Introduction
The Filoviridae family of RNA viruses
includes the highly contagious and lethal Ebola virus, which was
initially identified in 1976 in Sudan and the Democratic Republic
of the Congo (formerly Zaire), where it was responsible for two
epidemics of severe hemorrhagic fever that occurred at the same
time (1). The first-recorded
epidemic of the disease caused by the virus, termed Ebola virus
disease (EVD), occurred close to the Ebola River; thus, the virus
was named Ebola. Up to 90% case fatality rates have been observed
during certain outbreaks of the virus, which is regarded as one of
the most lethal viral pathogens worldwide (2). The Zaire, Sudan, Bundibugyo, Reston,
and Tai Forest ebolaviruses constitute the five species of Ebola
viruses that are currently known to exist (3). A linear, negative-sense RNA genome is
contained in filamentous, pleomorphic viral particles that encase
the encapsulated Ebola virus (4).
On January 11, 2023, the most recent Ebola virus
illness outbreak, was declared. According to the World Health
Organization (WHO), the epidemic began in September, 2022 in
Mubende District, Uganda, and was the fifth known outbreak caused
by the Sudan ebolavirus species. There were 142 confirmed cases, 55
fatalities and 22 suspected cases in the epidemic (5). The West African Ebola epidemic, which
began on March 23, 2014, in Guinea and was caused by the Zaire
ebolavirus species, was the largest Ebola outbreak in history. In
terms of worldwide public health, that epidemic resulted in ~28,652
total cases, 11,325 fatalities and a mortality rate of 40-70%
(6). Guinea, Liberia and Sierra
Leone were countries that were mostly affected by the epidemic; as
these countries were already afflicted by war and poverty, the
provision of essential care and containment measures was difficult
(7). Aside from the casualties
themselves, the outbreak had an impact on other healthcare
services, such as maternity and child health, the treatment of
human immunodeficiency virus (HIV) and running vaccine programs
(8). Ebolaviruses are considered to
be zoonotic viruses, which means they are suspected to be
transferred from an animal to a human via direct contact with
bodily fluids from infected animals, or through contact with items
contaminated by these bodily fluids. Humans, once infected, can
further transmit the illness by coming into close touch with body
fluids, such as blood or saliva, or contaminated items and surfaces
(9). A spillover event occurs when
a disease, such as a virus, spreads from one species to another
species that is not its reservoir host (10). Understanding spillover occurrences
is critical for preventing the spread of emerging infectious
illnesses. It is considered that three species of fruit bats of the
Pteropodidae family are the natural reservoir host of the
Zaire ebolavirus, following various modeling and serological
experiments (11). The Ebola virus
enters the human body through mucous membranes or skin breaches.
Once in the body, the virus infects immune cells, particularly
macrophages and dendritic cells, and other cell types such as liver
cells, endothelial cells and fibroblasts (12). The incubation period for EVD can
range from 2 to 21 days, with an average of 5 to 7 days. The virus
replicates in the body of the host throughout this asymptomatic
incubation phase, and the host can only transmit the infection when
symptoms appear (13). Typical
symptoms of EVD in the early stages include fever, headache,
jaundice, myalgia and gastrointestinal symptoms such as nausea,
vomiting and diarrhea (13).
Infected patients may experience complications such as internal
bleeding or multi-organ malfunction as the illness advances
(13). For diagnosis, the virus can
be detected using various techniques, such as enzyme-linked
immunosorbent assay (ELISA), reverse transcription-polymerase chain
reaction (RT-PCR), serological tests and on-field rapid diagnostic
kits (14). The handling of
patients and patient samples requires the use of personal
protective equipment by medical personnel and stringent
decontamination regimes, while the study of viral samples requires
biosafety level 4-grade facilities (15).
The Ebola virus particle or virion is a complex
structure comprised of multiple unique components. The
single-stranded RNA genome, which runs ~19,000 base pairs in
length, is at the heart of the virion. This genome is encircled by
viral structural proteins, which join together to form the inner
ribonucleoprotein (RNP) complex (16). The nucleoprotein (NP) and the
RNA-dependent RNA polymerase (L) are two key components of the RNP
complex. These proteins perform critical functions in viral
replication, ensuring that the virus can reproduce and propagate
inside the host cells (17). The
viral envelope, which is comprised of two viral glycoproteins, GP1
and GP2, surrounds the RNP complex. The glycoproteins interact with
host cell surface receptors, such as C-type lectin family
receptors, to enable attachment of the virus and membrane fusion
for subsequent entrance into cells (18). Knowledge of the Ebola virus
structure and replication cycle provides a foundation for the
development of prophylactic and therapeutic strategies. Notable
approved anti-Ebola vaccines include rVSV-ZEBOV-GP (ERVEBO),
Ad5-EBOV and rVSV/Ad5, all expressing the ebolavirus glycoprotein
as the immune system-stimulating antigen (19-21).
Treatment strategies encompass various types, such as the
convalescent serum of infected individuals, therapeutic
interferons, gene therapies such as small interfering RNAs that
target viral genes and lastly, inhibitors and monoclonal antibodies
(22). Inhibitors may target
various host machinery or viral enzymes crucial for the ebolavirus
life cycle; ion channel inhibitors target cell ion channels to
inhibit viral entry and viral entry inhibitors target host or viral
enzymes crucial for viral attachment, viral fusion and entry
(23). Moreover, monoclonal
antibodies have been approved for the treatment of EVD; Inmazeb and
the more recent Ebanga both target the ebolavirus glycoprotein,
thus inhibiting viral entry and attenuating viral spread across
host cells (24,25). Lastly, nucleotide analogues, such as
Remdesivir, become incorporated into nascent RNA molecules during
the viral RNA synthesis and thus interfere with viral replication
(26).
The threat of Ebola virus remains, as demonstrated
by the recurrent outbreaks even well into 2023, reaffirming the
need for safe and effective prophylactic measures and treatments.
