Introduction
Lymphoplasmacytic lymphoma (LPL)/Waldenström's
macroglobulinemia (WM) is a rare indolent B-cell
lymphoproliferative disorder (B-LPD), characterized by clonal
lymphoplasmacytic proliferation in the bone marrow and massive
secretion of monoclonal immunoglobulin M (IgM) (1). WM accounts for 90-95% of all LPL
cases, predominantly affecting the elderly population with an
annual incidence of ~3 per million adults (2-4).
Clinical manifestations are heterogeneous, with nearly 30% of
patients being asymptomatic initially at early stages and do not
require immediate treatment (1).
By contrast, symptomatic patients may present with a variety of
signs, the most common of which include fatigue and anemia caused
by bone marrow infiltration (1).
However, certain patients develop hepatosplenomegaly,
lymphadenopathy or life-threatening hyperviscosity syndrome
characterized by dizziness, fatigue, weakness and blurred vision
(1). Due to its insidious onset
and non-specific initial symptoms, numerous patients first present
to non-hematology departments, leading to diagnostic delay and
compromised prognosis (5).
Therefore, identifying early, recognizable diagnostic clues is
critical.
A major diagnostic challenge lies in WM's
overlapping features with other mature B-LPDs and the lack of
distinctive early warning signs. Cold agglutination-an uncommon but
insightful laboratory clue-rarely receives routine clinical
emphasis. Pathogenic cold agglutinins in WM are mostly monoclonal
IgM antibodies targeting erythrocyte I/i antigens, inducing
spontaneous agglutination at room or lower temperatures. Cold
agglutination not only triggers autoimmune hemolytic anemia via
high-titer monoclonal IgM binding to erythrocyte I/i antigens but
also severely distorts routine hematological test results,
mimicking primary autoimmune hemolytic anemia or idiopathic
cytopenia (6,7). This laboratory interference readily
leads to misdiagnosis and delays recognition of the underlying WM.
Given that WM cases presenting with cold agglutination as the
inaugural and pivotal diagnostic clue are rare, this misdiagnosis
risk is amplified, underscoring the critical need for heightened
clinical and laboratory awareness of this atypical presentation
(6).
Definitive WM diagnosis requires excluding other
B-LPDs via integrated clinical, histopathological, laboratory,
immunophenotypic and genetic data (4,8-10).
Core criteria include bone marrow lymphoplasmacytic infiltration,
serum monoclonal IgM elevation, characteristic immunophenotypic
profiles by flow cytometry (FCM) and the highly specific MYD88
innate immune signal transduction adaptor (MYD88) L265P mutation
(2,4,11).
For patients presenting with marked IgM elevation (>40 g/l) and
severe hyperviscosity syndrome, therapeutic plasma exchange (TPE)
is the first-line emergency intervention to rapidly reduce
circulating IgM levels, followed by bortezomib-dexamethasone
chemotherapy to control the underlying clonal proliferation
(1,12). The present study reported on a
63-year-old male patient with WM where cold agglutination-induced
test interference served as the decisive initial diagnostic clue.
Through integration of systematic laboratory assessments,
immunophenotyping and genetic analyses, the diagnosis of WM was
confirmed. Due to the markedly elevated IgM (>40 g/l) and
hyperviscosity syndrome, emergency TPE was implemented as the
initial intervention, followed by bortezomib plus dexamethasone
chemotherapy, achieving satisfactory clinical outcomes. This case
highlights cold agglutination as a pivotal diagnostic clue for WM,
emphasizes that integrating cellular immunophenotyping with MYD88
L265P mutation detection is critical for definitive diagnosis and
validates that early sequential plasma exchange and chemotherapy
effectively improve clinical manifestations in such patients.
Case report
Case presentation
A 63-year-old man was admitted to the Department of
Hematology, People's Hospital of Longhua (Shenzhen, China) in
August 2023, presenting with cough, dizziness, weakness, fatigue,
blurred vision and severe anemia. The symptoms had worsened
(dizziness and fatigue) with paroxysmal cough and expectoration
following an upper respiratory tract infection 1 month prior. An
ophthalmological consultation due to blurred vision confirmed
bilateral retinal hemorrhage, bilateral macular edema and bilateral
ametropia. After a series of examinations, the patient was
diagnosed with WM. Owing to the patient's concerns about treatment
efficacy, he was transferred to the Department of Hematology,
Shenzhen People's Hospital (Shenzhen, China) for further treatment.
The patient was managed with a stepwise strategy of symptomatic
plasma exchange followed by bortezomib plus dexamethasone
chemotherapy, resulting in satisfactory clinical symptom remission
and resolution of cold agglutination.
Diagnostic workup
Upon admission, a series of laboratory examinations
were performed to make a definite diagnosis. Of note, initial
hematology analysis triggered a clot detection alarm, with the
EDTA-anticoagulated blood specimen showing fine sand-like
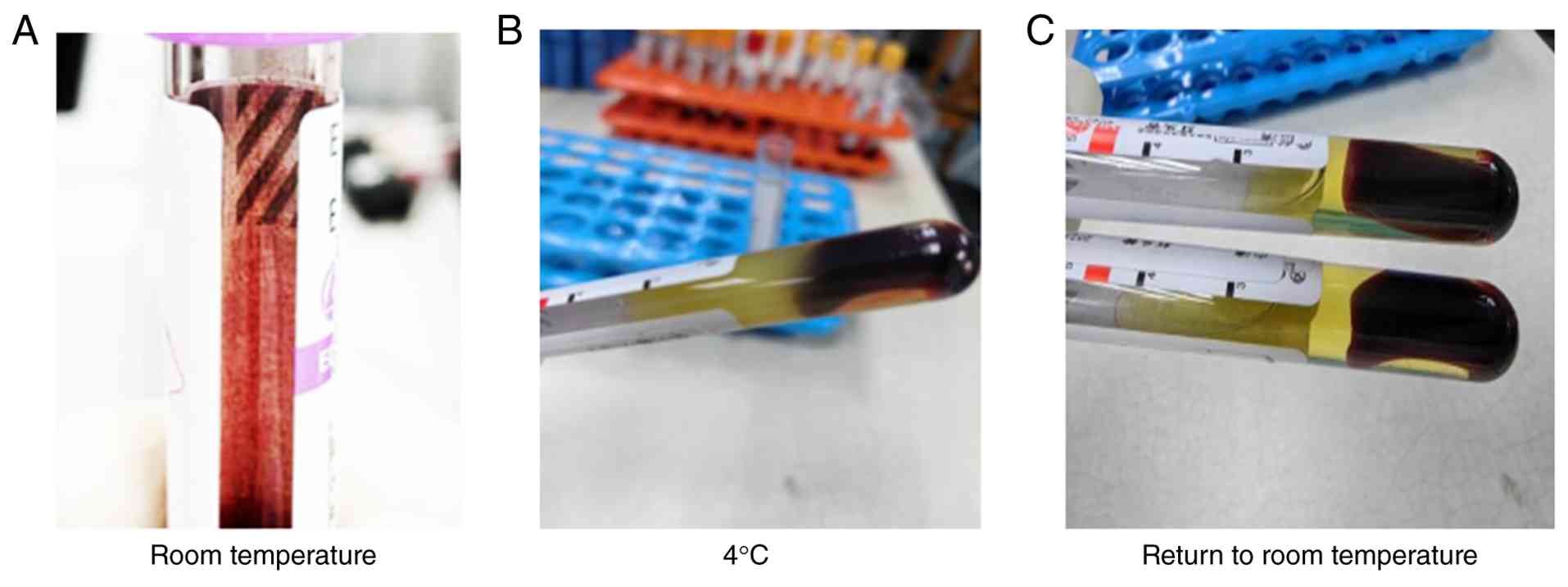
erythrocyte agglutination adherent to the tube wall (Fig. 1A). Following centrifugation, the
serum stored at 4˚C rapidly formed semisolid gel-like aggregates
(Fig. 1B), which gradually
liquefied at room temperature (~25˚C) (Fig. 1C), confirming severe cold
agglutination.
