Introduction
RNA polymerase II binds to the promoter of a gene
and it assembles the transcription mechanism by gathering general
transcription factors (GTFs), creating the pre-initiation complex
to initiate transcription. Transcription is regulated by
cis-regulatory elements, including the promoter. Distal
elements can exert a positive effect on transcription (termed
enhancers) or a negative effect (termed silencers) (1-3).
Transcription factors (TFs) are proteins that bind to such
regulatory regions of genes recognising multiple specific DNA
sequence motifs, termed TF binding sites (TFBSs) (4). There are >1,600 human TFs
catalogued (5), controlling
processes of cell type specification, developmental patterning
(6), as well as specific biological
pathways (7). The binding affinity
of a TF is dependent on its DNA-binding domain and the specific
sequence of nucleotides which is targeted (8). Potential binding sites are predicted
based on matches to a consensus sequence, often allowing for
certain mismatches. The methods originally used for searching
genome sequences to predict TFBSs were based on position weight
matrices (PWMs), also known as position-specific scoring matrices
(PSSMs) (9), derived from the
multiple sequence alignment of experimentally verified DNA target
motifs. A weight matrix is a two dimensional array of values that
represent the score for finding each of the four nucleotides at
each position in the DNA sequence (10). The main databases for the collection
of PWMs are TRANSFAC (11) and
JASPAR (12). The majority of
algorithms used for searching for PWMs in genomic sequences are
MATCH (13) and FIMO (14). To simulate TFBS motif intricacies,
more complex models have been proposed in recent years (15-18)
and, in particular, hidden Markov models (HMMs) (19) have been applied successfully for
TFBS prediction (20,21). To account further for TFBS length
variability and interactions between nucleotides, HMM-based TF
flexible models (TFFMs) (22) were
developed, by consulting already existing JASPAR models. They can
be graphically represented with a sequence logo, in a manner
similar to PWMs (23).
Comparing the relative order of gene orthologs in
the human and mouse genomes has revealed that a long-range sequence
organisation has been preserved to a large extent from their last
common ancestor (24).
Approximately 80% of the common genes can be matched between the
two organisms with the addition of a high rate of conservation of
nucleotides (25). Genes that share
close evolutionary associations are likely to possess similar
functions and, likewise, functionally similar cis-regulatory
elements have been shown to be conserved between species (1,26).
Therefore, it can be considered that the majority of functional
TFBS sequences are conserved between human and mouse.
DNase I is an endonuclease that cleaves DNA adjacent
to pyrimidine nucleotides. In order for DNase I to cleave a DNA
strand, it needs to be able to access it. Within cells, TFs
displace histone octamers, unwinding the tightly packed chromatin
structure. Through the technique of DNase I hypersensitivity (DH)
assays (27) and its evolutions
(28), DH sites, which denote the
open and accessible areas that DNase I can operate on, are
discovered. Analysing the whole genome accessibility landscape
using DNase I, yields DH maps that denote parts of the genome that
are probably transcriptionally active. From these data, it is
possible to discover potential cell-type specific TFBSs, as the DH
areas originate from open chromatin regions that contain regulatory
elements of active genes (29-31).
Predicting TFBSs in regulatory sequences of a gene
of interest requires some of the aforementioned computational
tools. The present study introduces TFBSPred, a TFBS prediction
webtool which eliminates false discoveries by using novel TFFM
searches on cell type-specific open chromatin regions, which are
conserved between Homo sapiens and Mus musculus.
Data and methods
Data collection
Using in-house PHP scripts, the present study
downloaded and processed various Homo sapiens and Mus
musculus genomic data, such as gene symbols and annotations
from HGNC (32) and MGI (33), as well as gene stable IDs,
transcript stable IDs and transcription start site (TSS)
information from Ensembl (34)
through BioMart (35). To determine
the open chromatin regions of each biological replicate of each
cell line and/or treatment, BroadPeak (36) DH data (31) for human hg19 and mouse mm9 genomes
were downloaded from the UCSC Genome Browser database (37). The LiftOver tool (38) was used to update the DH data
coordinates to those of the latest versions of the two genomes
(hg38 for human and mm10 for mouse), removing unmapped or duplicate
regions. DH data for each replicate, cover, on average, 3.1 and
3.6% of the human and mouse genomes, respectively (Table SI).
Furthermore, ENCODE (39) common cell type details from UCSC
Genome Browser were collected and parsed. To determine the
conserved sequences between Homo sapiens and Mus
musculus, Ensembl Compara (40)
pairwise alignments in multiple alignment format (MAF) for the
latest versions of the two genomes were downloaded, which were
constructed using the LASTZ (41)
alignment program. When parsing all MAF files, all pairwise
alignments were converted into FASTA format and their genomic
coordinates were extracted. The alignments cover, on average, 32.8
and 35.6% of the human and mouse genomes, respectively (Table SII). All aforementioned data were
stored in a relational database using the MySQL management system
(https://www.mysql.com/) on a Linux Ubuntu 64-bit,
16-core (Intel(R) Xeon(R) CPU E5-2650 v3), 64 GB memory virtual
machine, which is provided by GRNET (https://okeanos-knossos.grnet.gr/).
The TF and TFFM data were procured from the latest
version of the JASPAR database (http://jaspar.genereg.net/). From the TFFMs available,
only the detailed trained ones that belonged to vertebrates were
retained. A total of 462 TFFMs are included.
Web interface
The website is hosted on an Apache2 HTTP server. It
was developed in HTML5, which was validated by HTML validator addon
(https://www.gueury.com/mozilla/).
Bootstrap styling library (https://getbootstrap.com/) was used for the website
design. JavaScript was used to implement the selection checkboxes
and autocompletion fields. All scripts were written in PHP
scripting language.