Research on the Ebola virus is made challenging by the requirement
for austere safety measures in the laboratory setting, to prevent
accidental transmission of the pathogen (17). Furthermore, the drug development
process itself is a time-consuming and costly process from target
identification to lead development and optimization, with a
fraction of drugs under development finally making it to market
(27). Data abundance marks the
current state of scientific knowledge; a trove of structural,
genetic, chemical and molecular data is harnessed and is available
in public databases (28,29). Machine learning and deep learning
solutions, additionally, are being increasingly used in the context
of the drug development process (30). A risk prediction model for lymph
node metastasis in Ewing's sarcoma (ES) has been created using
clinicopathological data leading to the design of a clinical-use
service to improve detection of this metastasis in patients with ES
(31). Similarly, a model was
trained with medical data from patients with kidney cancer to aid
in the stratification for the risk of developing bone metastasis
and inform personalized treatment decisions (32).
Emerging technologies, such as semantic analysis or
neural networks can be employed in tasks such as target
identification or drug development against single-stranded RNA
viruses (28). The use of
computational methods has the potential to accelerate the
preclinical drug development of novel antivirals, reducing costs
and removing the need for biosafety measures. The present study
describes a computational pipeline for the generation of new
molecules targeting the Ebola virus polymerase, employing neural
network architectures, chemical data and structural bioinformatics.
The use of public big data of existing anti-virals enables the
training of models on critical features of anti-viral agents, which
in turns informs the design of novel candidates by the neural
network. The in silico assessment of the neural network
designed ligands by computational bioinformatics and the design of
pharmacophores further compresses these features and creates a
profile of antiviral potential, capable of informing further
experiments. The flexibility and scalability of the pipeline
components enhances and accelerates research on the preclinical
stage, where scale and safety are all the more relevant when faced
with the challenge of viral epidemics and pandemics.
Data and methods
Construction of pipeline
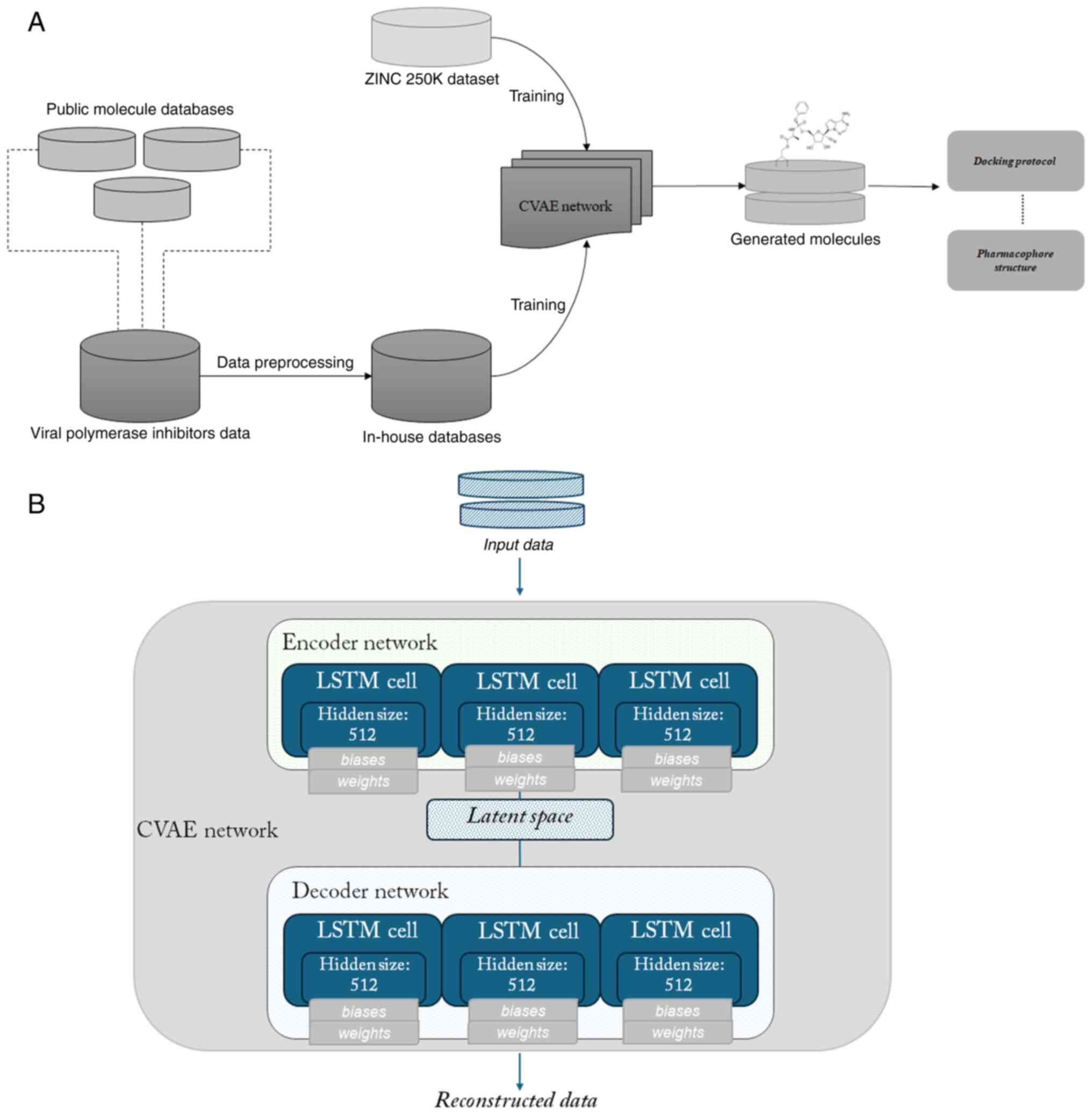
The overview of the pipeline is summarized in
Fig. 1A and is as follows: Public
chemical databases were employed towards building an in-house
chemical database of existing compounds which target viral
polymerases. A neural network was designed and trained on the
in-house database to learn the underlying features and
distribution. Subsequently, the latent space defined by the network
was explored to generate new molecules with a set of desired
properties. The resulting novel ligands were further filtered and a
conformational search was subsequently carried out to generate
three-dimensional conformations of the molecules, in preparation
for a docking simulation. A high throughput screening protocol was
used to dock the ligands against the previously experimentally
determined, three-dimensional structure of the Ebola virus
polymerase-VP35 complex, and lead performing ligands served as the
basis for pharmacophore construction.