To eliminate the interference of cold agglutination
and obtain valid hematological parameters, the specimens were
incubated at 37˚C for 30 min and analyzed immediately. Key findings
included severe anemia, as evidenced by a decreased red blood cell
count (1.73x1012/l; reference range:
4.1-5.8x1012/l), hemoglobin concentration (54 g/l;
reference range: 125-174 g/l), hematocrit (17.4%; reference range:
40-50%), white blood cells (5.1x109/l; reference range:
3.5-9.5) and platelets (109x109/l; reference range:
125-350x109/l). The biochemical analysis revealed
markedly elevated total protein (131 g/l; reference range: 63-82
g/l), globulin (92 g/l; reference range: 24-35 g/l), hypercalcemia
(serum Ca2+: 3.15 mmol/l; reference range: 2.1-2.69
mmol/l) and impaired renal function [creatinine (143 µmol/l;
reference range: 58-110 µmol/l); creatinine clearance (45 ml/min;
reference range: >80 ml/min)]. Additional abnormalities included
elevated β2-microglobulin (6.16 mg/l; reference range: 1.3-3.0
mg/l); 24-h urinary total protein (24-UMTP) (1,018.8 mg/24 h;
reference range: 50-80 mg/24 h), markedly elevated IgM (83.6 g/l;
reference range, 0.3-2.2 g/l), decreased IgG (6.05 g/l; reference
range, 8.6-17.4 g/l) and IgA (0.55 g/l; reference range, 1.0-4.2
g/l).
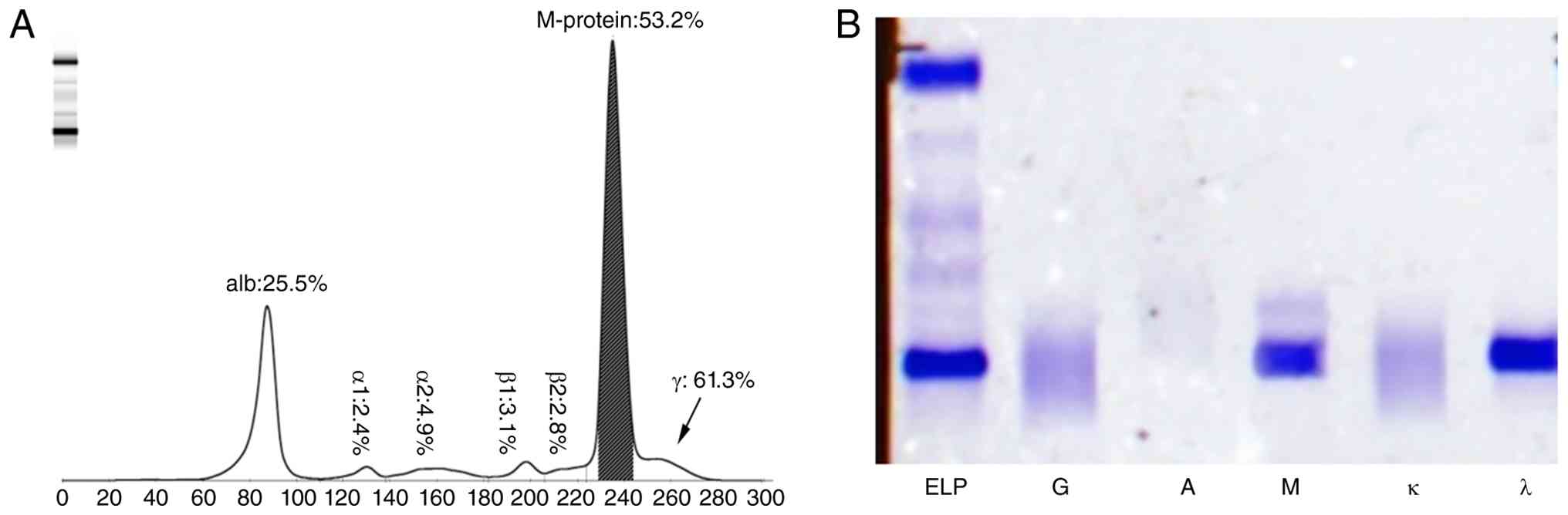
Given the markedly elevated serum IgM level, serum
protein electrophoresis was performed via capillary electrophoresis
using the HYDRAGEL PROTEIN(E) kit (cat. no. PN4140; Sebia)
according to the manufacturer's protocol, which demonstrated an
elevated γ-globulin fraction (61.3% of total protein) and a
distinct monoclonal protein peak accounting for 53.2% of the
γ-globulin fraction (Fig. 2A). The
agarose gel immunofixation electrophoresis using the HYDRAGEL IF
kit (cat. no. PN4309; Sebia) according to the manufacturer's
protocol to further identified the monoclonal component as IgM-λ in
the serum (Fig. 2B). In addition,
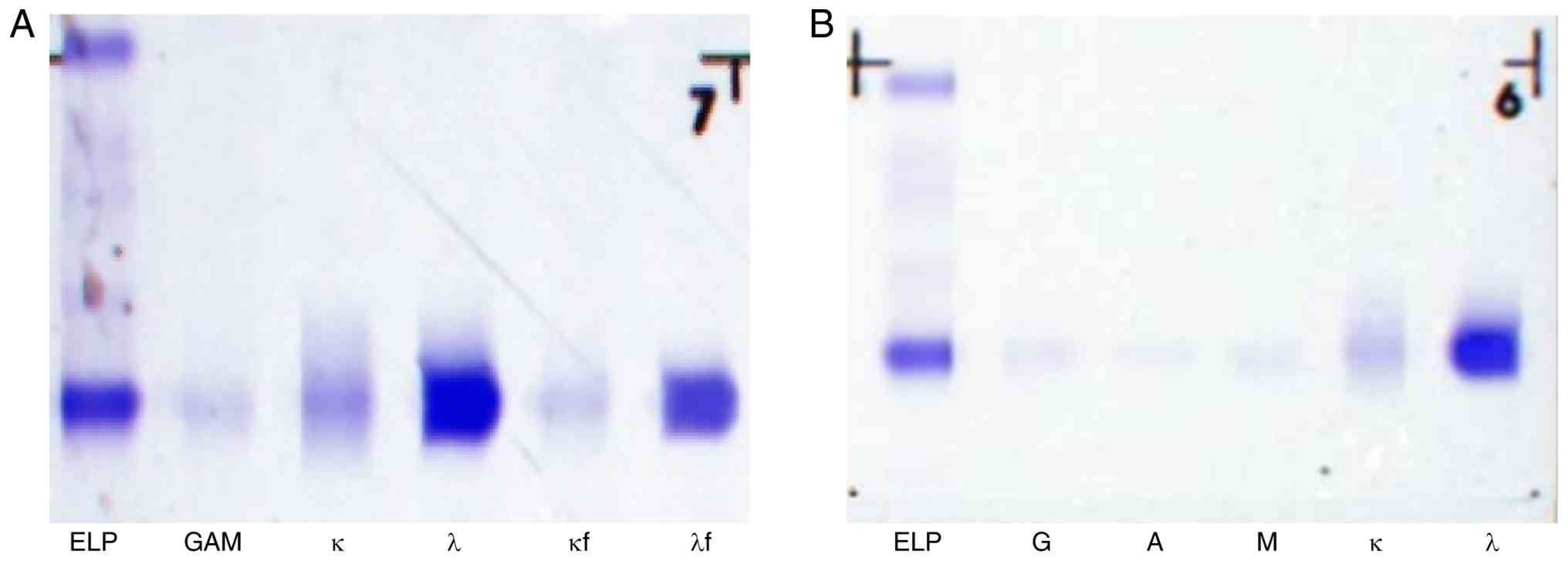
urine protein components were analyzed by agarose gel
immunofixation electrophoresis using the aforementioned HYDRAGEL IF
kit. Distinct precipitation bands were detected in both the λ light
chain and free λ light chain lanes (Fig. 3A), indicating positive urine
Bence-Jones protein of the free λ light chain type. Urine
immunofixation electrophoresis also revealed an abnormal monoclonal
band in the λ lane, indicating that the monoclonal immunoglobulin
was of the free λ light chain type (Fig. 3B). All electrophoresis experiments
were performed on the Sebia HYDRASYS 2 SCAN FOCUSING automated
electrophoresis system (Sebia) in accordance with the
manufacturer's reagent protocols.
