Introduction
E-cadherin (CDH1) is a well-known suppressor of
invasion/metastasis and an important Ca2+-dependent
adhesion molecule that mediates cell-cell contact and is important
for tissue morphogenesis and cell polarity (1). Owing to its critical function in
intercellular adhesion, E-cadherin has been assumed to act as a
tumor suppressor negatively regulating several critical steps of
invasion and metastasis (2).
E-cadherin expression is frequently suppressed or reduced in
carcinoma tissues of the breast and liver and in many carcinoma
cell lines derived from the colon, stomach and prostate (3). The loss of E-cadherin function
induced by promoter methylation was associated with the metastasis
and invasion of tumors. Studies using animal models and human
hepatocellular carcinoma (HCC) tissues have shown that
hypermethylation is associated with decreased E-cadherin expression
but also with microvascular invasion and recurrence of HCC
(4,5). Transcriptional or
post-transcriptional down-regulation may be the mechanism of
underexpression of E-cadherin in HCC (6). The decrease or loss of E-cadherin
expression is observed in HCC as well, particularly in poorly
differentiated cancers (7,8). E-cadherin plays a role in cancer
progression, and its therapeutic restoration as a strategy to
suppress metastasis has recently been considered (9). The presence of the HBx protein, which
is one of the crucial factors in HCC, was found to be involved in
this pathway and may be associated with the hypermethylation of the
E-cadherin promoter (10).
Over the past few years, many epigenetic drugs have
been discovered and found to effectively reverse DNA methylation
and histone modification aberrations that occur in cancer (11). 5-Azacytidine (5′-aza) and
5-aza-2′-deoxycytidine lead to the inhibition of DNA methylation
and induce gene expression via the blocking of DNA
methyltransferases (DNMTs). Trichostatin A (TSA), one of the
effective HDAC inhibitors, re-establishes normal histone
acetylation patterns and reactivates silenced tumor suppressor
genes. These discoveries have led to the possibility of ‘epigenetic
therapy’ as a treatment option, and epigenetic agents are defined
as a legitimate set of targets for therapeutic approaches to
cancer. In the present study, we investigated
E-cadherin-up-regulating drugs, proposing a schema for restoring
E-cadherin by targeting its epigenetic mechanism.
Materials and methods
Cell culture and 5′-aza/TSA
treatment
The human HCC cell line SMMC-7721 and human
hepatocellular pericarcinoma cell line QSG-7701 (Cell Bank
Shanghai, P.R. China) were maintained by serial passage in
RPMI-1640 (Gibco, USA) containing 10% heat-inactivated newborn
bovine serum, and incubated at 37°C in an atmosphere containing 5%
CO2. Cells were cultured in medium containing 120 ng/ml
of TSA (Sigma, USA) or DMSO for 24 h. For 5′-aza (Sigma) treatment,
cells were plated and treated with 0, 10, 50 and 100 μmol/l 5′-aza
for up to 2 days.
Transfection of DNMT1 siRNA, DNMT3A siRNA
and DNMT3B siRNA into the HCC cell line SMMC-7721
SMMC-7721 cells were transfected with DNMT1 siRNA
(pMT1), DNMT3A siRNA (pMT3A) and DNMT3B siRNA (pMT3B) constructs,
and their scramble sequences as control, respectively, using
Transfectamine™ 2000 transfection reagent (Invitrogen, USA) as in
our previous studies (12,13). Cells were grown and selectively
cultured in 0.4 mg/ml Geneticin (Life Technologies, USA) for 2
months after the initial transfection. SMMC-7721 cells transfected
with pMT1, pMT3A and pMT3B were labeled as 7721-pMT1, 7721-pMT3A
and 7721-pMT3B; those transfected with DNMT scramble sequence were
labeled as 7721-sMT1, 7721-sMT3A and 7721-sMT3B.
Semi-quantitative reverse
transcription-PCR (RT-PCR)-detected expression of genes
The expression of the tumor suppressor gene
E-cadherin and of DNMTs was analyzed by semi-quantitative RT-PCR.