Creation of in-house chemical database
and data pre-processing
In the era of big data and high-throughput
processes, vast amounts of chemical data are available in openly
accessible databases. ChEMBL constitutes a widely-known, public
database of curated chemicals, containing more than two million
preclinical drugs with associated bioactivities, ~14,000 clinical
candidate drugs and >4,000 clinical drugs (33). Its sister database, SureChEMBL,
aggregates similar data (34). The
Kyoto Encyclopedia of Genes and Genomes (KEGG) Drug database
collects drug data across three different continents in a
systematic manner (35). The Virus
Pathogen Database and Analysis Resource (ViPR) hosts multi-omics
data regarding various viral families, combining data from
archives, as well as data submitted by scientific groups and
virologists, rendering it a valuable source of information
(36). PubChem, supported by the
National Library of Medicine of NIH, constitutes a principal
information source for medicinal chemistry and drug discovery, with
substance, compound and bioassay records available along with
various metadata (37).
Modules were built in Python to use the Application
Programming Interface (API) of ChEMBL, PubChem, KEGG and SureChEMBL
to collect data of compounds which target viral polymerases. The
selection of the data sources was steered by a set of defined
criteria, such as the scope and coverage of the data, the public of
private nature of the datasets, the format of the data, as well the
availability of an API to enable the efficient sourcing. The
chemical data were collected in simplified molecular input line
entry system (SMILES) format. SMILES is used to convert the
three-dimensional structure of a chemical's into a string of
symbols that can be used as input in neural network training tasks
or other bioinformatic analyses (38). In the case of ViPR, where the API
availability is not straightforward, the collection of compounds
was executed in a semi-manual manner. The ChEMBL Python module, for
instance, queries the ChEMBL molecule endpoint against a set of
ChEMBL target identifiers of interest (such as the influenza
polymerase, CHEMBL4523676) to collect compounds against that
target, along with metadata such as compound preferred names, max
phase, molecular formula, molecular weight and the canonical SMILES
string. The module can be found in the following repository:
https://github.com/IoDiakou/Automatic-collection-of-drugs-for-specific-target-from-ChEMBL.
Analogous modules were constructed for all the aforementioned
databases and the collections of compounds queried from each
database were finally collected into an in-house SQLite database of
viral inhibitors. SQLite offers flexible and scalable storage,
which enables the preprocessing and handling of the data (39).
Upon establishing the database, preprocessing was
carried out using the Python RDKit interface, which allows the easy
manipulation and transformation of chemical strings (40). Functions were used to filter noisy
compounds out of the in-house database; examples include mixtures,
non-organic salts, organometallic compounds, polymers. Considerably
lengthy molecules were also filtered out, as lengthy strings which
exceed a threshold of ~120 characters can hinder deep learning and
cheminformatics tasks. Furthermore, to handle the existence of
duplicate records, as a direct result of sourcing inhibitors from
public databases where overlap may occur, a deduplication step was
also carried out. Lastly, molecular properties of interest were
calculated for each entry of the filtered and deduplicated
compounds database.
Neural network architecture design and
training
Generative probabilistic models based on neural
networks have gained traction over the past decade, with generative
adversarial nets and variational autoencoders (VAEs) as two
prominent examples, being applied to various tasks (41). The neural network learns a
distribution p(x) across data points and allows sampling, thus
carrying out the generative portion (42). The architecture used herein is a
subtype of VAE, termed a conditional variational autoencoder
(CVAE); the model samples from the generative distribution upon a
set condition, to generate novel data points and simultaneously
predict properties. All data handling and machine learning tasks
were carried out using Python; the neural network model design and
deployment was carried out using the TensorFlow framework, which
offers scalability and reliability for machine-learning tasks
(43). The VAE consists of two
units, the encoder, which maps input data points to a distribution
over the low-dimensional, latent space, and the decoder, which maps
the points of the aforementioned latent space back into the data
space. During training, the loss function implemented aims to
balance two objectives. The reconstruction loss reflects how well
the decoded data points match their original inputs and is
calculated by the mean squared error (44). The regularization part of the loss
function encourages the high similarity between the distribution
which the neural network learns, and an a priori distribution,
commonly a standard Gaussian distribution.
The encoder component of the network comprises of a
list of Long short-term memory (LSTM) cells, with each LSTM cell in
the list having a specific hidden size, and all LSTM cells are
wrapped into a multi-layered LSTM cell. An analogous structure of
LSTM cells is defined for the decoder. LSTMs belong to the umbrella
group of recurrent neural networks and constitute, one might say,
an evolution of them (45). A
schematic of the configuration and cell structure of the neural
network is presented in Fig. 1B.
The designed neural network was trained sequentially on the ZINC
250K dataset and then the in-house database of viral polymerase
inhibitors, to learn the representations and the characteristics
underlining these viral inhibitors. The ZINC database and
established subsets of it are commonly used in docking experiments
or other string-based chemical research approaches; the 250K subset
of the ZINC database has been used in Kaggle applications and other
autoencoder-related articles (46).
Generation and filtering of novel
molecules
Upon being fine-tuned on the dataset of existing
viral polymerase inhibitors, the CVAE was tasked with generating
novel molecules in SMILES format. The generation was steered
towards molecules which would exhibit the properties within the
‘neighborhood’ of an established small-molecule viral polymerase
inhibitor. Both the training and the sampling stage were carried
out in VS Code, using a system comprising of a quad-core Intel Core
i7-3770 processor and an NVIDIA GeForce GXT 1650 architecture for
GPU acceleration, which is crucial for machine-learning tasks.
The dataset of generated molecules was stored in a
new SQLite table instance for the subsequent pipeline steps of
re-filtering and docking experiments. RDKit functions and SMARTS
patterns were used to flag compounds with high similarity to the
starting in-house dataset, or with low likelihood of being
synthesized or proceeding further during a conventional hit
identification stage, such as fully acyclic compounds.
