Introduction
Myelodysplastic syndrome (MDS) comprises a group of
clonal hematopoietic malignancies characterized by dysplastic
hematopoiesis, an increased risk of progression to acute myeloid
leukemia (AML), and persistent single-stage or multilineage
cytopenias. The incidence of MDS in children is significantly lower
than that in adults and accounts for <5% of all hematological
cancers in children, with an estimated annual incidence of 1-4 per
1,000,000 children (1). In
contrast to adult MDS, the pediatric type of MDS is frequently
associated with underlying inherited bone marrow failure syndromes
(IBMFs) or a history of high-dose alkylating agent exposure.
Consequently, pediatric MDS has distinct characteristics in terms
of diagnosis, clinical presentation, laboratory findings,
therapeutic management and prognosis (2,3).
Refractory cytopenia of childhood (RCC) represents the most
prevalent subtype of pediatric MDS and accounts for ~50% of cases.
This condition is defined by persistent peripheral blood cytopenias
(including thrombocytopenia, anemia, and/or neutropenia) and may
manifest as pancytopenia (4).
The established indications for initiating treatment
in RCC include the following: i) Transfusion dependency; ii)
clinically significant neutropenia, which is defined as an absolute
neutrophil count (ANC) <1.0x109/l; and iii) the
presence of high-risk chromosomal abnormalities, such as monosomy
7, deletion 7q or complex karyotypes (≥2 aberrations). For patients
with RCC who do not meet these criteria (specifically those with a
normal karyotype and no evidence of a predisposition syndrome),
observational follow-up is considered an appropriate management
strategy, as supported by relevant studies (5).
For eligible patients with RCC who meet the
aforementioned indications, the primary treatment modalities
include chemotherapy, immunosuppressive therapy (IST) and
allogeneic hematopoietic stem cell transplantation (HSCT).
Allogeneic HSCT remains the cornerstone of curative therapy and
should be prioritized for all pediatric patients with MDS,
particularly those exhibiting high-risk features, including
monosomy 7, complex karyotypes, transfusion dependence or severe
neutropenia with a significant risk of infection (2).
By contrast, IST may be considered for patients with
hypocellular BM and associated cytopenias. Although IST yields
favorable outcomes in a subset of adult patients with MDS
(particularly those with low-risk, hypocellular subtypes), it has
also demonstrated efficacy in pediatric RCC, with reported response
rates ranging from 63 to 76% and a more favorable adverse effect
profile than that found in adults (6).
In recent years, epigenetic therapies [particularly
DNA methyltransferase (DNMT) inhibitors such as decitabine. have
been increasingly utilized in adult patients with MDS, with a
considerable proportion of patients achieving clinical benefits
(7). However, clinical experience
with these agents in treating pediatric patients with MDS remains
limited, and robust clinical data from this population are
scarce.
The present case report describes the use of
decitabine in a pediatric patient with RCC who did not respond to
prior immunosuppressive and supportive therapies. It also discusses
the clinical implications of this approach for similar refractory
cases where HSCT is not feasible.
Case report
In September 2015, an 8-year-old girl was admitted
to Lishui Central Hospital (Lishui, China) due to recurrent skin
petechiae and ecchymoses that had persisted for 3 weeks. The
patient's medical history was otherwise unremarkable. A physical
examination revealed scattered dark red petechiae and ecchymoses on
the limbs and trunk, with no hepatosplenomegaly observed. A
complete blood count revealed pancytopenia, with the following
parameters being reported: Hemoglobin concentration, 92 g/l (normal
range, 115-150 g/l); platelet count, 22x109/l (normal
range, 125-350 x109/l); white blood cell count,
2.3x109/l (normal range, 3.5-9.5x109/l); ANC,
1.2x109/l (normal range, 1.8-6.3x109/l);
lymphocyte count, 1.0x109/l (normal range,
1.1-3.2x109/l); and reticulocyte count,
70x109/l (normal range, 25-75x109/l). No
blast cells were detected in the peripheral blood or BM
aspirate.