Total RNA was purified with TRIzol (Invitrogen). First-strand
complementary DNA (cDNA) was synthesized from 2 μg of total RNA
using Oligo(dT)18 primer and M-MLV reverse transcriptase
(Invitrogen). β-actin was used as an internal control. Each PCR was
repeated with at least three different cDNA preparations and three
independent PCRs for each cDNA with β-actin co-amplification. The
primer sequence of each gene and the PCR conditions are listed in
Table I.
 | Table I.Primers and annealing temperature of
genes analyzed by RT-PCR. |
Table I.
Primers and annealing temperature of
genes analyzed by RT-PCR.
| Gene | Primers (5′-3′) | Temperature (°C) | Amplicon size
(bp) |
|---|
|
E-cadherin | F:
GGTGGGTGACTACAAAATCAATCT | 58 | 310 |
| R:
TTCTCCGCCTCCTTCTTCATCATA | | |
| DNMT1 | F:
CCGAGTTGGTGATGGTGTGTAC | 61 | 324 |
| R:
AGGTTGATGTCTGCGTGGTAGC | | |
| DNMT3A | F:
TATTGATGAGCGCACAAGAGAGC | 65 | 110 |
| R:
GGGTGTTCCAGGGTAACATTGAG | | |
| DNMT3B | F:
GACTTGGTGATTGGCGGAA | 64 | 270 |
| R:
GGCCCTGTGAGCAGCAGA | | |
| HBx | F:
TTCTTCGTCTGCCGTTCC | 54 | 201 |
| R:
TCGGTCGTTGACATTGCT | | |
| ACTB | F:
AAAGACCTGTACGCCAACAC | 61 | 220 |
| R:
GTCATACTCCTGCTTGCTGAT | | |
Antibody and Western blotting
The protein concentration of each extract was
quantitated using the BCA assay (Pierce, USA). Total protein (2–40
μl) was electrophoresed on 7–15% SDS-polyacrylamide gel and
transferred to polyvinylidene fluoride membranes (PVDF; Amersham)
electrophoretically. Western blotting was performed with the mouse
monoclonal anti-E-cadherin or the mouse monoclonal anti-β-actin
(Sigma) antibodies and detected by Super Signal chemiluminescence
substrate (Pierce). β-actin protein levels were used as a control
for equal protein loading.
Methyl-specific PCR (MSP) for promoters
of E-cadherin
Genomic DNA was obtained from cell lines and
modified with sodium bisulfite as described previously (14). Sodium bisulfite-treated genomic DNA
from 7721-sMT1 and 7721-pMT1 were specifically amplified by
methylated and unmethylated primers of E-cadherin as reported
previously (15).
Colony formation assay
Cells (1×103) were evenly plated onto
6-well plates in medium containing 10% FBS and incubated at 37°C in
5% CO2. After 14 days of incubation, colony growth on
the plates was assayed by the visual counting of the colonies.
Cells were then fixed in methanol and stained using crystal violet
solution to evaluate foci formation. Experiments were performed in
triplicate during two independent repetitions.
Transfection with HBx construct
Cells were transfected with 4 μg of the
pcDNA4/TO-HBx construct (a gift from Professor X.Y. Guan,
University of Hong Kong) and the control pcDNA4/TO using
Lipofectamine™ 2000 transfection reagent (Invitrogen) for 36 h.