Docking and pharmacophore
generation
To investigate the potential efficacy of the novel
molecules as inhibitors for the Ebola polymerase, docking
experiments were carried out in molecular operating environment
(MOE); docking is a popular technique in computational drug
discovery for the study of ligand-target interactions (47). To transition from the generated
molecules, which are in a 2-dimensional flat string format, to a
three-dimensional molecule, conformational search protocols were
carried out. The complex of EBOV polymerase and cofactor VP35 with
suramin (PDB id: 7YET) served as the docking structure, following
structure preparation steps of assigning formal charges, energy
minimization and extraction of the suramin molecule in complex. The
complexed suramin additionally served as a baseline for the binding
site region as well as for interactions with surrounding residues
and binding affinities. High-throughput screening constitutes a
robust protocol for the rapid assay of candidate compounds,
allowing for the time and computational resource-efficient
evaluation of large sets of chemical data against a target
(48). Pharmacophore modeling was
carried out on the generated molecules with optimal binding scores
and interactions; pharmacophores can serve as a framework for
virtual screening efforts, allowing the identification of motifs
and interactions of interest underlying activity (49).
Results
Construction of in-house chemical
database
In the stage of database creation, API queries were
built against public databases, such as PubChem and ViPR, for the
search and retrieval of information related to molecules with
inhibitory activity against viral polymerases. Prior to filtering,
the in-house database of compounds contained 12,369 molecules;
following deduplication and removing out-of-scope molecules, such
as mixtures, non-organic salts and molecules >120 characters,
the database contained 11,960 unique molecule entries. For each
SMILES molecule, relevant molecular properties were calculated,
namely molecular weight, logP and topological polar surface area
(TPSA), serving as additional descriptors and features for the
training of the machine-learning model. The TPSA represents the
total of the contributions to the molecule (typically van der
Waals) surface area of polar atoms, e.g., nitrogen, oxygen or
associated hydrogens (50). It
constitutes a popular molecular descriptor for tasks of evaluating
transport ability, penetrative ability (such as through the
blood-brain barrier) and absorption within the intestine (50).
Neural network training
The SMILES input molecular data are one-hot encoded
into their vector embedding, within the first layers of the
encoding unit, along with the corresponding molecular properties
previously calculated for each data entry. The latent
representation is created in the encoder unit and subsequently
serves as input for the decoder unit; the decoded vector generated
by the decoder unit is funneled through a transformative layer of
equal size to the original one-hot encoding vector of the encoding
unit, to ensure the faithful reconstruction of the input. Softmax
activation is applied to the vectors. To set the basis for the
faithful reconstruction of encoded SMILES, the neural network was
first trained for 10 epochs on the ZINC 250K dataset, using Adam as
the optimizer, while also implementing an early-stopping criterion.
Subsequently, the model was fine-tuned on the in-house database of
existing inhibitors using ten-fold augmentation, to prepare for the