To perform a TFBS search on TFBSPred, the users are
guided through a number of online steps, using a web wizard. As
input to TFBSPred, the users initially select an organism between
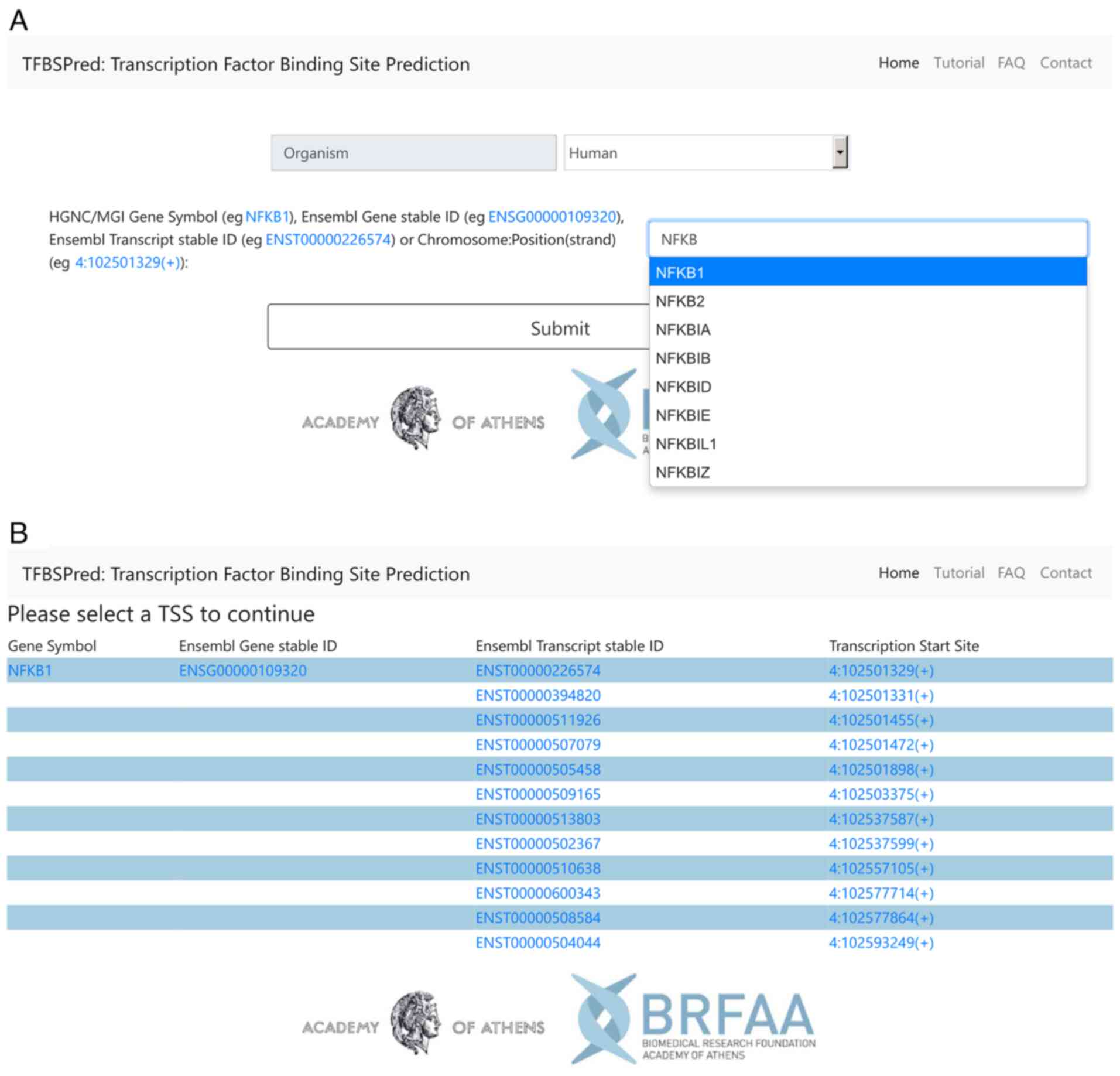
human or mouse on the main page (Fig.
1A) and consequently, they type an autocompleted HGNC or MGI
gene symbol, respectively, or an Ensembl Gene or Transcript stable
ID, or an exact hg38 or mm10 genomic location in the form of
Chromosome: Location(Strand), e.g., 4:102501329(+). In the case a
gene symbol or Ensembl Gene ID is provided, the users are
redirected to the TSS selection page of the gene of interest
(Fig. 1B). The TSS selection page
provides links to the TSSs of the input gene and to related Ensembl
pages. If an Ensembl Transcript ID is used as input, a single link
to a TSS will appear on the TSS selection page. By clicking on one
of the links to a TSS, the users proceed to the cell type filtering
page. The users can directly reach this page from the main page, if
they use an exact genomic coordinate input, to perform a TFBS
search on a region containing a specific genomic location, instead
of a region of a gene promoter.
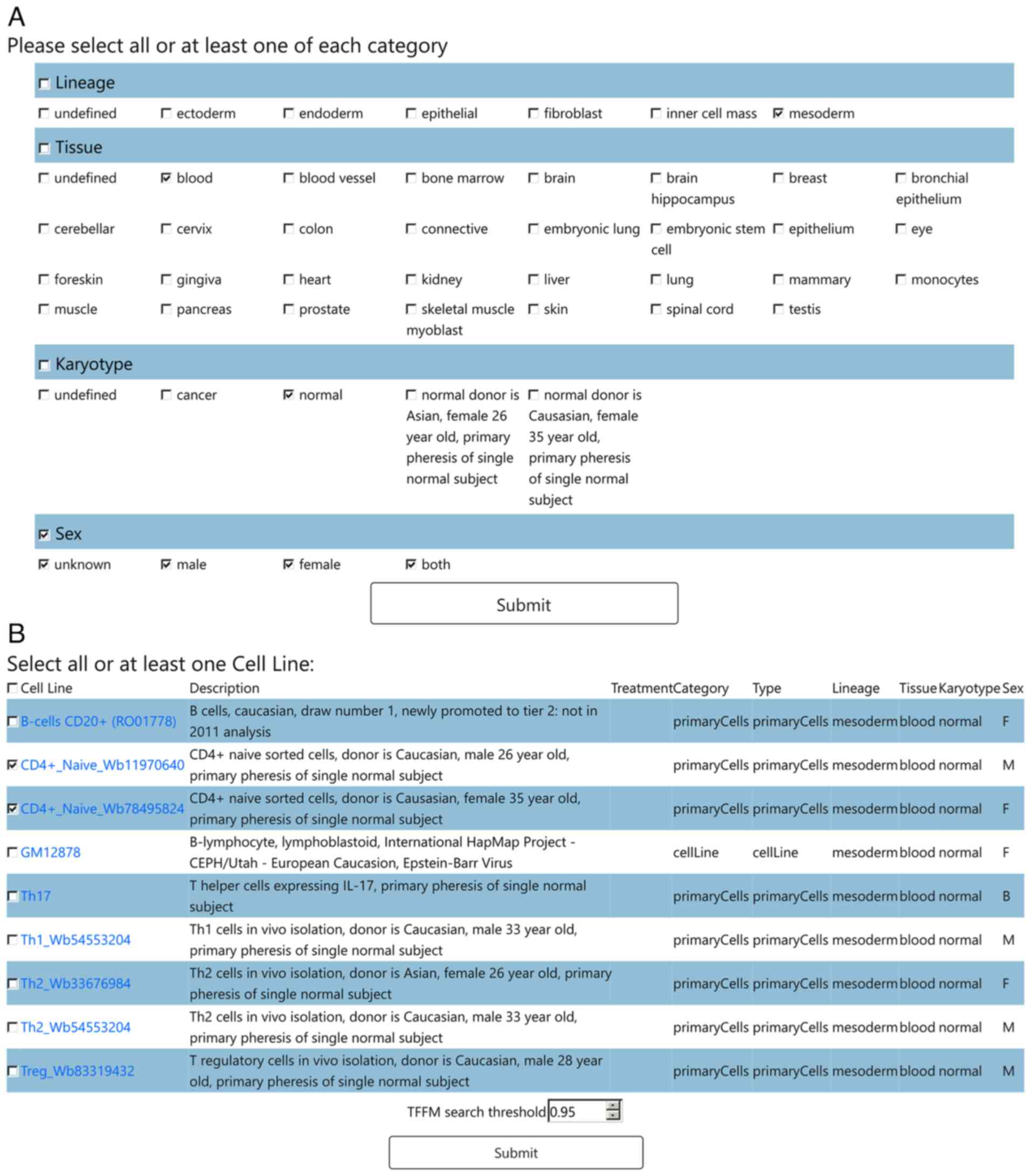
On the cell type filtering page (Fig. 2A), the users filter the cell types
whose open chromatin region will be used for analysis, by checking
at least one of the boxes of each of four categories: Lineage,
Tissue, Sex and Karyotype. By submitting these selections, the
users proceed to the cell line selection page (Fig. 2B), where only filtered cell lines
are displayed, along with relevant details from Encode. The users
check at least one of the boxes of the cell lines. Alternatively,
if the users wish to identify all TFs which bind to the promoter
region of their gene of interest, irrespective of the tissue
context, they need to check all boxes of the cell type filtering
and cell line selection pages. To conduct TFFM searches, a TFFM
threshold must be specified at the bottom of the cell line
selection page. The threshold value ranges from 0.60 to 1, and the
higher the number, the stricter the TFBS search is. By submitting
the selected cell lines and cut-off value, the TFBS prediction page
appears, after a few minutes. A conserved open chromatin region
whose extent depends on the cell lines selected, is depicted as a
pairwise alignment between human and mouse, with the top sequence
belonging to the organism that was initially selected (Fig. 3A). The pairwise alignment follows
the ‘pair’ format (http://emboss.sourceforge.net/docs/themes/AlignFormats.html#pair)
and can also be downloaded in FASTA format, to be used as input for
downstream analyses. The TSS or specified genomic location is
marked with an arrow highlighted red, indicating its orientation of
transcription. The predicted TFBSs are displayed above their
corresponding location in each pairwise alignment with consecutive
arrows indicating their binding orientation. Detailed TFBS
prediction results can be shown, located below (Fig. 3B). The predicted TFs are sorted
alphabetically by their gene symbol, which links to its
corresponding JASPAR entry. Each TF contains one or more discovered
binding sites presented as a human and mouse pairwise alignment in
‘pair’ format, indicating the actual genomic coordinates of each
binding site.
TFFM search
TFBSPred TFBS prediction is based on TFFM searches