Results
Treatment with 5′-aza/TSA up-regulates
E-cadherin expression
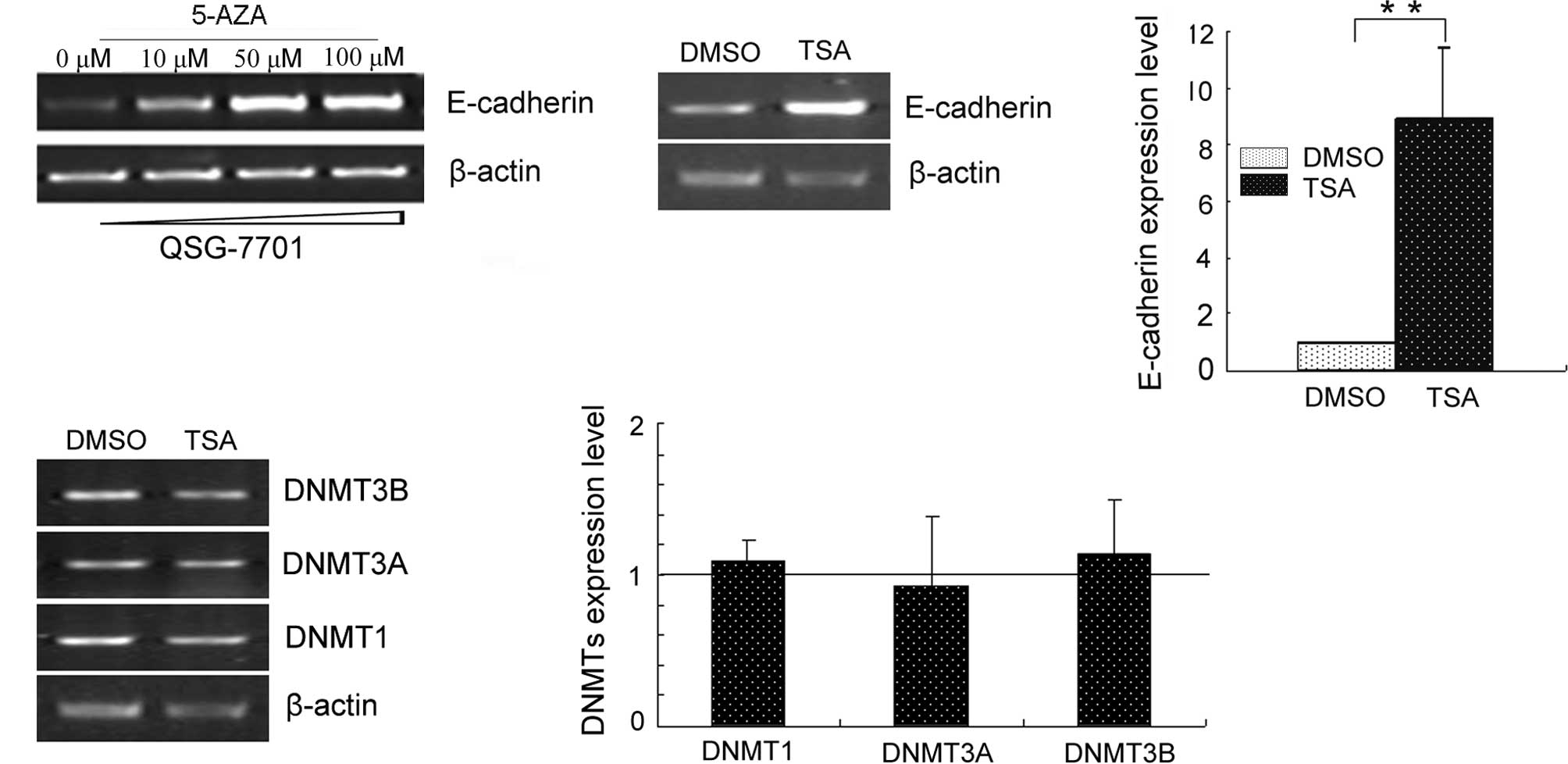
After cells were treated with 5′-aza or TSA,
semi-quantitative RT-PCR was performed on E-cadherin expression
(Fig. 1). Both 5′-aza and TSA
treatments up-regulated E-cadherin expression. 5′-aza restored
E-cadherin in a dose-dependent manner. TSA-regulated E-cadherin
expression did not occur through DNMTs.