generative step of the pipeline.
Novel molecule generation, docking and
pharmacophore design
The latent space constructed between the encoder and
decoder unit served as the chemical space for the generation of
novel molecules. The latent space was sampled for the generation of
1,400 novel molecules along with prediction of molecular properties
for the generated molecules, resulting in sets of SMILES,
properties. SMARTS patterns were employed, post-deduplication, for
the removal of invalid SMILES strings from the resulting dataset.
As a last step the generated molecules were evaluated for
sufficient dissimilarity against the existing inhibitors within our
in-house database, which served as the fine-tuning set.
To investigate the binding behavior and interactions
between the generated molecules and the target of interest, herein
the ebolavirus polymerase, the structure of the ebolavirus
polymerase complex was first prepared in MOE in anticipation of the
docking experiments. The employed structure consists of the Ebola
virus L polymerase complexed with the co-factor VP35, an accessory
protein that is an essential part of the replication unit, with
supplementary roles as an antagonist of the host immune response
and virulence factor (51). VP35,
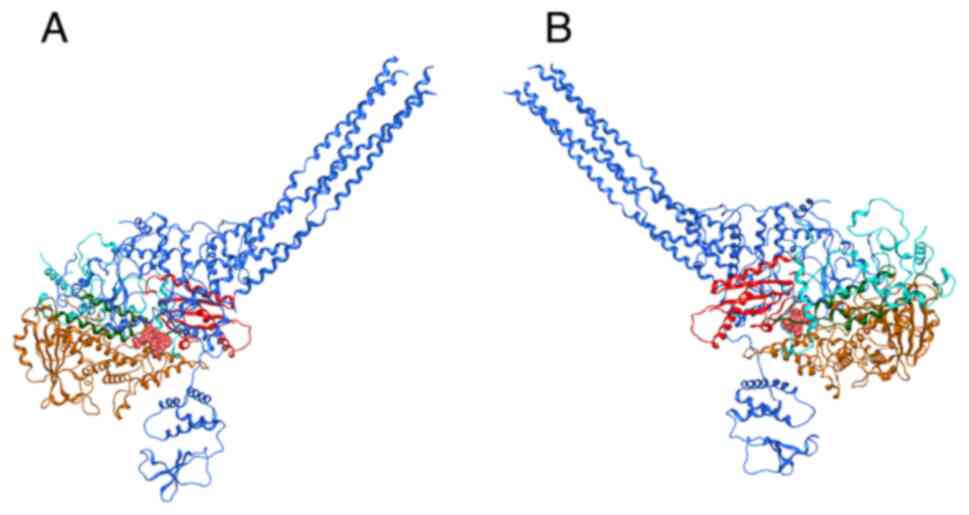
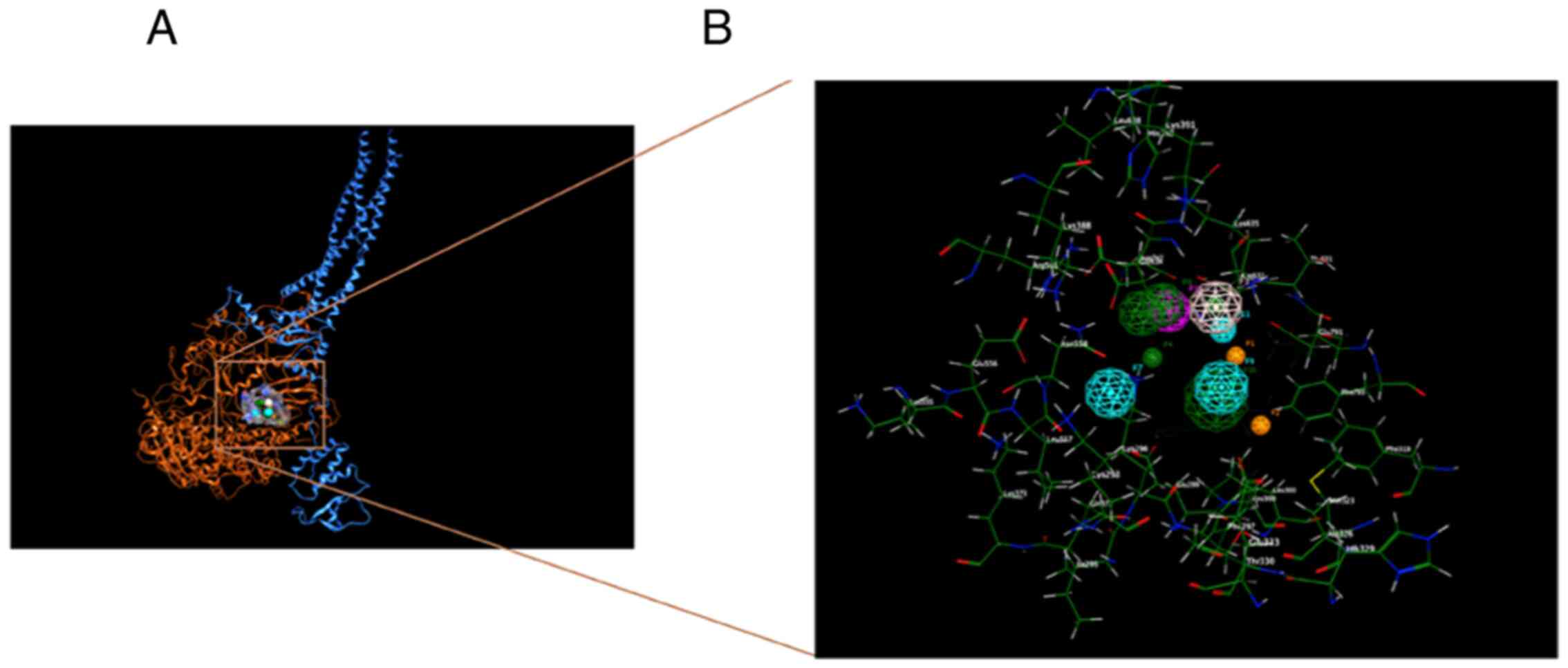
which is illustrated by the dark blue ribbon formation in Fig. 2A, interacts with the Ebola virus L
protein (Fig. 2A with red, yellow
and cyan ribbons for the different polymerase domains) to form the
replication complex for the generation of viral genome strands
during the viral cycle. In the employed three-dimensional structure
(PDB ID: 7YET), suramin is additionally bound to the complex
(52) (Fig. 2 in pink sphere representation to
denote the span of its molecular surface).
The generated molecules underwent a protocol of
conformational search within MOE to build a library of
three-dimensional conformations, which are crucial for the
exploration of the structural space of the binding site during the
docking experiments. Detailed conformational search cycles were
performed, with 100 iterations threshold per molecule for each
conformation, and a set of ten conformations per molecule within
the dataset. The suramin molecule which is bound to the active site
of the Ebola virus L-VP35 complex served as a guideline for the
identification of the site for docking, using dummy atoms to define
the site and the receptor and solvent as the receptor parameter.
The ligand database containing the conformations of the novel
molecules was then docked against the site (53).
The docking results were collected in a MOE
molecular database (mdb) file for further analysis, which
encompassed the evaluation of the results on multiple levels. The
docking scores and position of suramin within the site, upon using
the same docking protocol and parameters, served as a baseline for
the comparison of the scoring results of the novel molecules as
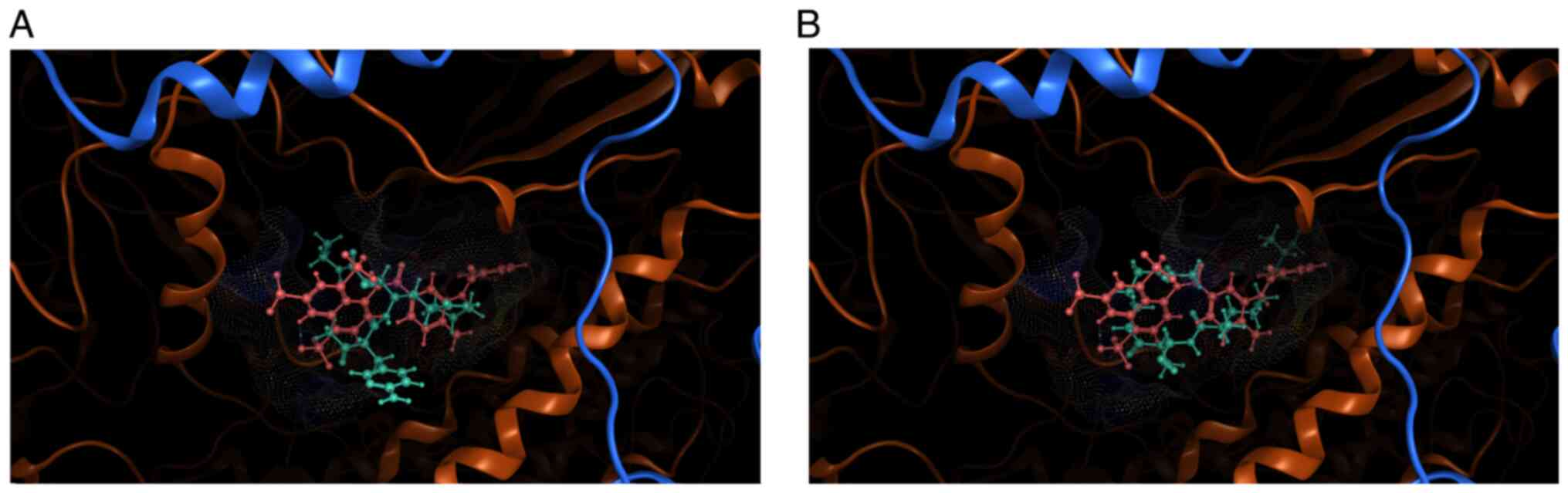
well as for visual comparisons. Examples of docking positions of
the novel molecules are illustrated in Fig. 3, with the positioning of suramin
(pale pink coloring) for visual comparison. Energy minimization was
additionally carried out to optimize the poses, monitoring the
shift in the positioning of the docked conformer. This is
exemplified in Fig. 3, where the
positioning of the ligand (light blue) relative to the binding
pocket and position of suramin (in pale pink) has shifted following
energy minimization cycles.