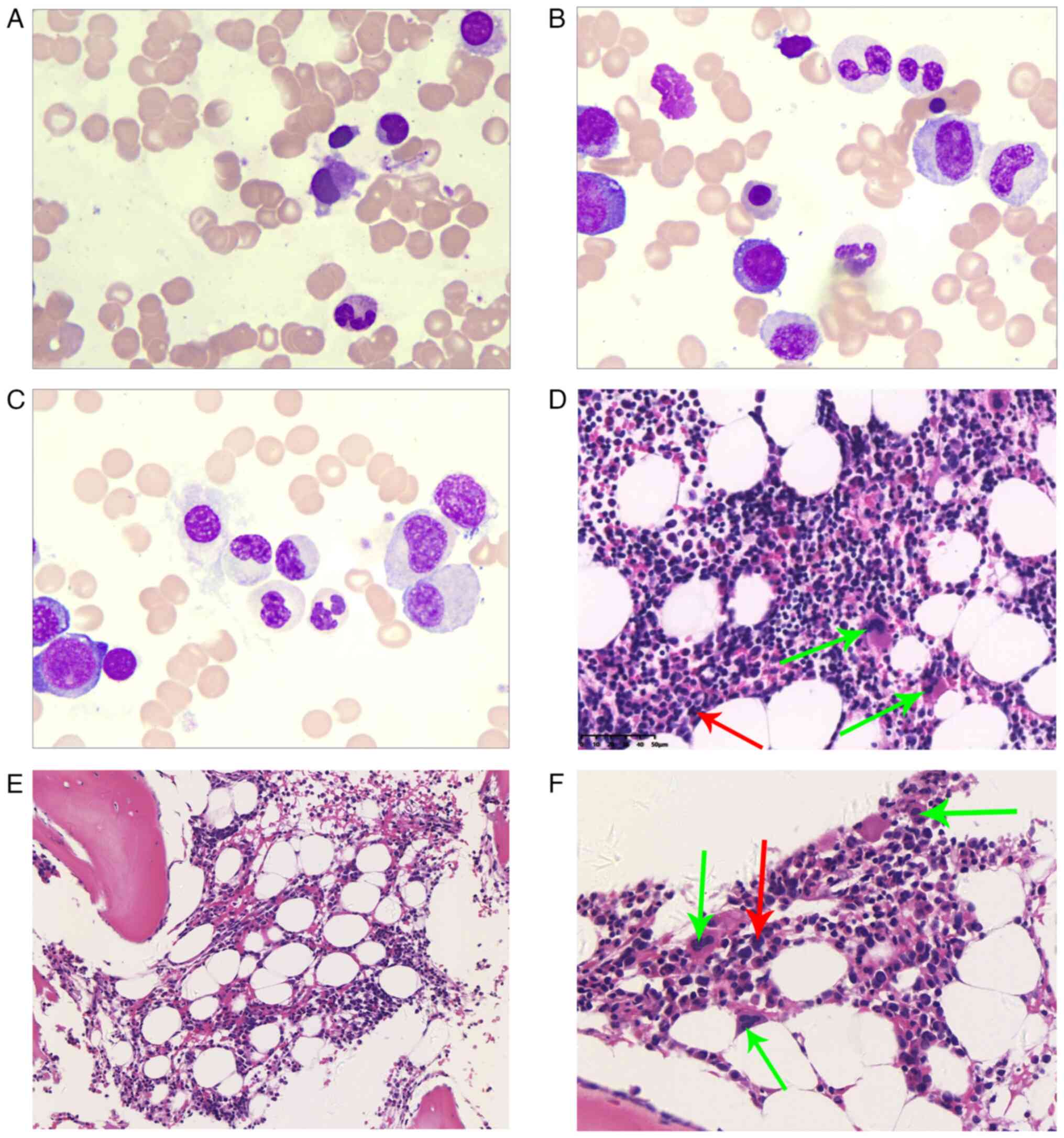
BM aspiration revealed dysplastic hematopoiesis,
which was characterized by small megakaryocytes and partial
neutrophil maturation arrest (Fig.