Morphological examination is a crucial approach for
evaluating hematological disorders (8). In the present study, Wright-Giemsa
staining (Solution A, cat. no. BA4017C; Solution B, cat. no.
BA4017D; Baso Diagnostics Inc.) was performed to assess the
morphology of peripheral blood and bone marrow specimens, according
to the manufacturer's protocol. Briefly, smears were stained with
solution A for 1 min, mixed with solution B and incubated at room
temperature for 3-10 min. After rinsing and air-drying, the smears
were examined under a light microscope with x100 oil immersion.
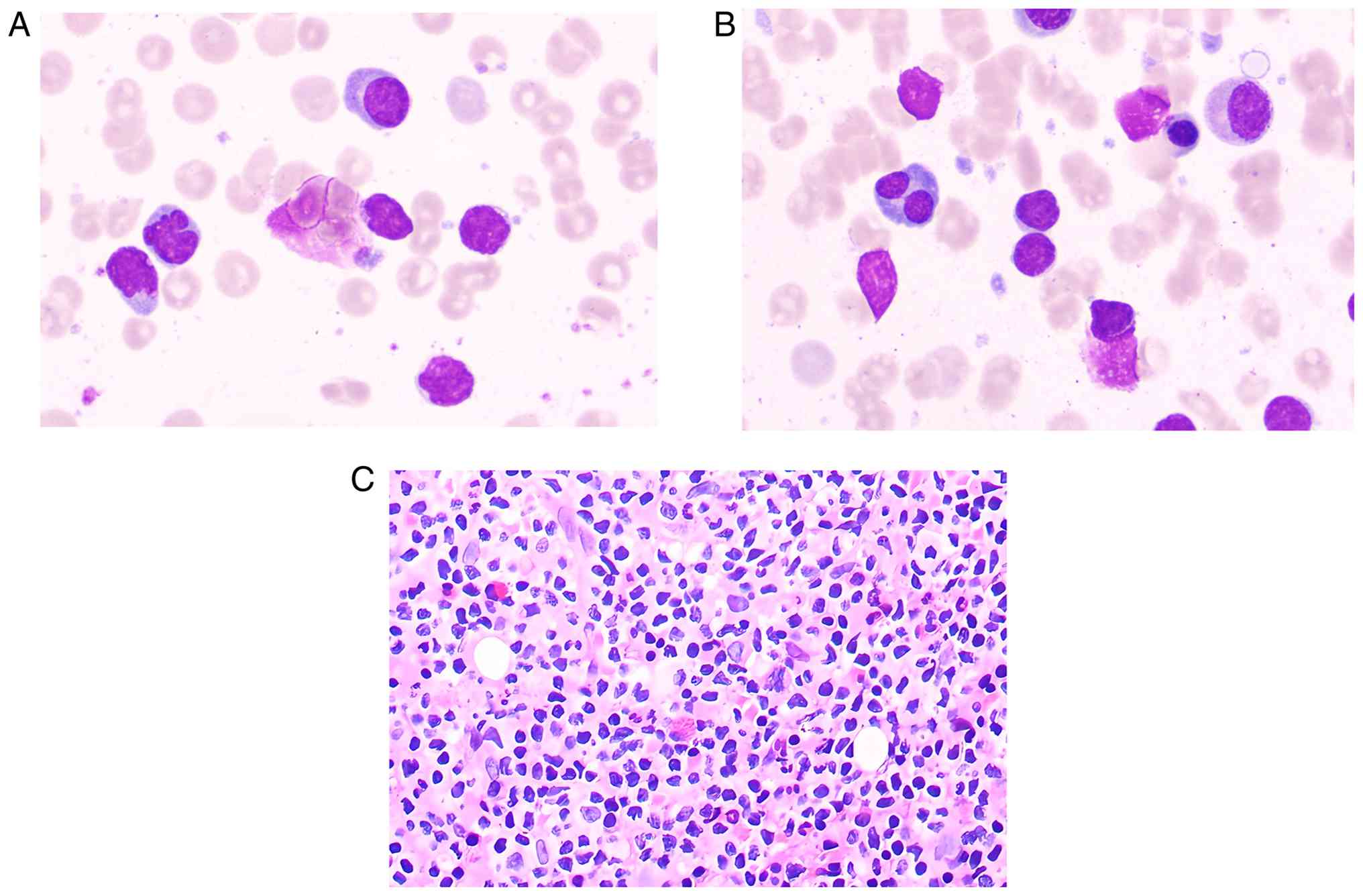
Peripheral blood smear showed rouleaux formation and atypical
lymphocytes (Fig. 4A). Bone marrow
cytology also demonstrated rouleaux formation and an increased
lymphocyte proportion with frequent plasmacytoid lymphocytes
(Fig. 4B). Furthermore,
hematoxylin and eosin (H&E) staining was used to evaluate the
cellular morphology of bone marrow biopsy specimens. Briefly,
specimens were fixed in 10% formalin for 12 h at room temperature,
embedded in paraffin and cut into 5-µm section. The sections were
then deparaffinized (xylene I: 5 min; xylene II: 5 min), hydrated
through graded ethanol (100% for 3 min, 90% for 3 min, 80% for 2
min and 70% for 2 min), stained with hematoxylin for 5 min,
differentiated in 1% hydrochloric alcohol for 3 sec, blued in tap
water for 15 min, counterstained with 0.5% eosin for 10-15 sec,
dehydrated through graded ethanol (80% for 5 sec, 90% for 15 sec,
95% for 30 sec and 100% for 5 min), cleared in xylene (xylene I: 5
min; xylene II: 5 min) and mounted with neutral balsam; all at room
temperature. Morphological observation was carried out using an
Olympus BX53 microscope (Olympus Corporation). It revealed focal
collagenous fibrosis of the medullary stroma, a diffuse lymphocytic
infiltrate exhibiting plasma cell differentiation and small
clusters or focal aggregates of plasma cells. The bone marrow was
extensively infiltrated by B-cell lymphomatous cells, which
accounted for ~80% of nucleated cells, while abnormal plasma cells
comprised 10%, with only 10% residual normal hematopoietic elements
remaining (Fig. 4C). These
morphological features are consistent with B-cell lymphoma, which
is characterized by lymphoplasmacytic infiltration and suppression
of normal hematopoiesis (1,8).
To identify the specific B-cell lymphomatous
subtype, immunophenotyping by FCM was performed on bone marrow
cells (13-15).
Fresh bone marrow aspirate (anticoagulated with EDTA) was subjected
to red blood cell lysis (BD Pharm Lyse™; cat. no.