Furthermore, the ligand interaction functionality
within MOE allowed the examination of the specific interactions
between the atoms of the molecules and crucial residues around the
binding site of the viral polymerase. Conformational and spatial
characteristics, such as steric hindrances, were also taken into
account during the analysis of the docking results; structural
characteristics, such as the absence or presence of highly cyclic
groups, can serve as additional indicators of the molecules'
nature, such as their potential toxicity. Fig. 4 depicts this type of interactions
information for the same docked molecule as in Fig. 3, termed mol_2 for brevity.
The interaction diagram illustrated in Fig. 4, on the bottom left panel, confirms
that mol_2, for instance, forms several hydrogen bonds with the
receptor ebolavirus protein complex. Key residues involved in these
interactions are Lys 788, Lys 293, Val 559 and Lys 296. The
hydrogen bonds, indicated by green dashed lines, suggest that these
interactions are key for stabilizing the ligand-receptor complex.
The interaction table provides quantitative details, with
interaction energies ranging from -0.9 to -5.1 kcal/mol. These
values indicate the strength of the hydrogen bonds, with the
interaction between mol_2 atom O6 and EBOV polymerase Lys 788 being
the most significant due to its lowest energy (-5.1 kcal/mol). The
distances of these interactions (around 3.2-3.5 Å) are typical for
hydrogen bonds, further validating the strength and specificity of
these interactions. Top performing molecules as identified during
this evaluation process were further optimized with molecular
dynamics simulations without restraints, monitoring their shifts in
positions as in the example of Fig.
3. Lastly, pharmacophore modeling was carried out via the
pharmacophore building utility of MOE, calculating consensus
features from the bound ligands wherever meaningful.
To carry out the union of consensus pharmacophores
generated by different schemes, for each scheme, the individual
pharmacophores were first generated per ligand member among the top
100 scoring docked ligands. The R-strength value was enabled, and
each feature was adjusted with an appropriate assignment of
weights, after assessing its importance in the set of overall
interactions between the ligand and the receptor residues.
Pharmacophore data were saved in the pharmacophore format of MOE,
to be subsequently used to build the consensus pharmacophore for
the scheme by combining the individual pharmacophore features from
all ligands. As illustrated in Figs.
5 and 6, the unified
pharmacophore features span the pocket defined by the docking site,
reflecting interactions between the novel ligands and key residues
of the viral enzyme. Overall, the binding site environment is
comprised of a mix of hydrophobic and polar/charged residues. This
diverse environment supports a range of interactions, from
hydrophobic and aromatic interactions to hydrogen bonding and ionic
interactions. Key residues of the receptor harboring potential
interactions with features of the ligand are highlighted in yellow
boxes in depicted Fig. 6, colored
dark green and in stick figure representation as captured in MOE.
Notably, Lys393, Glu398, Val583, Glu394 and Glu399 are highlighted,
as these are key residues for binding interactions, highlighted by
the analyzed interactions of suramin as well. Aromatic
interactions, as highlighted in the generated pharmacophores, play
a pivotal role in the ligand binding, particularly through π-π
stacking interactions.
The unified pharmacophore reflects a comprehensive
interaction profile, capable of forming multiple types of
interactions within the receptor binding site, spanning aromatic,
hydrophobic, hydrogen bonding and potential metal ion coordination
interactions. Furthermore, this rich set of binding and chemical
feature information can be employed in the subsequent design or
screening of candidates against the specific target, employing the
availed pharmacophoric features as quick guidelines for the design,
screening or optimization of compounds and leads. Within the
context of the pipeline described herein, the data which were
collected during the docking and pharmacophore step of the pipeline
were fed back into the SQLite database, linked as metadata to the
SMILES molecules and further enhancing the knowledgebase.
Discussion
The pipeline described herein constitutes a scalable
approach for the discovery of novel potentially active anti-Ebola
agents, harnessing the rapidly increasing power of machine learning
capabilities and publicly accessible chemical data. Integral
processes of the drug design challenge, such as the hit
identification and lead optimization, can be supported by
state-of-the-art computational methods, which have the potential of
bringing vetted candidate molecules to the preclinical and
laboratory stage, effectively reducing operational costs and time,
both of which are crucial in the drug development cycle. This is
all the more relevant when the target of interest is a virus,
which, as exhibited by the recent COVID-19 epidemic, can grow into
a global issue rapidly. Through the implementation of rapid and
effective computational pipelines which combine string-based
formats of drugs and active compounds, with the power of robust
neural networks, a shift can be supported towards the adoption of a
proactive stance in the development of safe and effective
anti-virals.
Such a pipeline is marked by key features of
scalability and repurposing. The training dataset and the target of
interest are vessels, able to adopt different identities, depending
on the task at hand. What is described herein as the design and
evaluation of candidate, novel polymerase inhibitors targeting the
EBOV polymerase can be efficiently translated into the generation
of novel immunosuppressants in the context of Lupus, for instance,
or the generation of novel inhibitors against a metabolic enzyme
implicated in metabolic disorders. Furthermore, the neural network
architecture itself constitutes a flexible parameter of great
impact; tuning different parameters such as the network cell of
choice or the optimization algorithm can in turn yield different
generative behaviors, rendering the generation of novel data
points, or molecules, more or less restrictive or more or less
imaginative. One must note that there are inherent challenges in
using SMILES as a representation of chemical information for the
design and deployment of deep neural networks, including limited
ability to reflect stereochemistry and challenges in encoding and
interpreting complex molecules. Canonical SMILES can be employed to
address the aspect of ambiguity, while the combination of SMILES
with state-of-the-art tokenization techniques may enhance
performance (54).