Depletion of DNMT1 induced E-cadherin
expression via demethylation of the promoter
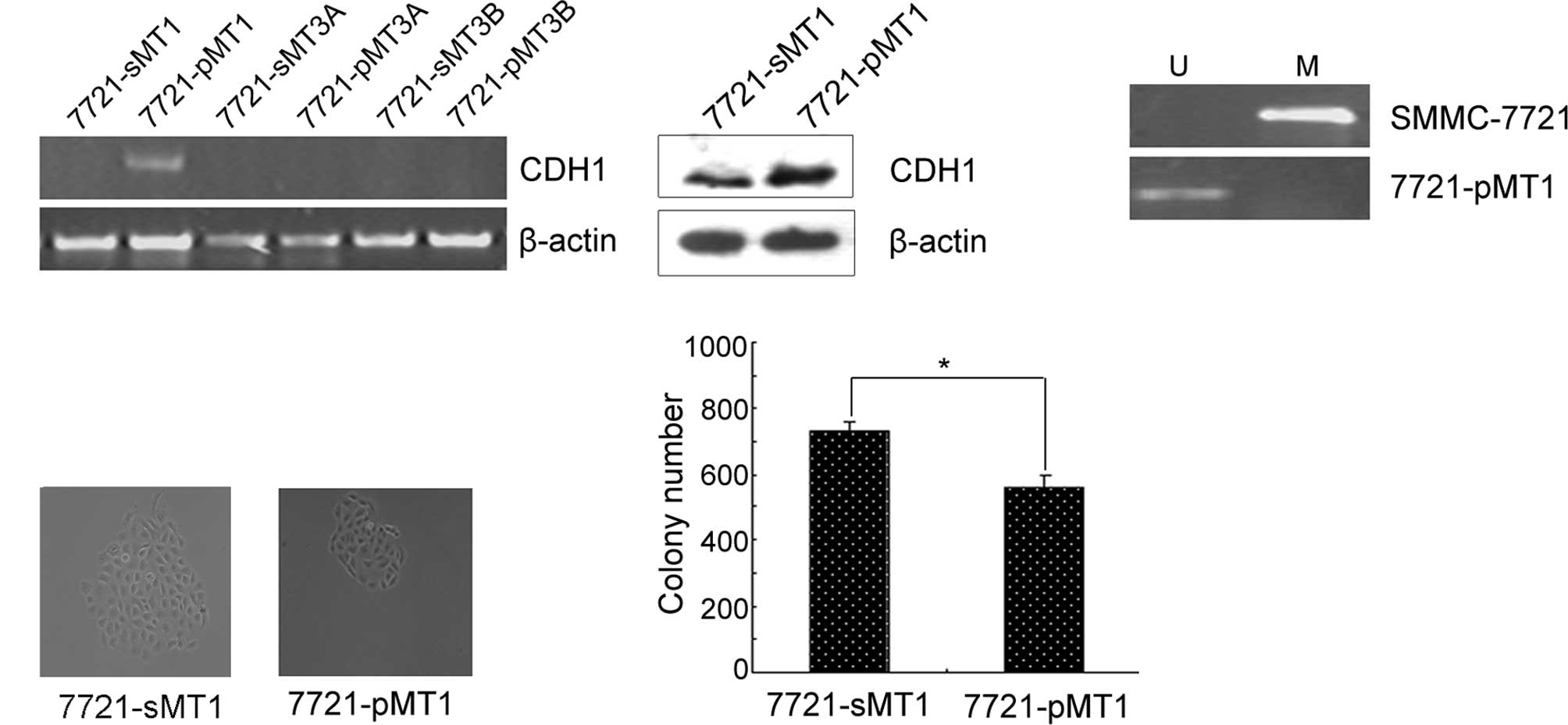
In order to determine which DNMTs play a major role
in reducing the expression of E-cadherin, we detected the
expression of E-cadherin in 7721-pMT1, 7721-pMT3A and 7721-pMT3B
cells. RT-PCR results showed that the knockdown of DNMT1 restored
E-cadherin expression, whereas the knockdown of DNMT3A or DNMT3B
did not (Fig. 2A). In
DNMT1-depleted HCC cells, E-cadherin expression was upregulated at
the protein level and coincided with the transcriptional level.
These results indicated that the E-cadherin gene may be one of the
direct targets of DNMT1. We next investigated whether the
up-regulated expression of E-cadherin induced by DNMT1 RNAi would
be reflected in the promoter methylation status of the genes.