555899; BD Biosciences) and mononuclear cells were resuspended in
PBS at 1x106 cells/ml. Surface staining with
fluorochrome-conjugated antibodies (cat. nos and dilutions in
Table SI) was performed for 20
min on ice. Intracellular light chains were assessed after
fixation/permeabilization (cat. no. 554722; BD Biosciences) using
anti-cytoplasmic (c)κ-FITC and anti-cλ-PE. Internal negative
controls were used for both fluorescence compensation and gating. T
cells (CD3+CD19-) and B cells
(CD3-CD19+) served as reciprocal negative
controls, with compensation adjusted to achieve proper alignment of
populations in all dot plots. Data acquisition was performed on a
BD FACSCanto II (50,000-100,000 events/tube) and analyzed with
FlowJo v10.8.1 (both from BD Biosciences). Gating employed
CD45/SSC-A for initial lymphocyte discrimination, followed by
CD19-positive B-cell enrichment for immunophenotypic
characterization. The results revealed an increased lymphocyte
proportion (60.4% of nucleated cells, P4), with CD19-positive cells
(P9) accounting for ~72.2% of lymphocytes, suggesting the presence
of abnormally mature B lymphocytes. Furthermore, the
immunophenotypes of these abnormally mature B lymphocytes were
analyzed. These cells exhibited strong positivity for cλ, CD19,
CD20, CD79b and human leukocyte antigen-DR; partial positivity for
CD23, CD25, CD27 and CD200; and negativity for cκ, CD5, CD10, CD22,
CD38, CD103 and follicular mantle clone 7 (FMC7). Additionally,
plasma cells exhibiting strong CD38 expression were identified,
accounting for 0.30% of the cells (P10). Subsequent analysis of
CD27 and CD38 expression levels in plasma cells revealed that the
proportion of normal plasma cells was ~0.10% (P11), whereas
abnormal plasma cells constituted ~0.20% (P12). Abnormal plasma
cells showed strong expression of cλ, CD19, CD38 and CD138, partial
expression of CD20 and CD27, and absence of cκ, CD5, CD10 or CD56
(Fig. 5).
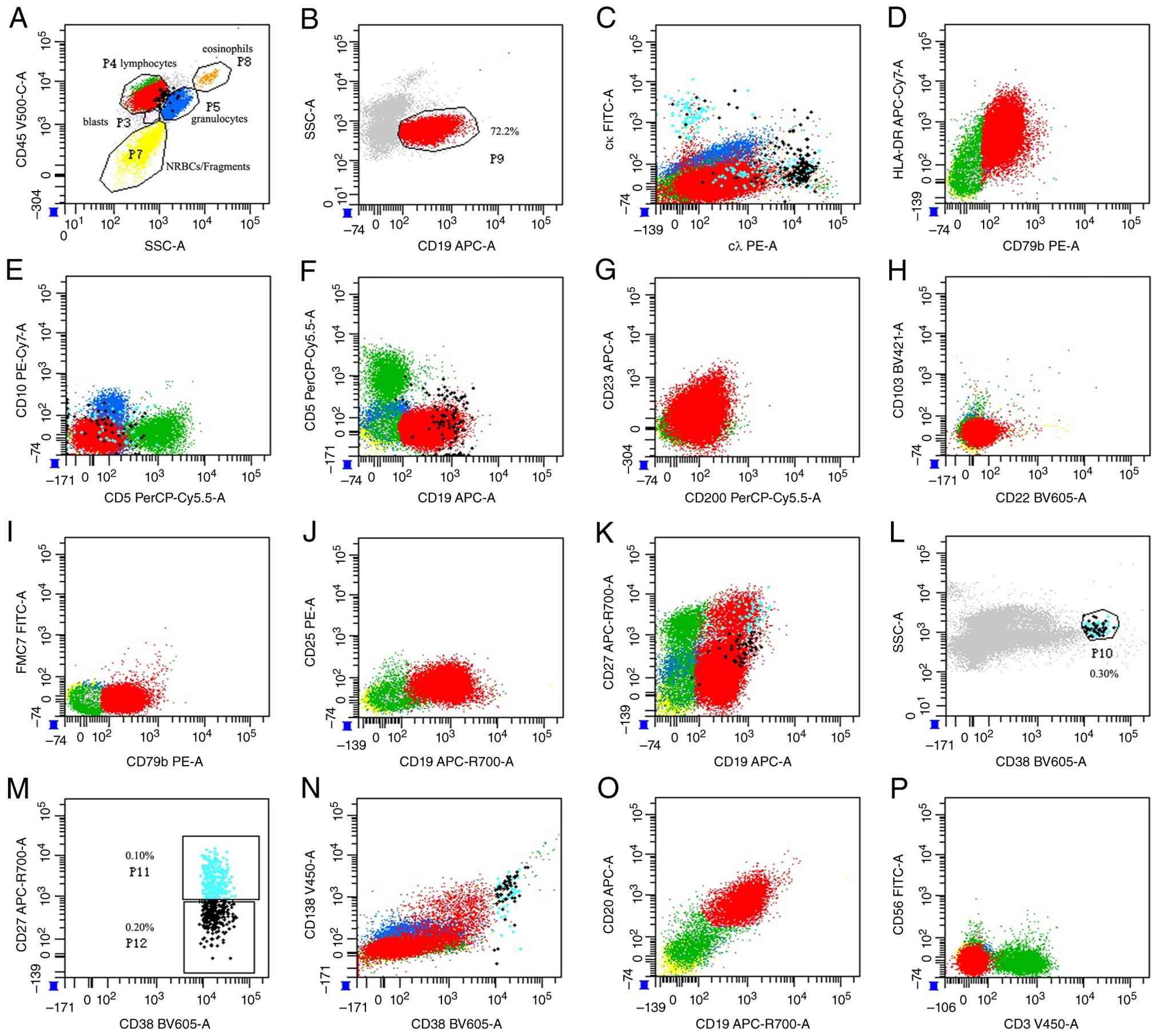 | Figure 5Immunophenotyping of bone marrow
cells by flow cytometry. (A) CD45/SSC-A gating strategy: Blasts
(P3), lymphocytes (P4), granulocytes (P5), NRBCs/fragments (P7),
eosinophils (P8). (B) CD19/SSC-A: CD19+ B cells
comprised 72.2% of lymphocytes (P9). (C) cκ/cλ: Both light chains
expressed. (D) HLA-DR/CD79b: Mature B-cell lineage confirmation.
(E) CD10/CD5: Germinal center and T-cell marker evaluation. (F)
CD5/CD19: CD5+CD19+ B-cell subset. (G)
CD23/CD200: follicular vs. marginal zone phenotype. (H) CD103/CD22:
CD22+CD103-/dim profile (excluding hairy cell
leukemia). (I) FMC7/CD79b: Mature B-cell phenotype. (J) CD25/CD19:
Activated B-cell subset (CD19+CD25+). (K)
CD27/CD19: Naive (CD27-) and memory (CD27+)
subsets. (L) SSC/CD38: Total plasma cells (P10: 0.30%). (M)
CD27/CD38: Normal plasma cells (P11: 0.10%,
CD38++CD27+) and abnormal plasma cells (P12:
0.20%, CD38++CD27dim). (N) CD138/CD38: Plasmacytic
differentiation. (O) CD20/CD19: Mature B cells
(CD19+CD20+) vs. plasmacytic elements
(CD19+CD20dim). (P) CD56/CD3: Absence of T/NK-cell
contamination. ++, bright positive; +, positive; -, negative; dim,
partial expression/weak positive; SSC-A, side scatter-area; NRBCs,
nucleated red blood cells; cκ, cytoplasmic κ light chain; cλ,
cytoplasmic λ light chain; HLA-DR, human leukocyte antigen-DR
isotype. |
Genetic testing plays a crucial role in accurately
diagnosing WM and distinguishing it from other hematologic
malignancies (2). IgM multiple
myeloma (IgM-MM) often harbors the t(11;14) (q13; q32)
translocation or other 14q32 [immunoglobulin heavy chain (IGH)]
rearrangements, whereas the MYD88 L265P mutation is detected in
>90% of WM cases (1,3,14,16).
