban. 52:11–15.
2021.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
107
|
Yu M, Chen F, Wang H, Fu Q, Yan L, Chen Z,
Li H, Jia M, Yang D, Hua X, et al: Endoplasmic reticulum stress
mediates nickel chloride-induced epithelial-mesenchymal transition
and migration of human lung cancer A549 cells through Smad2/3 and
p38 MAPK activation. Ecotoxicol Environ Saf.
249(114398)2023.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Abraham AO and Panda PK: Itraconazole
induced congestive heart failure, A case study. Curr Drug Saf.
13:59–61. 2018.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Guo H, Huang RR, Qu SS, Yao Y, Chen SH,
Ding SL and Li YL: FAM134B deletion exacerbates apoptosis and
epithelial-to-mesenchymal transition in rat lungs exposed to
hyperoxia. iScience. 27(110385)2024.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Wang S, Xu F, Liu H, Shen Y, Zhang J, Hu L
and Zhu L: Suppressing endoplasmic reticulum stress alleviates
LPS-induced acute lung injury via inhibiting inflammation and
ferroptosis. Inflammation. 47:1067–1082. 2024.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Pao HP, Liao WI, Tang SE, Wu SY, Huang KL
and Chu SJ: Suppression of endoplasmic reticulum stress by 4-PBA
protects against hyperoxia-induced acute lung injury via
up-regulating Claudin-4 expression. Front Immunol.
12(674316)2021.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Li J, Wang Z, Nan X, Yin M and Fang H:
Hotspots and frontier trends of diabetic associated cognitive
decline research based on rat and mouse models from 2012 to 2021: A
bibliometric study. Front Neurol. 13(1073224)2022.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Kurmus Ferik O, Akbuga K, Tolunay H, Aslan
T, Eren M, Erkan AF, Ekici B, Akgul Ercan E and Kervancıoglu C:
Poor nutritional status is associated with arrhythmic events on
24-hour holter recording. Med Princ Pract. 31:368–375.
2022.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Sanada J, Kamei S, Shimoda M, Tatsumi F,
Kimura T, Obata A, Kohara K, Nakanishi S, Kaku K, Mune T and Kaneto
H: Amelioration of hypercalcemia by cinacalcet treatment in a
subject with relapsing acquired hypocalciuric hypercalcemia: A case
report. Medicine (Baltimore). 100(e27579)2021.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Bastarache JA and Blackwell TS:
Development of animal models for the acute respiratory distress
syndrome. Dis Model Mech. 2:218–223. 2009.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Matute-Bello G, Frevert CW and Martin TR:
Animal models of acute lung injury. Am J Physiol Lung Cell Mol
Physiol. 295:L379–L399. 2008.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Tiba MH, McCracken BM, Leander DC,
Colmenero CI, Nemzek JA, Sjoding MW, Konopka KE, Flott TL, VanEpps
JS, Daniels RC, et al: A novel swine model of the acute respiratory
distress syndrome using clinically relevant injury exposures.
Physiol Rep. 9(e14871)2021.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Ballard-Croft C, Wang D, Sumpter LR, Zhou
X and Zwischenberger JB: Large-animal models of acute respiratory
distress syndrome. Ann Thorac Surg. 93:1331–1339. 2012.PubMed/NCBI View Article : Google Scholar
|