Therefore, we determined the methylation status of the promoter
using MSP as shown in Fig. 2B. The
results showed that the restoration of E-cadherin was significantly
associated with its promoter demethylation in the 7721-pMT1 cell
line. Subsequently, colony formation assays were performed using
the 7721-pMT1 and control cell lines. Compared to the random
control, siRNA-treated HCC cells with knocked down DNMT1 expression
exhibited a significantly decreased colony size in the colony
formation assays (Fig. 2C).
HBx leads to the promoter
hypermethylation of the E-cadherin gene by activating DNA
methyltransferase-1
Previous immunohistochemical studies of E-cadherin
expression in HBV-related HCC have demonstrated the significant
down-regulation of E-cadherin expression in tumor tissues compared
with adjacent non-tumor tissues (16). Although the pathogenesis of
HBV-related HCC has not been fully described, evidence suggests
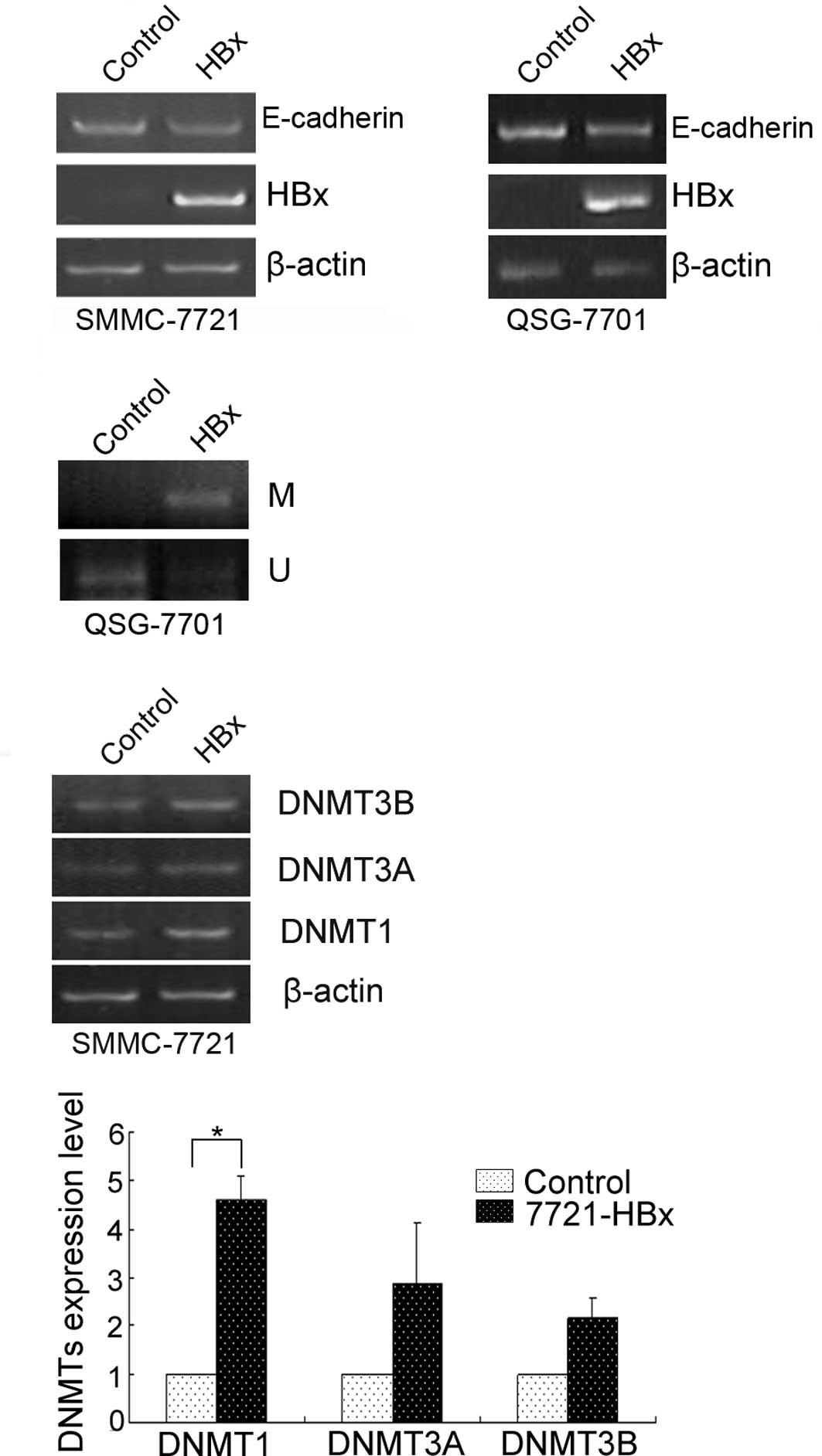
that HBx plays a crucial role in the pathogenesis of HCC (17). Therefore, we first investigated
whether HBx represses E-cadherin expression in cultured human liver
cells. For this purpose, we transfected the transiently
pcDNA4/TO-HBx construct into QSG-7701 and SMMC-7721 cells. As a
result, the E-cadherin mRNA level was reduced by HBx (Fig. 3A), suggesting that HBx represses
E-cadherin expression. The repression of E-cadherin by HBx was
significantly associated with its promoter methylation (Fig. 3B) and increased DNMT1 (Fig. 3C).