1A-C). A BM biopsy confirmed the presence of hypocellular
myeloid hematopoietic tissue (~50%), with a normal
granulocyte-erythrocyte ratio. Granulocytic cells at all stages
were present, accompanied by myelodysplasia of neutrophils. The
erythroid lineage was dominated by intermediate and late
erythroblasts. Megakaryocytes were not scarce and were composed
mainly of lobulated forms; small megakaryocytes (with
hyperchromatic nuclei, small cell bodies and few lobes) were easily
detectable and scattered in distribution. Lymphocytes and plasma
cells were scattered. Reticulin staining revealed a myelofibrosis-0
grade, defined as no significant fibrosis according to the European
Consensus/WHO grading system (8)
(Fig. 1D-F). Flow cytometry
immunophenotyping (Data S1;
Fig. S1) revealed no aberrant
blast population, and conventional cytogenetic analysis revealed a
normal karyotype. Next-generation sequencing (NGS) of a targeted
gene panel (including ASXL1, BCOR, DNMT3A, EZH2, IDH1, IDH2, JAK2,
KRAS, NRAS, RUNX1, SF3B1, TET2 and TP53, among other genes)
revealed no significant somatic mutations.
Further investigations were performed to exclude
underlying conditions. Genetic testing for genes indicating IBMFs
(including FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF,
FANCG, FANCL, FANCM, SAMD9, SAMD9L, GATA2 and DKC1) yielded
negative results. A chromosomal breakage test for Fanconi anemia
was also negative. Additional workup ruled out parvovirus B19
infection, vitamin B12 deficiency and folate deficiency as causes
of the hypoplastic BM. Serum antinuclear antibody and rheumatoid
factor results were negative, and serum ferritin levels were within
the normal range.
On the basis of these comprehensive findings, a
definitive diagnosis of RCC was established. Human leukocyte
antigen typing within the family failed to identify a matched
donor. Given an initial ANC >1.0x109/l and the
absence of high-risk cytogenetic features, a watch-and-wait
strategy was initially adopted. However, at the 2-month follow-up,
the ANC had decreased to <1.0x109/l, and the patient
had become platelet transfusion-dependent. The patient's parents
declined HSCT due to concerns over procedural risks and financial
burden.
Consequently, IST with cyclosporine A (CsA) at 150
mg daily (5 mg/kg/day) was initiated alongside transfusion support.
However, after 2 weeks, the patient developed severe drug-induced
liver injury, as evidenced by markedly elevated liver enzymes
[alanine aminotransferase, 1,178 U/l (normal range, 10-40 U/l);
aspartate aminotransferase, 1,090 U/l [normal range, 10-40 U/l)].
CsA was promptly discontinued, and IST was deemed intolerable.
Following the recovery of liver function, decitabine
treatment was initiated in January 2016 at a dosage of 10
mg/m2/day for 5 days per 28-day cycle. Hematological
improvement was observed after two cycles, with a maximum
hemoglobin concentration >60 g/l and a platelet count of
~40x109/l. The transfusion frequency was decreased
accordingly, and the patient achieved transfusion independence by
the third cycle.
However, after the third cycle, the patient
developed a severe secondary infection accompanied by high-grade
fever (peak temperature, 39.5˚C). Management included
administration of granulocyte colony-stimulating factor at 5
µg/kg/day subcutaneously for 14 days and recombinant human
thrombopoietin at 300 U/kg/day subcutaneously for 12 days to
support hematopoietic recovery, along with meropenem at 20 mg/kg
intravenously every 8 h for a 10-day course for anti-infective
therapy. The infection was successfully controlled, and the body
temperature normalized within ~2 weeks. A period of severe
neutropenia (<0.5x109/l) persisted for ~6 weeks, with
blood counts recovering by the seventh week. Transfusion
independence was maintained throughout this period.
The patient completed a total of eight cycles of
decitabine. Complete peripheral blood recovery was achieved after
the fourth cycle, with a platelet count of 100x109/l and
an ANC of 1.7x109/l (Table
I). The post-treatment evaluation included the collection of BM
aspirates in June 2017 (Fig. 2),
March 2018 and March 2019, all of which confirmed stable disease
without evidence of progression. Repeated flow cytometry analyses
during this period yielded negative results. Given the sustained
normalization of the peripheral blood counts, a follow-up BM biopsy
was deemed unnecessary. As of the most recent follow-up on August
21, 2024, the patient continues to maintain a stable clinical
status under routine outpatient surveillance (every 3 to 6
months).
 | Table IPeripheral blood indicator changes
across different treatment cycles. |
Table I
Peripheral blood indicator changes
across different treatment cycles.