Accordingly, the MYD88 L265P mutation was detected by
allele-specific polymerase chain reaction (AS-PCR) at Guangzhou
Huayin Medical Laboratory Center (Guangzhou, China). The following
primers were used: Forward 5'-CCTTGGCTTGCAGGTGC-3', reverse
5'-AGGATGCTGGGGAACTCTTT-3', and the following probes: Mutant
(FAM-MGB) 5'-AAGCGACCGATCC-3', and wild-type (VIC-MGB)
5'-AAGCGACTGATCC-3'. Each 20-µl reaction contained 10 µl 2X qPCR
Mix [with uracil-N-glycosylase (UNG)], 0.4 µl each primer (10 µM),
0.2 µl each probe (10 µM), 2-5 µl template DNA and nuclease-free
water to a final volume of 20 µl. The thermal cycling conditions
were as follows: 50˚C for 2 min, 95˚C for 10 min; 45 cycles of 95˚C
for 15 sec and 60˚C for 60 sec. Fluorescence was acquired at FAM
(mutant allele) and VIC/HEX (wild-type allele) channels at the end
of each cycle. In this patient, genetic testing confirmed
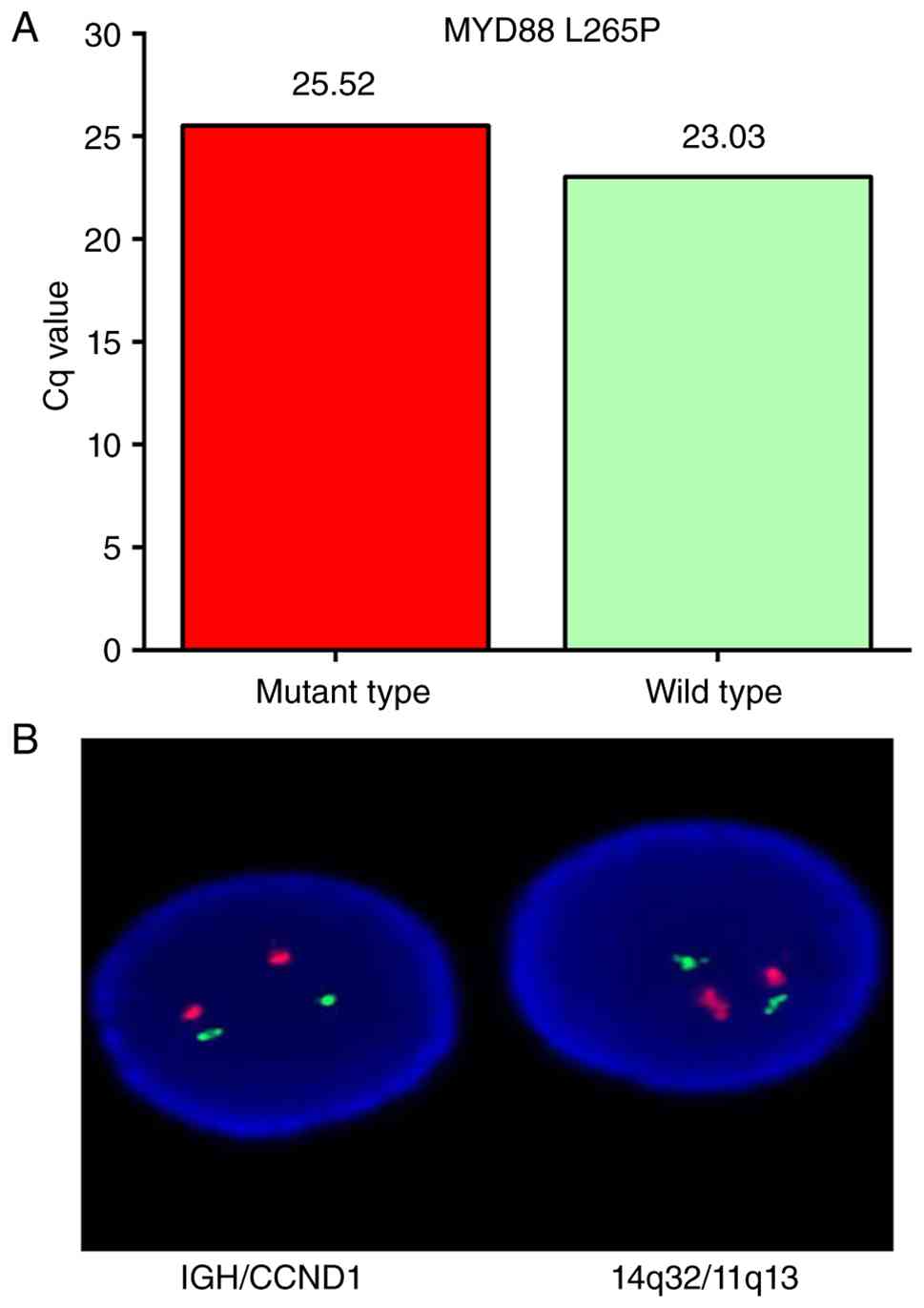
positivity for the MYD88 L265P mutation. AS-PCR showed a Cq value
of 25.52 for the mutant allele and 23.03 for the wild-type allele
(Fig. 6A). Subsequently,
fluorescence in situ hybridization (FISH) was performed on
bone marrow samples to detect the t(11;14) (q13;q32) translocation
using the Vysis LSI IGH/cyclin D1 (CCND1) DF FISH Probe Kit (cat.
no. 08L58-020; Abbott Laboratories, Inc.), following the
manufacturer's instructions. FISH analysis, performed using an
Olympus BX53 fluorescence microscope (Olympus Corporation),
revealed a normal signal pattern in 199 out of 200 interphase
nuclei (99.5%), with two copies each of CCND1 and IGH and no
evidence of gene fusion, indicating the absence of the t(11;14)
(q13;q32) translocation (Fig.
6B).
Treatment process and efficacy
Due to the patient showing serum IgM >40 g/l,
complicated by hyperviscosity syndrome and cold agglutination, a
stepwise therapeutic approach was chosen in accordance with the
latest WM clinical practice guidelines (12). This strategy prioritized
symptomatic relief of hyperviscosity with TPE followed by
etiological chemotherapy targeting the underlying malignant
lymphoplasmacytic proliferation. Initially, one cycle of TPE
comprising five sessions was performed to rapidly mitigate acute
hyperviscosity-related manifestations-a key and potentially
life-threatening complication of WM-with fresh frozen plasma (FFP)
as replacement fluid. Specifically, 2,000 ml FFP was administered
in the first session, followed by 2,500 ml per session for each of
the subsequent four sessions. During the first TPE session, the
patient developed urticarial rash after FFP infusion, which
resolved with temporary infusion cessation and intravenous
dexamethasone (5 mg). Subsequent sessions were premedicated with
intravenous dexamethasone (5 mg) 30 min prior to TPE, with no
further allergic reactions. Upon completion of TPE, systemic
chemotherapy with a bortezomib-dexamethasone regimen was initiated,
consistent with frontline therapeutic recommendations for WM
(1,3,12),
to further control the disease and reduce tumor burden. The regimen
was administered as follows: 2 mg of bortezomib was subcutaneously
injected twice weekly (on days 1 and 4) for 2 consecutive weeks,
and 10 mg of dexamethasone sodium phosphate was intravenously
administered immediately after each bortezomib dose to enhance
therapeutic efficacy and minimize adverse events. Concurrently,
targeted supportive therapy was provided to manage other
accompanying symptoms and improve the patient's treatment
tolerance.