Discussion
It has been suggested that genetic alterations such
as loss of heterozygosity and mutations in tumor suppressor genes
accumulate during multistep hepatocarcinogenesis (18,19).
Recently, epigenetic alterations including histone deacetylation
and DNA methylation in promoter areas were also hypothesized to
play crucial roles in the development of HCC. DNA methylation
inhibitors including 5′-aza induce gene expression. 5′-aza was the
first epigenetic drug proposed for use in cancer therapeutics
(20). TSA alone or in combination
with 5′-aza is capable of reactivating the transcription of tumor
suppressor genes that are silenced by methylation-mediated
mechanisms in human cancer cells (21,22).
A number of genes involved in cell cycle- or apoptosis-regulation
were also up-regulated in hepatoma cell lines, as previously
reported (23).
Hypermethylation of CpG regions of the E-cadherin
promoter represents the most common cause for its inactivation and
has been observed in many malignancies, including HCC (24–27).
Reactivation of E-cadherin, proposed as a target of epigenetic
therapy for tumors (28), may be
effective in HCC. In the present study, we investigated for the
first time the epigenetic activation of E-cadherin by treatment
with 5′-aza in an HCC cell line, and found it to be dependent on
the administered dose of 5′-aza. Several studies have suggested
that 5′-aza restores the expression of silenced genes by selective
degradation or the partial influence of DNMT1 (30,31).
In our study, we found that the depletion of DNMT1 restored
E-cadherin gene expression by demethylating the promoter and
suppressed HCC cell colony formation.
As HBV is the main factor leading to HCC in Chinese
populations (29), HBx, an
important gene associated with HBV, was transfected into HCC cells
to evaluate the potential cause of inactivated E-cadherin in HCC
samples from Chinese patients. It was observed that some of the
tumor-associated genes, including IGFBP3, were epigenetically
silenced in HCC cell lines infected with the recombinant HBx
(32,33). This preliminary observation led to
further in vivo and in vitro analyses of this
characteristic abnormality of HBV-associated HCC. Few studies have
focused on the mechanisms of the promoter methylation of host genes
in association with HBV infection. Here, we showed that HBx
suppressed expression of the E-cadherin gene by activating DNMT1.
Our study regarding the epigenetic modulation of E-cadherin by HBx
may suggest a mechanism for the epigenetic silencing of tumor
suppressor genes in HBV-related HCC.
The findings presented in the present study
demonstrate that diverse epigenetic agents restore E-cadherin
expression. In addition, results obtained from studies involving
HBx-transfected HCC cell lines suggest that the inhibition of DNMT1
may be considered a strategy by which silenced E-cadherin in
HBV-related HCC may be inactivated.
Acknowledgements
The present study was supported by the
National Natural Science Foundation of China (nos. 30470950 and
30971605). We thank Dr Wu Dianqing for providing the
pSUPER-EGFP.