| Decitabine
cycles | WBCs
(x109/l) | RBCs
(x1012/l) | ANC
(x109/l) | HB, g/l | PLTs
(x109/l) |
|---|
| Cycle 1 | 1.05 (0.73,
1.28) | 2.16 (1.90,
2.89) | 0.30 (0.13,
0.65) | 70.0 (60.5,
89.0) | 18.5 (9.0, 53.5) |
| Cycle 2 | 0.80 (0.73,
1.10) | 1.98 (1.71,
2.23) | 0.10 (0.10,
0.30) | 61.5 (56.5,
70.8) | 12.0 (8.3, 36.0) |
| Cycle 3 | 1.00 (0.80,
1.70) | 2.23 (2.04,
2.55) | 0.10 (0.10,
0.23) | 69.5 (61.8,
76.3) | 21.5 (7.8, 34.3) |
| Cycle 4 | 1.70 (1.18,
1.95) | 2.79 (2.30,
3.10) | 0.40 (0.10,
0.60) | 86.5 (71.8,
103.3) | 39.0 (14.0,
67.0) |
| Cycle 5 | 3.10 (2.48,
4.63) | 3.72 (3.67,
3.78) | 1.20 (0.48,
1.93) | 125.5 (123.8,
131.3) | 86.5 (72.5,
111.8) |
| Cycle 6 | 4.10 (3.70,
4.63) | 3.71 (3.58,
3.87) | 1.55 (1.28,
1.83) | 126.0 (119.5,
130.3) | 114.5 (77.5,
145.8) |
| Cycle 7 | 4.20 (3.93,
4.98) | 4.12 (3.87,
4.19) | 1.75 (1.45,
2.38) | 135.0 (127.3,
135.8) | 152.0 (146.5,
180.0) |
| Cycle 8 | 5.00 (4.25,
5.38) | 4.12 (3.95,
4.17) | 2.30 (1.80,
3.70) | 139.5 (131.3,
143.3) | 205.5 (170.5,
235.3) |
| 2 years after
treatment | 5.1 (5.00, 5.20) | 4.23 (4.10,
4.25) | 2.80 (2.70,
2.82) | 138.0 (134.0,
140.0) | 264.0 (245.0,
268.0) |
Discussion
Childhood MDS represents a diverse category of
clonal hematopoietic diseases characterized by their rare
occurrence. RCC represents the most common subtype of pediatric MDS
and was first classified as a provisional entity in the 2008 World
Health Organization classification revision (9).
Pancytopenia accompanied by reduced BM cellularity
in children has diverse etiologies, with acquired severe aplastic
anemia, IBMFs and RCC representing the three most common
hematopoietic disorders (2). This
overlap results in specific challenges regarding the diagnosis of
RCC. The diagnosis of RCC is primarily based on morphological
criteria for MDS, which specifically include <5% blasts in the
BM, <2% blasts in the peripheral blood and the presence of
dysplastic features (which are most frequently observed in the
erythroid and megakaryocytic lineages) (1). A significant diagnostic hurdle
involves the phenotypic mimicry between RCC and certain IBMFs, such
as Fanconi anemia, which can present with similar morphological
characteristics (10).
In this context, ancillary techniques are crucial
for the differential diagnosis. Immunohistochemical staining of BM
biopsies for megakaryocytic antigens (such as CD61, CD41, and
CD42b) can aid in identifying micromegakaryocytes, which is a
feature that supports a diagnosis of RCC over aplastic anemia or
other conditions (11).
Cytogenetic abnormalities, which are detected in ~30% of RCC cases,
provide another diagnostic clue, with monosomy 7 being the most
frequent finding (12).
Flow cytometry is widely employed in MDS diagnosis
due to its ability to detect abnormal maturation patterns or
aberrant antigen expression that is indicative of dysplasia
(13). However, its utility in RCC
or low-grade MDS is limited, as the results are often observed to
be within normal limits. The advent of NGS has revealed a distinct
mutational landscape in childhood MDS (cMDS). Specifically,
germline mutations in genes such as SAMD9/SAMD9L and GATA2 are
detected in patients with MDS across the blast spectrum (14), whereas somatic mutations are more
common in patients with MDS with increased blasts. Notably, somatic
mutations in genes such as ASXL1, RUNX1, SETBP1 and KRAS are
frequently identified in ~46% of patients with secondary MDS or
IBMFs (15,16). By contrast, mutations that are
typically detected in adult MDS (such as TET2, DNMT3A, SF3B1, and
SRSF2) are typically absent in cMDS (17).