which are executed in the webtool backend, after the users have
submitted their data. To this end, the TFFM framework (22), as well as its prerequisites, the
general hidden Markov model (GHMM) (42) and Biopython (43) libraries, were installed.
To define the extent of the open chromatin region of
the genome of the organism of interest, which includes the selected
TSS or specific genomic location, the borders of the open chromatin
region broad peaks of all selected cell lines, which contain the
specified genomic coordinate are retrieved from the database and
merged together using BEDTools (44), defining the borders of a genomic
region which represents the union of all open chromatin regions.
The borders of the conserved genomic region which contains the
specified genomic coordinate are also retrieved. The conserved open
chromatin region is the cross section of the merged open chromatin
region and the conserved region, and its borders are calculated
using BEDTools. From the pairwise alignment of the conserved open
chromatin sequence, the corresponding sequence of the other
organism is determined. Both the human and mouse genomic sequences
are then searched upon with all TFFMs available, using an in-house
python script which outputs the matches above the user designated
cut-off value. TFBSPred parses all search results and only displays
as pairwise alignments the predicted TFBSs, which are conserved
between the human and mouse sequences.
Benchmarking TFBS prediction
webtools
TFBS predictions on various human genes were
performed using TFBSPred and other webtools, mostly using the
default settings. Gene names were submitted to TFBSPred.
FASTA-formatted gene regulatory sequences were submitted to
AliBaba2.1(45), TFBIND (46), SITECON (47), PROMO (48), MATCH (13) and STAMP (49). Both human and their corresponding
mouse sequences were submitted to ConSite (50), FOOTER (51) and rVista 2.0(52). To execute rVista 2.0, the integrated
zPicture (53) alignment program
was used to align the two sequences. Gene names were used as input
and both human and mouse gene promoter sequences were selected,
along with JASPAR Core Matrices with all available TFs, in
LASAGNA-Search 2.0(54). Gene names
were used as input, the exploration function was executed followed
by the visualisation function using the discovered TFs, in ConTra
v3(55). Finally, the motif
features discovered in the regulatory regions of genes in Ensembl
regulatory build (56) were also
identified.
Results
Interferon beta 1 (IFNB1)
enhanceosome
To assess the predictive capabilities of TFBSPred
and other TFBS prediction webtools, the IFNB1 enhanceosome
(57) was used. The enhanceosome is
an ~50 bp enhancer sequence of the IFNB1 gene and requires the
coordinate activation and DNA binding of the TFs ATF2/Jun, IRF3 and
IRF7 and NF-κB (57,58), effectively dividing the enhanceosome
into four positive regulatory domains (PRDI to PRDIV). Τhe exact
binding sites of each of these TFs have been experimentally
confirmed (57,59). PRDM1 has also been shown to bind
specifically to the PRDI domain of IFNB1 enhanceosome, with a
negative effect on transcription (60), while additionally playing a role in
recruiting co-repressor complexes required to silence gene
expression (61). With the default
0.90 TFFM threshold, TFBSPred discovered PRDM1, NF-κB (RELA, RELB
and NFKB1), IRF1, SPIB and TEAD2 TFBSs in the enhanceosome region.