References
|
1.
|
Takeichi M: Cadherin cell adhesion
receptors as a morphogenetic regulator. Science. 251:1451–1455.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Hirohashi S and Kanai Y: Cell adhesion
system and human cancer morphogenesis. Cancer Sci. 94:575–581.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Momparler RL and Bovenzi V: DNA
methylation and cancer. J Cell Physiol. 183:145–154. 2000.
View Article : Google Scholar
|
|
4.
|
Hazan RB, Qiao R, Keren R, Badano I and
Suyama K: Cadherin switch in tumor progression. Ann NY Acad Sci.
1014:155–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Calvisi DF, Ladu S, Conner EA, Factor VM
and Thorgeirsson SS: Disregulation of E-cadherin in transgenic
mouse models of liver cancer. Lab Invest. 84:1137–1147. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Huang GT, Lee HS, Chen CH, Sheu JC, Chiou
LL and Chen DS: Correlation of E-cadherin expression and recurrence
of hepatocellular carcinoma. Hepatogastroenterology. 46:1923–1927.
1999.PubMed/NCBI
|
|
7.
|
Wei Y, van Nhieu JT, Prigent S,
Srivatanakul P, Tiollais P and Buendia MA: Altered expression of
E-cadherin in hepatocellular carcinoma: correlations with genetic
alterations, beta-catenin expression and clinical features.
Hepatology. 36:692–701. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Ihara A, Koizumi H, Hashizume R and
Uchikoshi T: Expression of epithelial cadherin and alpha- and
beta-catenins in nontumoral livers and hepatocellular carcinoma.
Hepatology. 23:1441–1447. 1996.PubMed/NCBI
|
|
9.
|
Howard EW, Camm KD, Wong YC and Wang XH:
E-cadherin upregulation as a therapeutic goal in cancer treatment.
Mini Rev Med Chem. 8:496–518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Liu J, Lian Z, Han S, Waye MM, Wang H, Wu
MC, Wu K, Ding J, Arbuthnot P, Kew M, Fan D and Feitelson MA:
Downregulation of E-cadherin by hepatitis B virus X antigen in
hepatocellullar carcinoma. Oncogene. 25:1008–1017. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Yoo CB and Jones PA: Epigenetic therapy of
cancer: past, present and future. Nat Rev Drug Discov. 5:37–50.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Fan H, Zhao Z, Quan Y, Xu J, Zhang J and
Xie W: DNA methyltransferase 1 knockdown induces silenced CDH1 gene
reexpression by demethylation of methylated CpG in hepatocellular
carcinoma cell line SMMC-7721. Eur J Gastroenterol Hepatol.
19:952–961. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Xu J, Fan H, Zhao ZJ, Zhang JQ and Xie W:
Identification of potential genes regulated by DNA
methyltransferase 3B in a hepatocellular carcinoma cell line by RNA
interference and microarray analysis. Yi Chuan Xue Bao.
32:1115–1127. 2005.PubMed/NCBI
|
|
14.
|
Herman JG, Graff JR, Myohanen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Zhang H, Xiao W, Liang H, Fang D, Yang S
and Luo Y: Demethylation in the promoter area by the antisense of
human DNA MTase gene. Zhonghua Zhong Liu Za Zhi. 24:444–447.
2002.PubMed/NCBI
|
|
16.
|
Kanai Y, Ushijima S, Hui AM, Ochiai A,
Tsuda H, Sakamoto M and Hirohashi S: The E-cadherin gene is
silenced by CpG methylation in human hepatocellular carcinomas. Int
J Cancer. 71:355–359. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Feitelson M: Hepatitis B virus infection
and primary hepatocellular carcinoma. Clin Microbiol Rev.
5:275–301. 1992.PubMed/NCBI
|
|
18.
|
Murakami Y, Hayashi K and Sekiya T:
Aberration of the tumor suppressor p53 and retinoblastoma in human
hepatocellular carcinomas. Cancer Res. 51:5520–5525.