During one month of treatment, serial monitoring of
clinical symptoms and core laboratory parameters were conducted, as
shown in Table I. Before
treatment, the patient presented with severe hyperviscosity
syndrome and multiple organ damage, characterized by a markedly
elevated serum IgM level (83.6 g/l), total protein (131 g/l) and
globulin (92 g/l). Concurrently, the patient exhibited renal
impairment (creatinine: 143 µmol/l; creatinine clearance rate: 45
ml/min; 24-UMTP: 1,018.8 mg/24 h), hypercalcemia (calcium: 3.15
mmol/l), and high tumor burden (β2-microglobulin: 6.16 mg/l,
reference range: 1.3-3.0 mg/l). Immediately after TPE, the serum
IgM level rapidly decreased by 48.5% to 43.04 g/l, with a
concurrent reduction in total protein (from 131 to 99.3 g/l) and
globulin (from 92 to 61 g/l), indicating immediate relief of
hyperviscosity. Renal function also showed rapid improvement, with
creatinine decreasing to 96 µmol/l, and hypercalcemia was corrected
to 2.47 mmol/l within the normal range. At 1 month after
bortezomib-based chemotherapy, sustained therapeutic effects were
observed. The serum IgM level remained lower than before treatment
(from 83.6 to 59.9 g/l) and the β2-microglobulin level dropped to
<0.191 mg/l, demonstrating profound suppression of the
underlying WM clone. Renal function was further improved, with
creatinine normalized to 94 µmol/l and creatinine clearance rate
increased to 74.32 ml/min. Hypercalcemia was completely corrected,
with the calcium level stabilizing at 2.22 mmol/l. The 24-UMTP
decreased from 1,018.8 to 867 mg/24 h, indicating ongoing recovery
of renal injury.
 | Table IComparison of key indicators before
and after treatment. |
Table I
Comparison of key indicators before
and after treatment.
| Index | Before
treatment | After therapeutic
plasma exchange | After
chemotherapy | Reference
range |
|---|
| Calcium,
mmol/l | 3.15 | 2.47 | 2.22 | 2.1-2.69 |
| Total protein,
g/l | 131 | 99.3 | 98.3 | 63-82 |
| Albumin, g/l | 38.7 | 38.0 | 33.3 | 35-50 |
| Globulin, g/l | 92 | 61 | 65 | 24-35 |
| IgG, g/l | 6.05 | 10.75 | 7.61 | 8.6-17.4 |
| IgA, g/l | 0.55 | 1.59 | 0.62 | 1.0-4.2 |
| IgM, g/l | 83.6 | 43.04 | 59.9 | 0.3-2.2 |
| Creatinine,
µmol/l | 143 | 96 | 94 | 58-110 |
| Urea, mmol/l | 8.0 | 8.62 | 8.31 | 3.6-9.5 |
| Creatinine
clearance rate, ml/min | 45 | / | 74.32 | >80 |
| β2-microglobulin,
mg/l | 6.16 | / | <0.191 | 1.3-3.0 |
| 24-UMTP, mg/24
h | 1,018.8 | / | 867 | 50-80 |
Follow-up
After discharge, a structured follow-up plan was
established, consisting of regular monthly visits with hematology
and ophthalmology specialists to monitor disease activity.
Follow-up evaluations included key indicators such as physical
examinations, complete blood counts, serum biochemical tests and
immunoglobulin quantification. However, follow-up was prematurely
terminated after the first month (November 2023) because the
patient required re-admission to the hospital for treatment of a
pulmonary infection. Notably, the patient ultimately developed
severe pulmonary infection and was admitted to the intensive care
unit for management, thereafter progressing to multi-organ failure
in the setting of profound systemic immunocompromise. Regrettably,
in January 2025, the patient's family requested discharge and chose
to withdraw active treatment.
Discussion
LPL/WM is a rare, indolent small B-cell lymphoma
with plasma cell differentiation, accounting for <2% of all
non-Hodgkin lymphoma cases (12).
Pathologically, it is characterized by neoplastic proliferation of
small B lymphocytes, plasmacytoid lymphocytes and plasma cells that
overproduce monoclonal IgM, predominantly involving the bone marrow
with less frequent lymph node or spleen involvement (17,18).
Approximately 90-95% of LPL cases are classified as WM, whose
typical clinical manifestations include anemia, hyperviscosity
syndrome, renal impairment and less commonly organomegaly (1).
Cold agglutinin disease (CAD) is a relatively rare
condition, primarily mediated by IgM (with IgG-mediated forms being
rare), and serves as an important clinical indicator of underlying
WM (6,19). In the present case, cold
agglutination-induced laboratory interference served as the key
diagnostic clue for WM. The patient's samples displayed classic
temperature-dependent agglutination (gelation at 4˚C, liquefaction
at 25˚C)-a hallmark manifestation of WM-related CAD, accompanied by
peripheral blood erythrocyte rouleaux formation-findings that
further confirmed pathological cold agglutination (6). This phenomenon is attributed to
high-titer monoclonal IgM cold agglutinins (6). Notably, incubation of samples at 37˚C
for 30 min resolved this interference, uncovering severe anemia
(hemoglobin 54 g/l)-a manifestation attributable to the
cold-agglutinating activity of monoclonal IgM, which recognizes
specific erythrocyte antigen epitopes at temperatures <37˚C, and
induces chronic hemolytic anemia, as previously reported (6,19-21).
Thus, cold agglutination may serve as an early manifestation of
IgM-secreting lymphoproliferative disorders, providing important
clues for identifying potential patients.
Biochemical and immunoelectrophoretic findings
supported clonal IgM secretion: Elevated total protein (131 g/l),
globulin (92 g/l), inverted A/G ratio (0.42), marked serum IgM
elevation (83.6 g/l), a dominant IgM-λ paraprotein (53.2% of
γ-globulins) and urinary λ free light chains. While these findings
are consistent with WM, they are not disease-specific-similar
abnormalities may occur in IgM-type multiple myeloma (IgM-MM),
IgM-type monoclonal gammopathy of undetermined significance
(IgM-MGUS) and other B-LPDs (1,22).
Therefore, a comprehensive approach combining clinical, laboratory,
pathological, immunophenotypic and molecular testing data is
required to make a definitive distinction.
Cytomorphology represents the fundamental
histopathological basis for diagnosing hematological disorders
(23). Peripheral blood smear in
this patient showed erythrocyte rouleaux formation, lymphocytosis
and numerous plasmacytoid lymphocytes. Bone marrow biopsy revealed
extensive infiltration of B-lymphomatous cells (~80% of nucleated
cells), 10% abnormal plasma cells, and the remaining 10% being
residual normal hematopoietic cells, accompanied by focal
collagenous fibrosis and large clusters of lymphocytes exhibiting
plasmocytic differentiation. These findings, characterized by a
B-cell lymphoproliferative process dominated by lymphoplasmacytic
infiltration and marked suppression of normal hematopoiesis, are
consistent with the pathological features of B-LPDs. Given the
marked elevation of serum IgM-λ paraprotein in the patient, the
primary diagnostic consideration centered on monoclonal
IgM-secreting B-LPDs, specifically IgM-MM, WM and IgM-MGUS.
Rigorous differential diagnosis between WM, IgM-MGUS
and IgM-MM is essential, as these entities differ fundamentally in
clinical management and prognosis (1,24).
According to the LPL/WM clinical practice guidelines, IgM-MGUS was
readily ruled out in this patient (1,12).
Defined as an asymptomatic, premalignant plasma cell disorder,
IgM-MGUS requires serum monoclonal IgM <30 g/l, bone marrow
lymphoplasmacytic infiltration <10% and the absence of organ or
tissue-compromising manifestations or myeloma-defining end-organ
damage. By contrast, the patient presented with a markedly elevated
serum IgM level (83.6 g/l, far exceeding the 30 g/l threshold),
extensive bone marrow lymphoplasmacytic infiltration (80%
B-lymphomatous cells and 10% abnormal plasma cells) and overt
clinical symptoms (severe anemia, hyperviscosity syndrome)-findings
directly inconsistent with the asymptomatic, premalignant nature of
IgM-MGUS.
Differentiating WM, a subtype of LPL, from the rare
IgM-MM is clinically critical, with definitive distinction relying
on integrated morphological, immunophenotypic, cytogenetic and
molecular features (6,25-27).