ATF1 was also discovered among other TFs, when the threshold was
decreased to 0.62 (Fig. 4);
however, 26 false-positive TFBSs were also predicted (data not
shown, as the default cut-off value is not 0.62). The ConTra v3
exploration function predicted only two TFs, NF-κB and PRDM1.
AliBaba2.1 discovered five total TFBSs, including IRF8 and ATF2
sites but not NF-κB. LASAGNA-Search 2.0 discovered NF-κB and STAT1,
MATCH discovered NF-κB, IRF1 and IKZF1, and the single sequence
option of ConSite discovered NF-κB, TEAD1, IRF1 and IRF2. The
pairwise alignment option of ConSite only discovered the IRF2 site.
PROMO, TFBIND, rVista2.0 and SITECON discovered a large amount of
TFBSs. PROMO found TFBSs for all verified TFs; however, it
displayed 23 false-positive TFBSs. TFBIND discovered IRF-family
TFs, NF-κB and JUN, with 10 false-positive sites. rVista2.0
discovered NF-κB and IRF-family TFs (including IRF7), also having
10 false-positives, and SITECON only found IRF-family TFs, while
having 29 false-positive TFBSs. Although STAMP predicted less TFBSs
than those of the four previous tools, it only found NF-κB from the
verified TFs, presenting 7 false positive sites. Finally, FOOTER
did not succeed in predicting any TFBSs. Although the majority of
the experimentally verified TFBSs (including NFKB1, NFKB2, IRF3,
IRF7, JUN, ATF7 and PRDM1) were computationally predicted by the
Ensembl Regulatory Build of IFNB1 (ENSR00001147395), 53
false-positive TFBSs were also found (https://www.michalopoulos.net/tfbspred/erb.html).
Nevertheless, none of the putative TFBSs was marked as
‘experimentally verified’ in the Ensembl genome browser (all TFBSs
are coloured in grey), indicating that none of the TFBS hits was
supported by ChIP-Seq peak evidence.
FGA gene promoter
In its basal state, Fibrinogen alpha chain (FGA) is
regulated by HNF1 (-59 to -47 nucleotides from the TSS) and CEBP
(-142 to -134 nucleotides from the TSS) family proteins. In an
acute state, STAT3 additionally binds to the IL-6RE region,
upstream of FGA TSS (62). To
examine the tissue-specificity of TFBSPred, two analyses on FGA
were conducted: In the first, only endodermal normal cell lines
were used, while in the second analysis, only the HepG2
hepatoblastoma cell line was selected. In both cases, the FGA gene
was used as input. In the first analysis, TFBSPred discovered
CEBP-family factors (CEBPE, CEBPA, CEBPD), HNF1 (HNF1A, HNF1B) and
VDR. In the second analysis, a larger conserved open chromatin
region was displayed, due to cancer cells having an abnormal
regulation pattern. Multiple TFs of the STAT (STAT3, STAT4, STAT5A,
STAT5B) and TEAD (TEAD1, TEAD2, TEAD4) families, as well as Bcl6,
were additionally discovered further upstream of the TSS (Fig. 5).
 | Figure 5Conserved open chromatin region of
FGA as calculated by TFBSPred. The grey section of the sequence
denotes the expanded open chromatin region that appears when the
HepG2 cell line is selected. The TSS and an arrowhead showing its
orientation of transcription are highlighted in red. The
experimentally confirmed binding sites are portrayed with dashed
lines, while the TFBSs predicted by the selected webtools with
coloured continuous lines. When a TFBS is depicted above the
nucleotide sequence, the name of the TF corresponding to it is then
located above it. Respectively, if a TFBS is depicted below the
sequence, the name of the TF is then located below it. The name of
the TF is displayed once for multiple adjacent converging TFBS
lines for the same TF. STAT* includes STAT1, STAT3, STAT5A and
STAT5B factors and TEAD* includes TEAD1, TEAD2, TEAD3 and TEAD4
factors. HNF1b (highlighted in black) was predicted by ConTra v3 in
other species, but not in Homo sapiens. TFBS, transcription
factor binding site. |
Four other webtools that performed best for IFNB1
(single sequence ConSite, rVista2.0, PROMO and ConTra v3), were
also used to predict TFBSs for FGA (Fig. 5). The FASTA-formatted human sequence
which corresponds to the conserved acute-state open chromatin area
upstream to the FGA TSS, as calculated by TFBSPred (345 nucleotides
length), was used as input. ConSite predicted only the HNF1 site
with 35 false-positives. While rVista2.0 and PROMO all predicted
the experimentally verified TFs (HNF1, CEBP and STAT3), they also
found 30 and 80 false-positive TFBSs, respectively. ConTra v3
discovered STAT-family TFs and HNF1b, although the latter was not
found on the Homo sapiens sequence, but rather in other
organisms. TFBIND was also tested as a candidate webtool; however,
it was not included since it predicted >400 binding sites
(Table SIII).