The prognosis of cMDS is influenced by the blast
percentage, complex karyotypes and single-lineage dysplasia
(18). To the best of our
knowledge, there are no reports on decitabine monotherapy for cMDS,
and data on the effect of genetic mutations on prognosis are
scarce. A recent study showed that decitabine-based combination
therapy or bridging to transplantation improves survival (19). Long-term remission with
decitabine-based combination therapy has been reported in
non-transplanted patients (20).
Although the prognostic effect of specific genetic abnormalities
remains an area of active investigation, emerging evidence suggests
that SETBP1 mutations may be associated with shortened overall
survival (OS) time (17).
The management of RCC remains controversial and is
guided primarily by clinical and cytogenetic risk stratification.
Allogeneic HSCT is considered a curative option for children with
MDS, particularly those demonstrating a high risk of disease
progression (20). For patients
with low-risk RCC who are not transfusion dependent and who lack
significant neutropenia, a watch-and-wait strategy is generally
recommended (5). Conversely,
therapeutic interventions (such as immunosuppressive therapy) are
indicated for those with persistent neutropenia and/or transfusion
dependency for platelets or red blood cells (2). Despite these general principles, data
on effective treatment strategies for patients with RCC who are
ineligible for either HSCT or immunosuppressive therapy remain
limited.
Epigenetic therapies (particularly DNMT inhibitors
such as decitabine and azacitidine) are being increasingly utilized
in the treatment of adult MDS. Although traditionally indicated for
intermediate- to high-risk MDS, emerging evidence supports the use
of hypomethylating agents in low-risk MDS with multilineage
dysplasia, where they have demonstrated clinical benefit (21). Nevertheless, the optimal dosing and
scheduling of decitabine remain to be fully established.
The efficacy of decitabine has been evaluated across
different dosing schedules in clinical trials conducted at the MD
Anderson Cancer Center. The standard 3-day regimen of decitabine
was established through comparative studies of earlier dosing
schedules, and subsequent trials have confirmed the efficacy of an
alternative 5-day schedule (22,23).
In one randomized study on adult patients with MDS, two regimens
were compared, including 20 mg/m2 decitabine
administered daily for 3 days every 28 days vs. 20 mg/m2
decitabine administered weekly for 3 weeks every 28 days. The two
schedules demonstrated comparable efficacy, with an objective
response rate of 23%, no drug-related mortality and transfusion
independence being achieved in 67% of the patients. Moreover, the
median OS time was not reached, and 70% of the patients were alive
at 500 days, thereby supporting the use of low-dose decitabine in
this patient population (7).
However, the use of hypomethylating agents in
pediatric MDS remains limited. Emerging evidence suggests that
high-risk pediatric patients with MDS may benefit from
decitabine-containing conditioning regimens prior to treatment with
allogeneic HSCT. In one study involving 27 children with high-risk
MDS, decitabine-based conditioning was associated with an 84.8%
3-year OS rate, thus indicating excellent outcomes in this
population (19). Similarly,
another study including 20 children with MDS demonstrated that
compared with those receiving AML-type chemotherapy, the subgroup
receiving decitabine combined with a minimally myelosuppressive
regimen before transplantation achieved significantly better
survival, with a 4-year OS rate of 84.6±10%, compared with
66.7±27.2% in the decitabine alone group and 0.0±0.0% in the
chemotherapy group (20).
By contrast, data concerning the use of DNMT
inhibitors in non-transplanted pediatric patients with MDS remain
scarce. A report by Glasser et al (24) described the cases of two children
with secondary AML and MDS who achieved BM disease stabilization
following treatment with decitabine and vorinostat, highlighting a
potential role for epigenetic therapy outside the transplant
setting.
In the present patient, the primary clinical
manifestation was pancytopenia, without evidence of high-risk
features for progression to advanced MDS or AML. Treatment was
indicated due to transfusion dependency and significant
neutropenia. However, therapeutic options for RCC or cMDS beyond
IST and transplantation remain limited.