According to LPL/WM diagnostic guidelines (12), the typical immunophenotype of WM is
CD19(+), CD20(+), sIgM(+), CD22(+), CD25(+), CD27(+), FMC7(+),
CD23(-), CD5(-), CD10(-), usually CD38/CD138(+) and CD103(-), with
10-20% of patients also expressing CD5, CD10 or CD23(27). Consistent with this guideline
profile, the patient's bone marrow revealed a 60.4% lymphocyte
population, of which 72.2% were abnormally mature B cells-highly
expressing cλ, CD19 and CD20, partially expressing CD23, CD25 and
CD27, and negative for CD5, CD10 and CD103. Abnormal plasma cells,
accounting for 0.3% of nucleated cells, showed restricted cλ
expression, high CD38, CD138 and CD19 expression, partial CD27 and
CD20 expression, and negativity for CD5, CD10 and CD56. By
contrast, IgM-MM is characterized by clonal plasma cells, high CD38
and CD138 expression, CD19 and CD45 negativity, and frequent
osteolytic lesions (11,28). Gene testing provided decisive
evidence to further distinguish these two entities: The MYD88 L265P
mutation-a pathognomonic marker present in >90% of WM cases but
rare in IgM-MM (<5%) and absent in IgM-MGUS (1,2,29,30),
was detected in the patient. Additionally, t(11;14)(q13;q32)
translocations, a type of IGH translocation frequently observed in
IgM-MM (31,32), are uncommon in WM. Collectively,
the patient's immunophenotype, positive MYD88 L265P mutation,
negative 14q32 translocations and absence of pure plasma cell
proliferation or osteolytic lesions firmly confirm WM and exclude
IgM-MM, underscoring the clinical value of integrated testing for
differentiating these rare IgM-secreting disorders.
For patients with high-risk WM with a serum IgM
level of 83.6 g/l complicated by hyperviscosity syndrome and CAD,
the core of treatment lies in balancing the relief of acute
symptoms with long-term disease control. A guided strategy-initial
TPE to rapidly alleviate life-threatening acute manifestations,
followed by bortezomib and dexamethasone for etiological
treatment-is fully aligned with current authoritative guidelines
and clinical practice consensus (1,3,33).
TPE is universally recognized as the first-line
emergency intervention for patients with WM with symptomatic
hyperviscosity syndrome (34). Its
mechanism involves the rapid clearance of large amounts of
intravascular IgM, thereby promptly reducing serum viscosity and
alleviating microcirculatory dysfunction (34). In this case, following TPE
treatment, the patient's serum IgM level decreased from 83.6 to
43.04 g/l, accompanied by significant alleviation of hyperviscosity
syndrome-related symptoms (dizziness, fatigue, blurred vision) and
complete resolution of cold agglutination. This validates the
exceptional efficacy of TPE in rapidly reducing IgM burden and
reversing acute organ dysfunction. Notably, serum albumin remained
stable during treatment (38.7 to 38.0 g/l), demonstrating the good
safety profile of TPE when fresh frozen plasma is used as the
replacement fluid, which effectively maintains plasma colloid
osmotic pressure and prevents complications related to
hypoalbuminemia (35).
Although TPE alleviates acute symptoms, it does not
cure the disease, and IgM levels rebound rapidly. Therefore,
immediate follow-up with systemic therapy is crucial. In the
present study, a chemotherapy regimen centered on the proteasome
inhibitor bortezomib was selected. This choice is consistent with
the results of the WMCTG 05-180 clinical trial conducted by Treon
et al (36), which
demonstrated that the bortezomib-dexamethasone-rituximab regimen
achieved an overall response rate as high as 96% in treatment-naive
symptomatic patients with WM, significantly reducing tumor burden.
Although rituximab was not administered in this case, single-agent
bortezomib-dexamethasone still yielded significant, sustained
efficacy. Following treatment, serum IgM was stably controlled at
59.9 g/l, the key tumor burden marker β2-microglobulin decreased
markedly from 6.16 to <0.191 mg/l, and 24-UMTP declined from
1,018.8 mg to 867 mg. These findings confirm effective suppression
of malignant lymphoplasmacytic proliferation and improvement in
IgM-mediated renal injury (37),
in line with reports that bortezomib induces durable remission in
WM (37,38). Therefore, TPE combined with
bortezomib and dexamethasone is an effective and safe strategy for
patients with WM with severe hyperviscosity syndrome and CAD.
Nevertheless, because IgM levels remained above normal after
treatment, long-term follow-up is necessary, and subsequent
addition of BTK inhibitors (e.g., ibrutinib, zanubrutinib) may be
considered based on molecular typing and tolerability to achieve
deeper remission.
Nevertheless, this case study has several
limitations: i) As a single case report, it lacks statistical power
and generalizability; ii) follow-up was prematurely terminated due
to worsening symptoms, resulting in insufficient long-term data for
efficacy and prognosis evaluation; and iii) mechanistic exploration
is limited, and the causal relationships among related pathological
factors remain unclear. Future studies should incorporate larger
cohorts and in-depth molecular mechanistic investigations to
enhance reliability and applicability.
In conclusion, in this case, cold
agglutination-initially considered an analytical
interference-served as a key diagnostic clue for IgM-secreting
lymphoproliferative disorders. Integration of bone marrow
morphology, immunophenotyping and MYD88 L265P mutation detection
confirmed WM and excluded other IgM-secreting disorders, such as
IgM-MM and IgM-MGUS. For patients with IgM-related cold
agglutination, timely bone marrow examination, immunophenotyping
and MYD88 L265P testing are crucial for accurate WM diagnosis.
Early use of FFP as replacement fluid can rapidly relieve
hyperviscosity syndrome, and subsequent chemotherapy can
effectively control the underlying disease to improve clinical
manifestations. This case provides valuable clinical experience for
diagnosing and treating WM with atypical initial manifestations,
particularly cold agglutination.
Supplementary Material
Antibody list for flow cytometry.
Acknowledgements
Not applicable.
Funding
Funding: The authors hereby express their gratitude for the
support provided by the San Ming Project of Medicine in Shenzhen
(grant no. szzysm202311020), the National Flagship Department
Construction Project for Integrated Traditional Chinese and Western
Medicine [National Administration of Traditional Chinese Medicine
Comprehensive Integration Letter No. 221 (2024)] and the Team-based
Medical Science Research Program (grant no. 2024YZZ07) for this
research.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
WZ and YX performed the patient's laboratory
examinations, analyzed the data, and drafted the initial
manuscript. ZY, LZ and XJ participated in collecting the patient's
information and analyzing the data. TC and PM conceived the study,
acquired funding, conducted the investigation, and participated in
manuscript review and editing. All authors confirm the authenticity
of all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was conducted in accordance with the
Declaration of Helsinki and its amendments. According to Chinese
laws and regulations, a case report does not constitute clinical
research and thus does not require ethical review.
Patient consent for publication
Written informed consent for publication was
obtained from the patient and their family. This case report
includes all medical data, laboratory results, and clinical images
collected during the patient's outpatient and inpatient care, with
all personal identifiers removed to protect patient privacy.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gertz MA: Waldenström macroglobulinemia:
2025 update on diagnosis, risk stratification, and management. Am J
Hematol. 100:1061–1073. 2025.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao
Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al: MYD88
L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J
Med. 367:826–833. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Treon SP, Sarosiek S and Castillo JJ:
Diagnosis and management of Waldenstrom's macroglobulinemia.
Hematol Oncol. 43 (Suppl 2)(e70071)2025.PubMed/NCBI View Article : Google Scholar
|
|
4
|
McMaster ML: The epidemiology of
Waldenström macroglobulinemia. Semin Hematol. 60:65–72.
2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zorlu T, Kayer MA, Okumus N, Ulaş T, Dal
MS and Altuntas F: . Challenges, difficulties, and delayed
diagnosis of multiple myeloma. Diagnostics (Basel).
15(1708)2025.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kozub A, Nasiek A, Bohun N, Bednarczyk M,
Sędek Ł and Grosicki S: A rare combination: Cold agglutinin disease
followed by Waldenström macroglobulinemia-A case of early treatment
response. Diagnostics (Basel). 15(2654)2025.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Berentsen S: Diagnosis and management of
cold agglutinin disease. Hematology Am Soc Hematol Educ Program.