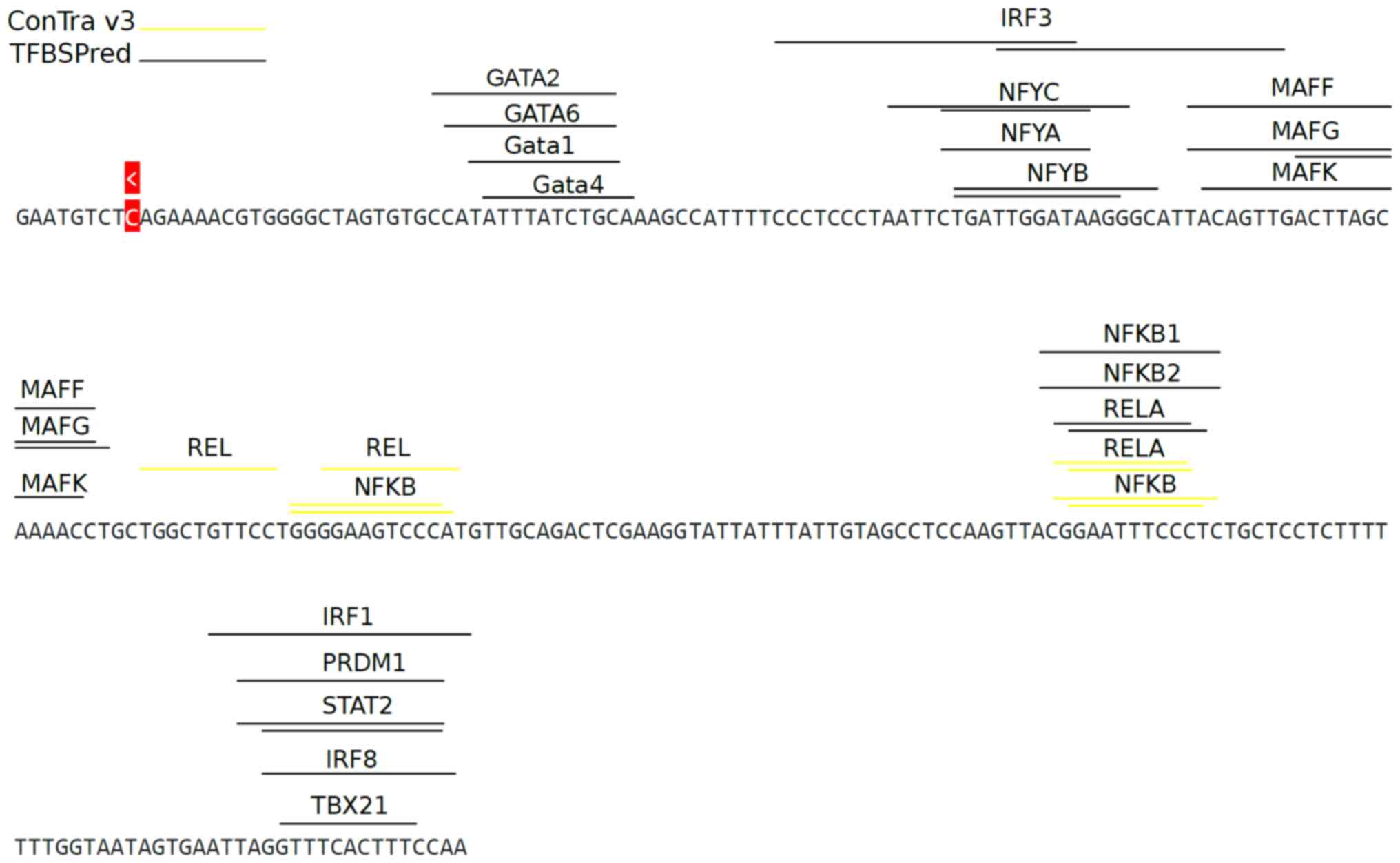
C-X-C motif chemokine ligand 10
(CXCL10) promoter
CXCL10 is a chemokine that is secreted during the
immune response. STAT1, NF-κB, AP1 and heat shock factors bind to
an ~230 bp region upstream from the TSS (63). Additionally, IRF3 binds to CXCL10
promoter during hepatitis C infection (64). The CXCL10 gene was used as input to
TFBSPred along with all the available cell-lines and default TFFM
settings. TFBSPred revealed several factors binding upstream of the
gene promoter, including members of the NF-κB (NFKB1, NFKB2 and
RELA) family, all the factors forming the NFY complex (NFYA, NFYB
and NFYC), as well as IRF1, IRF8, MAFG and STAT2. ConTra v3
exploration analysis discovered only NF-κB family-related TFs
(Fig. 6).
Discussion
The most popular approach for TFBS prediction,
PWM-based search, considers that the nucleotides of each position
exhibit independent participation in the DNA-protein interactions.
ConTra v3(55), a widely used
webtool, employs PWM-based technologies which use JASPAR and
TransFac PWMs to search a given genomic region for potential TFBSs.
Specifically, it uses FIMO for its exploration function and MATCH
for its visualisation function. The majority of other webtools,
such as TFBIND, PROMO and AliBaba2, use their own TFBS prediction
algorithms, all of which are based on PWM profiles. The Ensembl
Regulatory Build is a collection of regulatory features (denoted as
ENSR) across the whole genome. PWMs based on TF motifs which were
discovered through SELEX, were matched upon those regulatory
regions (34), using MOODS
(65), a PWM matching program.
However, in the case of the IFNB1 enhanceosome, the loose
parameters used for the execution of MOODS in Ensembl Regulatory
Build, resulted in low specificity, as >50 pseudo-positive TFBSs
were predicted. TFFMs, as a complete HMM-based approach, were
designed to address the confounding properties of nucleotide
composition, interpositional sequence dependence and variable
lengths observed in the recently emerging extensive experimental
data. They have been shown to perform more effectively than the
majority of PWM-based models (22),
while also being publicly available. TFBSPred is the only web based
TFBS prediction tool which, thus far, employs the HMM-based TFFM
algorithm. In the case of IFNB1, TFBSPred predicted an IRF1 site
which coincided with the experimentally verified IRF3 and IRF7
sites in PRDI (57), as well as
PRDM1, which was also discovered in the same coordinates. In
addition, it should be noted that TFBSPred cannot predict the IRF3
and IRF7 binding sites, as there is no TFFM model for these. The
TFBSPred search is connected to the available TFFM profiles from
JASPAR. As JASPAR is a continuously evolving project, TFBSPred will
incorporate future TFFMs, as well as updates to already existing
models. Finally, ATF2/c-Jun leucine zipper heterodimer, does not
bind tightly, as the c-Jun half-site is not fully complementary to
c-Jun binding and DNA is bent (57). Moreover, the majority of TFFMs for
dimer TFs are created for homodimers, rather than heterodimers.
Thus, model-based prediction algorithms could not predict this
heterodimeric factor. Thus, TFBSPred only predicted ATF1 when the
cut-off was lowered, indicating that it is possible to predict
difficult TFBS cases, when the TFFM threshold is sufficiently
decreased, although this should be used with caution, as it may
introduce false-positives.