Currently, the use of decitabine in cMDS has rarely
been reported and has been observed mainly in combination regimens
or as a bridge to transplantation, with no report on its use as
monotherapy. The present patient received 8 cycles of decitabine
monotherapy, with peripheral blood recovery observed after 4
cycles; moreover, disease stability has been maintained to
date.
To the best of our knowledge, there are no previous
reports of decitabine monotherapy for cDMS, and this is the first
case in which such monotherapy was used in this context. Moreover,
the present case has the longest follow-up reported to date, with
the patient continuing to demonstrate sustained remission with this
treatment approach. This case suggests that decitabine may
represent a viable treatment option for patients with low-risk cMDS
or RCC, particularly those who are not suitable candidates for
transplantation or who cannot tolerate immunosuppressive therapy.
However, further data from larger prospective studies are necessary
to confirm the safety and efficacy of epigenetic therapy (whether
administered as monotherapy or in combination regimens) in
pediatric patients with MDS.
Supplementary Material
Data S1
Figure S1
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be found
in the Genome Sequence Archive of the National Genomics Data
Center, China National Center for Bioinformation/Beijing Institute
of Genomics, Chinese Academy of Sciences under accession number
HRA015221 or at the following URL: https://ngdc.cncb.ac.cn/gsa-human/browse/HRA015221.
The remaining data generated in the present study may be requested
from the corresponding author.
Authors' contributions
ZF and JZ contributed to the conception of the
study. ZF collected the data and wrote the manuscript. AS, JZ and
NX analyzed patient data and revised the manuscript. ZF and JZ
confirm the authenticity of all the raw data. All authors have read
and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
parents of the patient for the publication of the present
study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hasle H: Myelodysplastic and
myeloproliferative disorders of childhood. Hematology Am Soc
Hematol Educ Program. 2016:598–604. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Locatelli F and Strahm B: How I treat
myelodysplastic syndromes of childhood. Blood. 131:1406–1414.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pastor V, Hirabayashi S, Karow A, Wehrle
J, Kozyra EJ, Nienhold R, Ruzaike G, Lebrecht D, Yoshimi A,
Niewisch M, et al: Mutational landscape in children with
myelodysplastic syndromes is distinct from adults: Specific somatic
drivers and novel germline variants. Leukemia. 31:759–762.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Passmore SJ, Chessells JM, Kempski H, Hann
IM, Brownbill PA and Stiller CA: Paediatric myelodysplastic
syndromes and juvenile myelomonocytic leukaemia in the UK: A
population-based study of incidence and survival. Br J Haematol.
121:758–767. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dufour C, Schwarz-Furlan S, Baumann I,
Rudelius M, Nöllke P, Lebrecht D, Ramamoorthy S, Rotari N, Karow A,
Hirabayashi S, et al: Long-term outcomes of patients with
refractory cytopenia of childhood under observation only. Blood
Adv. 9:4279–4285. 2025.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yoshimi A, Baumann I, Führer M,
Bergsträsser E, Göbel U, Sykora KW, Klingebiel T, Gross-Wieltsch U,
van den Heuvel-Eibrink MM, Fischer A, et al: Immunosuppressive
therapy with anti-thymocyte globulin and cyclosporine A in selected
children with hypoplastic refractory cytopenia. Haematologica.
92:397–400. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Garcia-Manero G, Jabbour E, Borthakur G,
Faderl S, Estrov Z, Yang H, Maddipoti S, Godley LA, Gabrail N,
Berdeja JG, et al: Randomized open-label phase II study of
decitabine in patients with low- or intermediate-risk
myelodysplastic syndromes. J Clin Oncol. 31:2548–2553.
2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Thiele J, Kvasnicka HM, Facchetti F,
Franco V, van der Walt J and Orazi A: European consensus on grading
bone marrow fibrosis and assessment of cellularity. Haematologica.
90:1128–1132. 2005.PubMed/NCBI
|
|
9
|
Garcia-Manero G: Myelodysplastic
syndromes: 2023 update on diagnosis, risk-stratification, and
management. Am J Hematol. 98:1307–1325. 2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H, Thiele J and Vardiman JW (eds): WHO
Classification of Tumours of Haematopoietic and Lymphoid Tissues.