1991.PubMed/NCBI
|
|
19.
|
Liao C, Zhao M, Song H, Uchida K, Yokoyama
KK and Li T: Identification of the gene for a novel liver-related
putative tumor suppressor at a high-frequency loss of
heterozygosity region of chromosome 8p23 in human hepatocellular
carcinoma. Hepatology. 32:721–727. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Constantinides PG, Jones PA and Gevers W:
Functional striated muscle cells from non-myoblast precursors
following 5-azacytidine treatment. Nature. 267:364–366. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yoshida M, Horinouchi S and Beppu T:
Trichostatin A and trapoxin: novel chemical probes for the role of
histone acetylation in chromatin structure and function. Bioessays.
17:423–430. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Zhu WG and Otterson GA: The interaction of
histone deacetylase inhibitors and DNA methyltransferase inhibitors
in the treatment of human cancer cells. Curr Med Chem Anti-Cancer
Agents. 3:187–199. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chiba T, Yokosuka O, Arai M, Tada M, Fukai
K, Imazeki F, Kato M, Seki N and Saisho H: Identification of genes
up-regulated by histone deacetylase inhibition with cDNA microarray
and exploration of epigenetic alterations on hepatoma cells. J
Hepatol. 41:436–445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Berx G, Cleton-Jansen AM, Strumane K, de
Leeuw WJ, Nollet F, van Roy F and Cornelisse C: E-cadherin is
inactivated in a majority of invasive human lobular breast cancers
by truncation mutations throughout its extracellular domain.
Oncogene. 13:1919–1925. 1996.
|
|
25.
|
Melki JR, Vincent PC, Brown RD and Clark
SJ: Hypermethylation of E-cadherin in leukemia. Blood.
95:3208–3213. 2000.PubMed/NCBI
|
|
26.
|
Tamura G, Yin J, Wang S, et al: E-cadherin
gene promoter hypermethylation in primary human gastric carcinomas.
J Natl Cancer Inst. 92:569–573. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Iwata N, Yamamoto H, Sasaki S, Itoh F,
Suzuki H, Kikuchi T, Kaneto H, Iku S, Ozeki I, Karino Y, Satoh T,
Toyota J, Satoh M, Endo T and Imai K: Frequent hypermethylation of
CpG islands and loss of expression of the 14-3-3 sigma gene in
human hepatocellular carcinoma. Oncogene. 19:5298–5302. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Nam JS, Ino Y, Kanai Y, Sakamoto M and
Hirohashi S: 5-aza-2′-deoxycytidine restores the E-cadherin system
in E-cadherin-silenced cancer cells and reduces cancer metastasis.
Clin Exp Metastasis. 21:49–56. 2004.
|
|
29.
|
Yu MC, Yuan JM, Govindarajan S and Ross
RK: Epidemiology of hepatocellular carcinoma. Can J Gastroenterol.
14:703–709. 2000.PubMed/NCBI
|
|
30.
|
Ghoshal K, Datta J, Majumder S, Bai S,
Kutay H, Motiwala T and Jacob ST: 5-Aza-deoxycytidine induces
selective degradation of DNA methyltransferase 1 by a proteasomal
pathway that requires the KEN box, bromo-adjacent homology domain
and nuclear localization signal. Mol Cell Biol. 25:4727–4741. 2005.
View Article : Google Scholar
|
|
31.
|
Palii SS, van Emburgh BO, Sankpal UT,
Brown KD and Robertson KD: DNA methylation inhibitor
5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage
that is distinctly influenced by DNA methyltransferases 1 and 3B.
Mol Cell Biol. 28:752–771. 2008.
|
|
32.
|
Park IY, Sohn BH, Yu E, Suh DJ, Chung YH,
Lee JH, Surzycki SJ and Lee YI: Aberrant epigenetic modifications
in hepatocarcinogenesis induced by hepatitis B virus X protein.
Gastroenterology. 132:1476–1494. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Zheng DL, Zhang L, Cheng N, Xu X, Deng Q,
Teng XM, Wang KS, Zhang X, Huang J and Han ZG: Epigenetic
modification induced by hepatitis B virus X protein via interaction
with de novo DNA methyltransferase DNMT3A. J Hepatol. 50:377–387.
2009. View Article : Google Scholar : PubMed/NCBI
|