To reduce the false discovery rate, ConTra v3 takes
advantage of the evolutionary conservation of regulatory elements
across various related species by expanding the PWM matching on a
multiple genome sequence alignment and subsequently visualising the
results of the TFBS predictions. However, it is up to the users
themselves to evaluate the importance of the conservation of each
predicted TFBS among the multitude of species presented. Other
webtools, such as rVista2.0 and ConSite, require the user to input
two homologous genomic sequences to search for conserved TFBSs by
creating a pairwise alignment where conserved TFBSs are
automatically determined and displayed. Similar to rVista2.0 and
ConSite, TFBSPred searches for conserved TFBSs in a pairwise
alignment. However, as opposed to those tools, TFBSPred uses
high-quality pairwise alignments from Ensembl Compara, which cover
~1/3 of each genome, while identifying homologous regions between
two genomic species is a complex task, which is not easily
performed by the average wet lab experimentalists. Another
difference is that TFBSPred only compares human and mouse
sequences. Although the ability to select between any combination
of species and set multiple parameters to search for TFBSs renders
rVista2.0 and ConSite more versatile, it markedly increases usage
complexity. FOOTER requires the user to input a human and a mouse
or rat genomic sequence, and predicts conserved TFBSs between the
two; however, its default parameters are not adequate for optimal
results. TFBPred requires only one human or mouse genomic sequence
as input, and automatically identifies its homologous region of the
other species: Mus musculus and Homo sapiens are
extensively studied model organisms which belong to the orders of
Rodentia and Primates, respectively. These orders which belong to
the mammalian superorder of Euarchontoglires, split 85-97 million
years ago (66), sharing a high
degree of genomic sequence conservation. Thus, predicted TFBSs
which are conserved between human and mouse are likely true
positives. Searching Compara-aligned human and mouse sequence pairs
and automatically discarding TFBSs which are not highly conserved,
offers the best possible TFBS results, without any further user
participation.
The boundaries of regulatory regions (promoters,
enhancers and insulators, as well as TF motifs in open chromatin
regions) were defined in Ensembl regulatory build, through a
variety of genome-wide experimental data from multiple epigenomic
consortia. Predicted TFBSs which are verified by ChIP-Seq can be
displayed in the Ensembl genome browser tracks with additional
information indicating cell-line/tissue specificity. Although this
is an extensive assortment of TFBSs, in the case of the IFNB1
enhanceosome, experimental verification has a low sensitivity, as
no experimentally verified TFBSs were identified. As opposed to
Ensembl regulatory build, ConTra v3 is unable to define on its own
the extent of regulatory regions and has no cell-line/tissue
specificity. ConTra v3 requires a user-specified region length to
search for TFBSs, with the arbitrary default value being 500 bps.
In addition, the majority of webtools perform TFBS prediction
solely on a FASTA-formatted genomic sequence the user selects. On
the other hand, TFBSPred automatically identifies the boundaries of
cell-line/tissue specific regions of open-chromatin which cover
around 3-4% of each genome, based on DH data of the selected cell
lines, and then searches for conserved TFBSs. Consequently, by
selecting certain tissues and cell types, TFBSPred can discover
potential cell-type specific TFBSs, as DH areas indicate open
chromatin regions that contain regulatory elements of active genes.
To examine the basal state of FGA, non-cancer cell lines were used
for a TFBSPred analysis: TFBSs for HNF1 and CEBP-family factors
were predicted, in accordance with experimentally confirmed sites.
To study the acute state of FGA, only Hep2G cancer cell line was
used as input for a TFBSPred analysis: Not only was a larger
conserved open chromatin region produced, but also STAT family
factors were predicted in this expanded open chromatin sequence of
the acute state, including experimentally confirmed STAT3. This
displays the potential for TFBSPred for tissue-specific
differential TFBS predictions, as an experimentally characterised
TFBS (STAT3) is predicted in the acute state (Hep2G cancer cell
line) and not in the basal state (non-cancer cell lines). R-based
BinDNase (67), also incorporates
DH data to increase PWM accuracy and has demonstrated the validity
of this approach.
ConTra v3 has a long execution period, particularly
if a number of PWMs are selected in a visualization analysis. On
the contrary, one of the main objectives of TFBSPred was the
provision of a rapid and user-friendly interface specifically
catering for wet lab biologists. Throughout the website, user
interaction is minimal, as the majority of the input fields and
selections are provided through TFBSPred database. The TFBSPred web
wizard begins with the selection of the TSS of a gene or a
chromosomal location in either human or mouse, followed by the
filtering and selection of cell types. Finally, the maximum
execution time does not exceed a couple of minutes.
To summarise, TFBSPred is a comprehensive and
easy-to-use TFBS prediction webtool, based on open chromatin
patterns, genomic conservation among human and mouse and HMM-based
searches. The present study demonstrated that the predictions of
TFBSPred were in accordance with both universal and tissue-specific
experimentally verified TFBSs and that in a number of cases, it
outperformed existing PWM-based webtools of similar function.
TFBSPred is expected to further improve as the TFFM quality and
quantity increases. TFBSPred may thus be a useful addition to the
experimental biologist community as it provides a working
hypothesis that can be experimentally verified.
Supplementary Material
Total open-chromatin region number,
average length, standard deviation of the length and percentage of
total genomic coverage of each cell line replicate of the DH data
for humans and mice.
Total conserved region number, average
length, standard deviation of the length and percentage of total
genomic coverage, of the pairwise alignment data for humans and
mice.
TFBS prediction results of TFBIND,
using FGA FASTA formatted sequence as input.
Acknowledgements
The authors wish to acknowledge that the webtool
host server is provided by GRNET.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
IM conceived the study. PGB and DAS were also
involved in the conception of the study. DH data were downloaded
and processed by CN. Multiple alignment between Homo sapiens
and Mus musculus data were downloaded and processed by KS.
VLZ downloaded the TFFM framework and TFFM profiles, and developed
the database and Graphical User Interface (GUI). IK assisted in the
website front end development. GAL performed the webtool
benchmarking. VLZ and IM confirm the authenticity of all raw data.