4th edition. IARC Press, Lyon, 2008.
|
|
11
|
Shimamura A and Alter BP: Pathophysiology
and management of inherited bone marrow failure syndromes. Blood
Rev. 24:101–122. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rudelius M, Weinberg OK, Niemeyer CM,
Shimamura A and Calvo KR: The international consensus
classification (ICC) of hematologic neoplasms with germline
predisposition, pediatric myelodysplastic syndrome, and juvenile
myelomonocytic leukemia. Virchows Arch. 482:113–130.
2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Baumann I, Führer M, Behrendt S, Campr V,
Csomor J, Furlan I, de Haas V, Kerndrup G, Leguit RJ, De Paepe P,
et al: Morphological differentiation of severe aplastic anaemia
from hypocellular refractory cytopenia of childhood:
Reproducibility of histopathological diagnostic criteria.
Histopathology. 61:10–17. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Westers TM, Ireland R, Kern W, Alhan C,
Balleisen JS, Bettelheim P, Burbury K, Cullen M, Cutler JA, Della
Porta MG, et al: Standardization of flow cytometry in
myelodysplastic syndromes: A report from an international
consortium and the European LeukemiaNet Working Group. Leukemia.
26:1730–1741. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kennedy AL and Shimamura A: Genetic
predisposition to MDS: Clinical features and clonal evolution.
Blood. 133:1071–1085. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sahoo SS, Pastor VB, Goodings C, Voss RK,
Kozyra EJ, Szvetnik A, Noellke P, Dworzak M, Starý J, Locatelli F,
et al: Clinical evolution, genetic landscape and trajectories of
clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat Med.
27:1806–1817. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kozyra EJ, Hirabayashi S, Loyola VBP,
Przychodzen B, Karow A, Catala A, De Moerloose B, Dworzak M, Hasle
H, Masetti R, et al: Clonal mutational landscape of childhood
myelodysplastic syndromes. Blood. 126(1662)2015.
|
|
18
|
Hasegawa D, Chen X, Hirabayashi S, Ishida
Y, Watanabe S, Zaike Y, Tsuchida M, Masunaga A, Yoshimi A, Hama A,
et al: Clinical characteristics and treatment outcome in 65 cases
with refractory cytopenia of childhood defined according to the WHO
2008 classification. Br J Haematol. 166:758–766. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ren Y, Liu F, Chen X, Zhang X, Zhao B, Wan
Y, Lan Y, Li X, Yang W, Zhu X and Guo Y: Decitabine-containing
conditioning improved outcomes for children with higher-risk
myelodysplastic syndrome undergoing allogeneic hematopoietic stem
cell transplantation. Ann Hematol. 103:1345–1351. 2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gao J, Hu Y, Gao L, Xiao P, Lu J and Hu S:
The effect of decitabine-combined minimally myelosuppressive
regimen bridged allo-HSCT on the outcomes of pediatric MDS from 10
years' experience of a single center. BMC Pediatr.
22(312)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Schwartz JR, Ma J, Lamprecht T, Walsh M,
Wang S, Bryant V, Song G, Wu G, Easton J, Kesserwan C, et al: The
genomic landscape of pediatric myelodysplastic syndromes. Nat
Commun. 8(1557)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kantarjian H, Issa JPJ, Rosenfeld CS,
Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C,
Ravandi F, et al: Decitabine improves patient outcomes in
myelodysplastic syndromes: Results of a phase III randomized study.
Cancer. 106:1794–1803. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kantarjian H, Oki Y, Garcia-Manero G,
Huang X, O'Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F,
Estrov Z, et al: Results of a randomized study of 3 schedules of
low-dose decitabine in higher-risk myelodysplastic syndrome and
chronic myelomonocytic leukemia. Blood. 109:52–57. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Glasser CL, Lee A, Eslin D, Marks L, Modak
S and Glade Bender JL: Epigenetic combination therapy for children
with secondary myelodysplastic syndrome (MDS)/acute myeloid
leukemia (AML) and concurrent solid tumor relapse. J Pediatr
Hematol Oncol. 39:560–564. 2017.PubMed/NCBI View Article : Google Scholar
|