Processes similar to the framework described herein,
such as the one proposed by van Tilborg and Grisoni (55), incorporate novel data acquisition to
perform iterative model improvement, parallel to the model's
execution of the task, which, in the study by van Tilborg and
Grisoni (55), is hit detection
through screening. Despite not incorporating this factor of ‘active
learning’, the proposed framework leverages the size and complexity
of the training data of anti-virals that have been designed and
assessed by the scientific and biomedical community, often in the
lab setting. This allows the model to learn features from
real-world examples, a ground truth of chemical information that
leads to precise, multifaceted, in-depth learning and subsequent
design of novel ligands. In summary, the proposed framework
reflects the significant potential which exists in the application
of machine-learning approaches in the field of computational drug
design. Leveraging big data and state-of-the-art algorithms
supports the effort of developing effective therapeutic agents,
under the scope of precision medicine and a proactive stance
against emerging viral threats.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The employed programming
code and datasets can be accessed in the first author's repository,
in the following web link: https://github.com/IoDiakou/anti-ebola-drugs-vae.
Authors' contributions
ID, GPC, DV and EE were involved in the design and
conceptualization of the study. ID, GPC, DV and EE contributed to
the collection and analysis of the data, as well as the design and
implementation of the pipeline described herein. ID, GPC, DV and EE
were involved in the writing and editing of the manuscript and in
the generation of relevant figures. ID, DV and EE confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
GPC is the Editor in Chief of the journal, and DV
and EE are Editors of the journal. However, they had no personal
involvement in the reviewing process, or any influence in terms of
adjudicating on the final decision, for this article. ID declares
that she has no competing interests.
References
|
1
|
Clegg J and Lloyd G: Ebola haemorrhagic
fever in Zaire, 1995. Curr Opin Infect Dis. 8:225–228. 1995.
|
|
2
|
Muyembe-Tamfum JJ, Mulangu S, Masumu J,
Kayembe JM, Kemp A and Paweska JT: Ebola virus outbreaks in Africa:
Past and present. Onderstepoort J Vet Res. 79(451)2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zheng H, Yin C, Hoang T, He RL, Yang J and
Yau SS: Ebolavirus classification based on natural vectors. DNA
Cell Biol. 34:418–428. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ajelli M, Merler S, Fumanelli L, Pastore
Y, Piontti A, Dean NE, Longini IM Jr, Halloran ME and Vespignani A:
Spatiotemporal dynamics of the Ebola epidemic in Guinea and
implications for vaccination and disease elimination: A
computational modeling analysis. BMC Med. 14(130)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Branda F, Mahal A, Maruotti A, Pierini M
and Mazzoli S: The challenges of open data for future epidemic
preparedness: The experience of the 2022 Ebolavirus outbreak in
Uganda. Front Pharmacol. 14(1101894)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kucharski AJ and Edmunds WJ: Case fatality
rate for Ebola virus disease in west Africa. Lancet.
384(1260)2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gostin LO and Friedman EA: A retrospective
and prospective analysis of the west African Ebola virus disease
epidemic: Robust national health systems at the foundation and an
empowered WHO at the apex. Lancet. 385:1902–1909. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Brolin Ribacke KJ, Saulnier DD, Eriksson A
and von Schreeb J: Effects of the West Africa ebola virus disease
on health-care utilization - A systematic review. Front Public
Health. 4(422)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Marí Saéz A, Weiss S, Nowak K, Lapeyre V,
Zimmermann F, Düx A, Kühl HS, Kaba M, Regnaut S, Merkel K, et al:
Investigating the zoonotic origin of the West African Ebola
epidemic. EMBO Mol Med. 7:17–23. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Plowright RK, Parrish CR, McCallum H,
Hudson PJ, Ko AI, Graham AL and Lloyd-Smith JO: Pathways to
zoonotic spillover. Nat Rev Microbiol. 15:502–510. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Koch LK, Cunze S, Kochmann J and Klimpel
S: Bats as putative Zaire ebolavirus reservoir hosts and their
habitat suitability in Africa. Sci Rep. 10(14268)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Osterholm MT, Moore KA, Kelley NS,
Brosseau LM, Wong G, Murphy FA, Peters CJ, LeDuc JW, Russell PK,
Van Herp M, et al: Transmission of ebola viruses: What We know and
what we do not know. mBio. 6(e00137)2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Velásquez GE, Aibana O, Ling EJ, Diakite
I, Mooring EQ and Murray MB: Time from infection to disease and
infectiousness for ebola virus disease, a systematic review. Clin
Infect Dis. 61:1135–1140. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bettini A, Lapa D and Garbuglia AR:
Diagnostics of ebola virus. Front Public Health.
11(1123024)2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Verbeek JH, Rajamaki B, Ijaz S, Sauni R,
Toomey E, Blackwood B, Tikka C, Ruotsalainen JH and Kilinc Balci
FS: Personal protective equipment for preventing highly infectious
diseases due to exposure to contaminated body fluids in healthcare
staff. Cochrane Database Syst Rev. 4(CD011621)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ghosh S, Saha A, Samanta S and Saha RP:
Genome structure and genetic diversity in the Ebola virus. Curr
Opin Pharmacol. 60:83–90. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Judson S, Prescott J and Munster V:
Understanding ebola virus transmission. Viruses. 7:511–521.
2015.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Nanbo A, Imai M, Watanabe S, Noda T,
Takahashi K, Neumann G, Halfmann P and Kawaoka Y: Ebolavirus is
internalized into host cells via macropinocytosis in a viral
glycoprotein-dependent manner. PLoS Pathog.
6(e1001121)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wolf J, Jannat R, Dubey S, Troth S,
Onorato MT, Coller BA, Hanson ME and Simon JK: Development of
pandemic vaccines: ERVEBO case study. Vaccines (Basel).