The manuscript was written by VLZ and IM. All authors contributed
to the revision of the work and, have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wittkopp PJ and Kalay G: Cis-regulatory
elements: Molecular mechanisms and evolutionary processes
underlying divergence. Nat Rev Genet. 13:59–69. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Zawel L and Reinberg D: Initiation of
transcription by RNA polymerase II: A multi-step process. Prog
Nucleic Acid Res Mol Biol. 44:67–108. 1993.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Liu X, Bushnell DA and Kornberg RD: RNA
polymerase II transcription: Structure and mechanism. Biochim
Biophys Acta. 1829:2–8. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Haberle V and Stark A: Eukaryotic core
promoters and the functional basis of transcription initiation. Nat
Rev Mol Cell Biol. 19:621–637. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lambert SA, Jolma A, Campitelli LF, Das
PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR and Weirauch MT:
The human transcription factors. Cell. 175:598–599. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lee TI and Young RA: Transcriptional
regulation and its misregulation in disease. Cell. 152:1237–1251.
2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Singh H, Khan AA and Dinner AR: Gene
regulatory networks in the immune system. Trends Immunol.
35:211–218. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Narlikar L and Hartemink AJ: Sequence
features of DNA binding sites reveal structural class of associated
transcription factor. Bioinformatics. 22:157–163. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Stormo GD: Modeling the specificity of
protein-DNA interactions. Quant Biol. 1:115–130. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Staden R: Computer methods to locate
signals in nucleic acid sequences. Nucleic Acids Res. 12:505–519.
1984.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Matys V, Kel-Margoulis OV, Fricke E,
Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M,
Hornischer K, et al: TRANSFAC and its module TRANSCompel:
Transcriptional gene regulation in eukaryotes. Nucleic Acids Res.
34 (Database Issue):D108–D110. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Fornes O, Castro-Mondragon JA, Khan A, van
der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M,
Baranašić D, et al: JASPAR 2020: Update of the open-access database
of transcription factor binding profiles. Nucleic Acids Res.
48:D87–D92. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kel AE, Gossling E, Reuter I, Cheremushkin
E, Kel-Margoulis OV and Wingender E: MATCH: A tool for searching
transcription factor binding sites in DNA sequences. Nucleic Acids
Res. 31:3576–3579. 2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Grant CE, Bailey TL and Noble WS: FIMO:
Scanning for occurrences of a given motif. Bioinformatics.
27:1017–1018. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhao Y, Ruan S, Pandey M and Stormo GD:
Improved models for transcription factor binding site
identification using nonindependent interactions. Genetics.
191:781–790. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Khamis AM, Motwalli O, Oliva R, Jankovic
BR, Medvedeva YA, Ashoor H, Essack M, Gao X and Bajic VB: A novel
method for improved accuracy of transcription factor binding site
prediction. Nucleic Acids Res. 46(e72)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Toivonen J, Kivioja T, Jolma A, Yin Y,
Taipale J and Ukkonen E: Modular discovery of monomeric and dimeric
transcription factor binding motifs for large data sets. Nucleic
Acids Res. 46(e44)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kulakovskiy I, Levitsky V, Oshchepkov D,
Bryzgalov L, Vorontsov I and Makeev V: From binding motifs in
ChIP-Seq data to improved models of transcription factor binding
sites. J Bioinform Comput Biol. 11(1340004)2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rabiner LR: A tutorial on hidden Markov
models and selected applications in speech recognition. Proceedings
IEEE. 77:257–286. 1989.
|
|
20
|
Xu D, Liu HJ and Wang YF: BSS-HMM3s: An
improved HMM method for identifying transcription factor binding
sites. DNA Seq. 16:403–411. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wu J and Xie J: Hidden Markov model and
its applications in motif findings. Methods Mol Biol. 620:405–416.
2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Mathelier A and Wasserman WW: The next
generation of transcription factor binding site prediction. PLoS
Comput Biol. 9(e1003214)2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Schneider TD and Stephens RM: Sequence
logos: A new way to display consensus sequences. Nucleic Acids Res.
18:6097–6100. 1990.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Frazer KA, Elnitski L, Church DM, Dubchak
I and Hardison RC: Cross-species sequence comparisons: A review of
methods and available resources. Genome Res. 13:1–12.
2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Emes RD, Goodstadt L, Winter EE and
Ponting CP: Comparison of the genomes of human and mouse lays the
foundation of genome zoology. Hum Mol Genet. 12:701–709.
2003.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dolan ME, Baldarelli RM, Bello SM, Ni L,
McAndrews MS, Bult CJ, Kadin JA, Richardson JE, Ringwald M, Eppig
JT and Blake JA: Orthology for comparative genomics in the mouse
genome database. Mamm Genome. 26:305–313. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Elgin SC: DNAase I-hypersensitive sites of
chromatin. Cell. 27:413–415. 1981.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Pipkin ME and Lichtenheld MG: A reliable
method to display authentic DNase I hypersensitive sites at
long-ranges in single-copy genes from large genomes. Nucleic Acids
Res. 34(e34)2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Boyle AP, Davis S, Shulha HP, Meltzer P,
Margulies EH, Weng Z, Furey TS and Crawford GE: High-resolution
mapping and characterization of open chromatin across the genome.
Cell. 132:311–322. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sabo PJ, Hawrylycz M, Wallace JC, Humbert
R, Yu M, Shafer A, Kawamoto J, Hall R, Mack J, Dorschner MO, et al:
Discovery of functional noncoding elements by digital analysis of
chromatin structure. Proc Natl Acad Sci USA. 101:16837–16842.
2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Thurman RE, Rynes E, Humbert R, Vierstra
J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H,
Vernot B, et al: The accessible chromatin landscape of the human
genome. Nature. 489:75–82. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Tweedie S, Braschi B, Gray K, Jones TEM,
Seal RL, Yates B and Bruford EA: Genenames.org. The
HGNC and VGNC resources in 2021. Nucleic Acids Res. 49:D939–D946.
2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Smith CL, Blake JA, Kadin JA, Richardson
JE and Bult CJ: Mouse Genome Database Group = Mouse Genome Database
(MGD)-2018: Knowledgebase for the laboratory mouse. Nucleic Acids
Res. 46:D836–D842. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Cunningham F, Achuthan P, Akanni W, Allen
J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et
al: Ensembl 2019. Nucleic Acids Res. 47:D745–D751. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kinsella RJ, Kahari A, Haider S, Zamora J,
Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P,
Kerhornou A, et al: Ensembl BioMarts: A hub for data retrieval
across taxonomic space. Database (Oxford).