2025:295–304. 2025.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Scarcella S and Tuccari G, Pizzimenti C,
Scarcella SC, Martini M, Granata F, Germanò A, Ieni A and Tuccari
G: Morphological, immunophenotypic and neuroradiological
characteristics of primitive B-large cell diffuse lymphoma of the
central nervous system: A retrospective cohort analysis. Oncol
Lett. 30(433)2025.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hobbs M, Fonder A and Hwa YL: Waldenström
macroglobulinemia: Clinical presentation, diagnosis, and
management. J Adv Pract Oncol. 11:381–389. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Puig N, Ocio EM, Jiménez C, Paiva B,
Miguel JFS and García-Sanz R: Waldenström's macroglobulinemia
immunophenotype. In: Waldenström's Macroglobulinemia. Leblond V,
Treon S and Dimoploulos M (eds). Springer International Publishing,
Cham, pp21-34, 2017.
|
|
11
|
Trojani A, Beghini A, Bossi LE, Stefanucci
MR, Palumbo C, Greco A, Frustaci A, Di Camillo B and Cairoli R:
Mutational landscape of bone marrow CD19 and CD138 cells in
Waldenström macroglobulinemia (WM) and IgM monoclonal gammopathy of
undetermined significance (IgM MGUS). Cancer Med.
13(e70525)2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kumar SK, Callander NS, Adekola K,
Anderson LD Jr, Baljevic M, Baz R, Campagnaro E, Castillo JJ,
Costello C, D'Angelo C, et al: Waldenström
macroglobulinemia/lymphoplasmacytic lymphoma, version 2.2024, NCCN
clinical practice guidelines in oncology. J Natl Compr Canc Netw.
22(e240001)2024.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Azoulay D, Tapuchi T, Ronen O, Akria L,
Cohen HI, Surio C, Chepa SR, Eshel E, Zarfati M, Stemer G and
Horowitz NA: Flow-cytometry assessment of DNA content and
immunophenotyping of immune-cells in lymph-node-specimens as a
potential diagnostic signature of aggressiveness in B-non-hodgkin
lymphomas. Ann Hematol. 103:4203–4210. 2024.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Avet-Loiseau H, Garand R, Lodé L,
Harousseau JL and Bataille R: Intergroupe Francophone du Myélome.
Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and
nonsecretory multiple myeloma variants. Blood. 101:1570–1571.
2003.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Craig FE and Foon KA: Flow cytometric
immunophenotyping for hematologic neoplasms. Blood. 111:3941–3967.
2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Varettoni M, Arcaini L, Zibellini S,
Boveri E, Rattotti S, Riboni R, Corso A, Orlandi E, Bonfichi M,
Gotti M, et al: Prevalence and clinical significance of the MYD88
(L265P) somatic mutation in Waldenstrom's macroglobulinemia and
related lymphoid neoplasms. Blood. 121:2522–2528. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Naderi N and Yang DT: Lymphoplasmacytic
lymphoma and Waldenström macroglobulinemia. Arch Pathol Lab Med.
137:580–585. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Berentsen S, D'Sa S, Randen U, Małecka A
and Vos JMI: Cold agglutinin disease: Improved understanding of
pathogenesis helps define targets for therapy. Hemato. 3:574–594.
2022.
|
|
19
|
Berentsen S: New insights in the
pathogenesis and therapy of cold agglutinin-mediated autoimmune
hemolytic anemia. Front Immunol. 11(590)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Brodsky RA: Complement in hemolytic
anemia. Blood. 126:2459–2465. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Berentsen S, Röth A, Randen U, Jilma B and
Tjønnfjord GE: Cold agglutinin disease: Current challenges and
future prospects. J Blood Med. 10:93–103. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Girard LP, Soekojo CY, Ooi M, Poon LM,
Chng WJ and de Mel S: Immunoglobulin M paraproteinaemias. Cancers
(Basel). 12(1688)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yadav DP, Kumar D, Jalal AS, Kumar A,
Singh KU and Shah MA: Morphological diagnosis of hematologic
malignancy using feature fusion-based deep convolutional neural
network. Sci Rep. 13(16988)2023.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ghafoor B, Masthan SS, Hameed M, Akhtar
HH, Khalid A, Ghafoor S, Allah HM, Arshad MM, Iqbal I, Iftikhar A,
et al: Waldenström macroglobulinemia: A review of pathogenesis,
current treatment, and future prospects. Ann Hematol.
103:1859–1876. 2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chehal A, Taher A and Shamseddine A: IgM
myeloma and Waldenstrom's macroglobulinemia: A distinct clinical
feature, histology, immunophenotype, and chromosomal abnormality.
Clin Lab Haematol. 25:187–190. 2003.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Cossarizza A, Chang HD, Radbruch A,
Abrignani S, Addo R, Akdis M, Andrä I, Andreata F, Annunziato F,
Arranz E, et al: Guidelines for the use of flow cytometry and cell
sorting in immunological studies (third edition). Eur J Immunol.
51:2708–3145. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sourdeau E, Boccon-Gibod C, Corneau A,
Costopoulos M, Bravetti C, Armand M, Chapiro E, Nguyen-Khac F, Davi
F, Blanc C, et al: Phenotypic profile of Waldenström
macroglobulinaemia B-cells: Establishment of a diagnosis scoring
system and clinico-biological correlations. J Cell Mol Med.
29(e70620)2025.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Riccardi F, Tangredi C, Dal Bo M and
Toffoli G: Targeted therapy for multiple myeloma: An overview on
CD138-based strategies. Front Oncol. 14(1370854)2024.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bagratuni T, Aktypi F, Theologi O, Sakkou
M, Verrou KM, Mavrianou-Koutsoukou N, Patseas D, Liacos C, Skourti
S, Papadimou A, et al: Single-cell analysis of
MYD88L265P and MYD88WT Waldenström
macroglobulinemia patients. Hemasphere. 8(e27)2024.PubMed/NCBI View
Article : Google Scholar
|
|
30
|
Yu X, Li W, Deng Q, Li L, Hsi ED, Young
KH, Zhang M and Li Y: MYD88 L265P mutation in lymphoid
malignancies. Cancer Res. 78:2457–2462. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Miura D, Narita K, Kuzume A, Tabata R,
Terao T, Tsushima T, Kobayashi H, Abe Y, Kitadate A, Takeuchi M, et
al: Clinical and prognostic impact of (11;14)(Q13;Q32)
translocation on patients with multiple myeloma. Blood.
134(5507)2019.
|
|
32
|
Fujishima T, Kobayashi T, Kobayashi I,
Kitadate A, Kameoka Y and Takahashi N: Successful salvage therapy
with pomalidomide, cyclophosphamide, and dexamethasone for IgM
myeloma with t (11;14) and Dim CD38 expression refractory to
daratumumab, lenalidomide, and dexamethasone. Intern Med.
64:3020–3026. 2025.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Grunenberg A and Buske C: How to manage
waldenström's macroglobulinemia in 2024. Cancer Treat Rev.
125(102715)2024.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Weaver A, Rubinstein S and Cornell RF:
Hyperviscosity syndrome in paraprotein secreting conditions
including waldenstrom macroglobulinemia. Front Oncol.
10(815)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Warner D, Duncan H, Gudsoorkar P and Anand
M: Indications and complications associated with centrifuge-based
therapeutic plasma exchange-a retrospective review. BMC Nephrol.
26(87)2025.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Treon SP, Ioakimidis L, Soumerai JD,
Patterson CJ, Sheehy P, Nelson M, Willen M, Matous J, Mattern J II,
Diener JG, et al: Primary therapy of Waldenström macroglobulinemia
with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial
05-180. J Clin Oncol. 27:3830–3835. 2009.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu Z, Jiang S, Gu J, Liu H, Song G and
Cao X: Bortezomib-based chemotherapy for patients with Waldenström
macroglobulinemia: A single-center experience. Ann Hematol.
102:167–174. 2023.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Leblond V, Morel P, Dilhuidy MS, Leleu X,
Soussain C, Leprête S, Dreyfus B, Dartigeas C, Mahé B, Anglaret B,
et al: A phase II Bayesian sequential clinical trial in advanced
Waldenström macroglobulinemia patients treated with bortezomib:
Interest of addition of dexamethasone. Leuk Lymphoma. 58:2615–2623.
2017.PubMed/NCBI View Article : Google Scholar
|