2011(bar030)2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
John S, Sabo PJ, Thurman RE, Sung MH,
Biddie SC, Johnson TA, Hager GL and Stamatoyannopoulos JA:
Chromatin accessibility pre-determines glucocorticoid receptor
binding patterns. Nat Genet. 43:264–268. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Casper J, Zweig AS, Villarreal C, Tyner C,
Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Karolchik D, et
al: The UCSC Genome Browser database: 2018 update. Nucleic Acids
Res. 46:D762–D769. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hinrichs AS, Karolchik D, Baertsch R,
Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA,
Hsu F, et al: The UCSC Genome Browser database: Update 2006.
Nucleic Acids Res. 34 (Database Issue):D590–D598. 2006.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Rosenbloom KR, Sloan CA, Malladi VS,
Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R,
Heitner SG, et al: ENCODE data in the UCSC Genome Browser: Year 5
update. Nucleic Acids Res. 41 (Database Issue):D56–D63.
2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Herrero J, Muffato M, Beal K, Fitzgerald
S, Gordon L, Pignatelli M, Vilella AJ, Searle SM, Amode R, Brent S,
et al: Ensembl comparative genomics resources. Database (Oxford).
2016(bav096)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Harris RS: Improved pairwise alignment of
genomic DNA (unpublished PhD thesis). The Pennsylvania State
University, 2007.
|
|
42
|
Schliepm A, Georgi B, Rungsarityotin W,
Costa IG and Schönhuth A: The general hidden markov model library:
Analyzing systems with unobservable states. In: Forschung und
wissenschaftliches Rechnen: Beiträge zum Heinz-Billing-Preis. 2004,
Gesellschaft f ür wissenschaftliche Datenverarbeitung. Kremer K and
Macho V (eds) Gesellschaft für wissenschaftliche Datenverarbeitung
mbH Göttingen, Göttingen, pp121-136, 2005.
|
|
43
|
Cock PJ, Antao T, Chang JT, Chapman BA,
Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B
and de Hoon MJ: Biopython: Freely available Python tools for
computational molecular biology and bioinformatics. Bioinformatics.
25:1422–1423. 2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Quinlan AR: BEDTools: The swiss-army tool
for genome feature analysis. Curr Protoc Bioinformatics.
47:11.12.1–34. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Grabe N: AliBaba2: Context specific
identification of transcription factor binding sites. In Silico
Biol. 2 (Suppl):S1–15. 2002.PubMed/NCBI
|
|
46
|
Tsunoda T and Takagi T: Estimating
transcription factor bindability on DNA. Bioinformatics.
15:622–630. 1999.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Oshchepkov DY, Vityaev EE, Grigorovich DA,
Ignatieva EV and Khlebodarova TM: SITECON: A tool for detecting
conservative conformational and physicochemical properties in
transcription factor binding site alignments and for site
recognition. Nucleic Acids Res. 32:W208–W212. 2004.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Messeguer X, Escudero R, Farre D, Nunez O,
Martinez J and Alba MM: PROMO: Detection of known transcription
regulatory elements using species-tailored searches.
Bioinformatics. 18:333–334. 2002.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Mahony S and Benos PV: STAMP: A web tool
for exploring DNA-binding motif similarities. Nucleic Acids Res.
35:W253–W258. 2007.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Sandelin A, Wasserman WW and Lenhard B:
ConSite: Web-based prediction of regulatory elements using
cross-species comparison. Nucleic Acids Res. 32:W249–W252.
2004.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Benos PV, Corcoran DL and Feingold E:
Web-based identification of evolutionary conserved DNA
cis-regulatory elements. Methods Mol Biol. 395:425–436.
2007.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Loots GG and Ovcharenko I: rVISTA 2.0:
Evolutionary analysis of transcription factor binding sites.
Nucleic Acids Res. 32:W217–W221. 2004.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Ovcharenko I, Loots GG, Hardison RC,
Miller W and Stubbs L: zPicture: Dynamic alignment and
visualization tool for analyzing conservation profiles. Genome Res.
14:472–477. 2004.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Lee C and Huang CH: LASAGNA-Search 2.0:
Integrated transcription factor binding site search and
visualization in a browser. Bioinformatics. 30:1923–1925.
2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kreft L, Soete A, Hulpiau P, Botzki A,
Saeys Y and De Bleser P: ConTra v3: A tool to identify
transcription factor binding sites across species, update 2017.
Nucleic Acids Res. 45:W490–W494. 2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Zerbino DR, Wilder SP, Johnson N,
Juettemann T and Flicek PR: The ensembl regulatory build. Genome
Biol. 16(56)2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Panne D: The enhanceosome. Curr Opin
Struct Biol. 18:236–242. 2008.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Merika M and Thanos D: Enhanceosomes. Curr
Opin Genet Dev. 11:205–208. 2001.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Panne D, Maniatis T and Harrison SC: An
atomic model of the interferon-beta enhanceosome. Cell.
129:1111–1123. 2007.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Keller AD and Maniatis T: Identification
and characterization of a novel repressor of beta-interferon gene
expression. Genes Dev. 5:868–879. 1991.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Elias S, Robertson EJ, Bikoff EK and Mould
AW: Blimp-1/PRDM1 is a critical regulator of type III Interferon
responses in mammary epithelial cells. Sci Rep.
8(237)2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Fish RJ and Neerman-Arbez M: Fibrinogen
gene regulation. Thromb Haemost. 108:419–426. 2012.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Vazirinejad R, Ahmadi Z, Kazemi Arababadi
M, Hassanshahi G and Kennedy D: The biological functions, structure
and sources of CXCL10 and its outstanding part in the
pathophysiology of multiple sclerosis. Neuroimmunomodulation.
21:322–330. 2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Brownell J, Bruckner J, Wagoner J, Thomas
E, Loo YM, Gale M Jr, Liang TJ and Polyak SJ: Direct,
interferon-independent activation of the CXCL10 promoter by NF-κB
and interferon regulatory factor 3 during hepatitis C virus
infection. J Virol. 88:1582–1590. 2014.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Korhonen JH, Palin K, Taipale J and
Ukkonen E: Fast motif matching revisited: High-order PWMs, SNPs and
indels. Bioinformatics. 33:514–521. 2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Kumar S, Stecher G, Suleski M and Hedges
SB: TimeTree: A resource for timelines, timetrees, and divergence
times. Mol Biol Evol. 34:1812–1819. 2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Kahara J and Lahdesmaki H: BinDNase: A
discriminatory approach for transcription factor binding prediction
using DNase I hypersensitivity data. Bioinformatics. 31:2852–2859.
2015.PubMed/NCBI View Article : Google Scholar
|