9(190)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Dolzhikova IV, Zubkova OV, Tukhvatulin AI,
Dzharullaeva AS, Tukhvatulina NM, Shcheblyakov DV, Shmarov MM,
Tokarskaya EA, Simakova YV, Egorova DA, et al: Safety and
immunogenicity of GamEvac-Combi, a heterologous VSV- and
Ad5-vectored Ebola vaccine: An open phase I/II trial in healthy
adults in Russia. Hum Vaccin Immunother. 13:613–620.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Li Y, Wang L, Zhu T, Wu S, Feng L, Cheng
P, Liu J and Wang J: Establishing China's national standard for the
recombinant adenovirus type 5 vector-based ebola vaccine (Ad5-EBOV)
virus titer. Hum Gene Ther Clin Dev. 29:226–232. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Malik S and Waheed Y: Tracing down the
updates on Ebola virus surges: An update on anti-ebola therapeutic
strategies. J Transl Int Med. 11:216–225. 2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lee JS, Adhikari NKJ, Kwon HY, Teo K,
Siemieniuk R, Lamontagne F, Chan A, Mishra S, Murthy S, Kiiza P, et
al: Anti-Ebola therapy for patients with Ebola virus disease: A
systematic review. BMC Infect Dis. 19(376)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Taki E, Ghanavati R, Navidifar T, Dashtbin
S, Heidary M and Moghadamnia M: Ebanga™: The most recent
FDA-approved drug for treating Ebola. Front Pharmacol.
14(1083429)2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Saxena D, Kaul G, Dasgupta A and Chopra S:
Atoltivimab/maftivimab/odesivimab (Inmazeb) combination to treat
infection caused by Zaire ebolavirus. Drugs Today (Barc).
57:483–490. 2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tchesnokov EP, Feng JY, Porter DP and
Götte M: Mechanism of inhibition of Ebola virus RNA-Dependent RNA
polymerase by remdesivir. Viruses. 11(326)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sun D, Gao W, Hu H and Zhou S: Why 90% of
clinical drug development fails and how to improve it? Acta Pharm
Sin B. 12:3049–3062. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vlachakis D: Genetic and structural
analyses of ssRNA viruses pave the way for the discovery of novel
antiviral pharmacological targets. Mol Omics. 17:357–364.
2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Papageorgiou L, Loukatou S, Sofia K,
Maroulis D and Vlachakis D: An updated evolutionary study of
Flaviviridae NS3 helicase and NS5 RNA-dependent RNA polymerase
reveals novel invariable motifs as potential pharmacological
targets. Mol Biosyst. 12:2080–2093. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Dara S, Dhamercherla S, Jadav SS, Babu CM
and Ahsan MJ: Machine learning in drug discovery: A review. Artif
Intell Rev. 55:1947–1999. 2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li W, Zhou Q, Liu W, Xu C, Tang ZR, Dong
S, Wang H, Li W, Zhang K, Li R, et al: A machine learning-based
predictive model for predicting lymph node metastasis in patients
with ewing's sarcoma. Front Med (Lausanne).
9(832108)2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dong S, Yang H, Tang ZR, Ke Y, Wang H, Li
W and Tian K: Development and Validation of a predictive model to
evaluate the risk of bone metastasis in kidney cancer. Front Oncol.
11(731905)2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Davies M, Nowotka M, Papadatos G, Dedman
N, Gaulton A, Atkinson F, Bellis L and Overington JP: ChEMBL web
services: Streamlining access to drug discovery data and utilities.
Nucleic Acids Res. 43:W612–W620. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Papadatos G, Davies M, Dedman N, Chambers
J, Gaulton A, Siddle J, Koks R, Irvine SA, Pettersson J, Goncharoff
N, et al: SureChEMBL: A large-scale, chemically annotated patent
document database. Nucleic Acids Res. 44:D1220–D1228.
2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Pickett BE, Sadat EL, Zhang Y, Noronha JM,
Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, et al: ViPR:
An open bioinformatics database and analysis resource for virology
research. Nucleic Acids Res. 40:D593–D598. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kim S, Chen J, Cheng T, Gindulyte A, He J,
He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, et al: PubChem in
2021: New data content and improved web interfaces. Nucleic Acids
Res. 49:D1388–D1395. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Polanski J and Gasteiger J: Computer
Representation of Chemical Compounds. Springer International
Publishing, pp1997-2039, 2017.
|
|
39
|
Silva Y, Almeida I and Queiroz M: SQL:
From Traditional Databases to Big Data. SIGCSE. 413–418. 2016.
|
|
40
|
Landrum G: RDKit. Open-Source
Cheminformatics Software. 2010. Available from: https://www.rdkit.org.
|
|
41
|
Goodfellow I, Pouget-Abadie J, Mirza M, Xu
B, Warde-Farley D, Ozair S, Courville A and Bengio Y: Generative
Adversarial Networks. Vol 27. Advances in Neural Information
Processing Systems, 2014.
|
|
42
|
Kingma DP and Welling M: An introduction
to variational autoencoders. ArXiv: Dec 11, 2019 (Epub ahead of
print).
|
|
43
|
Abadi M, Barham P, Chen J, Chen Z, Davis
A, Dean J, Devin M, Ghemawat S, Irving G, Isard M, et al:
TensorFlow: A system for large-scale machine learning. ArXiv: May
31, 2016 (Epub ahead of print).
|
|
44
|
Hodson TO: Root-Mean-Square Error (RMSE)
or Mean Absolute Error (MAE): When to use them or not.
Geoscientific Model Development. Vol 15. European Geosciences
Union, pp5481-5487, 2022.
|
|
45
|
DiPietro R and Hager G: Deep learning:
RNNs and LSTM. pp503-519, 2020.
|
|
46
|
Akhmetshin T, Lin A, Mazitov D, Ziaikin E,
Madzhidov T and Varnek A: HyFactor: Hydrogen-count labelled
graph-based defactorization Autoencoder. Comput Theor Chem: Dec 6,
2021 (Epub ahead of print).
|
|
47
|
Chemical Computing Group: Computer-Aided
Molecular Design. Molecular Operating Environment (MOE). 2024.
Available from: https://www.chemcomp.com/en/Products.htm.
|
|
48
|
Szymański P, Markowicz M and
Mikiciuk-Olasik E: Adaptation of high-throughput screening in drug
discovery-toxicological screening tests. Int J Mol Sci. 13:427–452.
2012.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Giordano D, Biancaniello C, Argenio MA and
Facchiano A: Drug design by pharmacophore and virtual screening
approach. Pharmaceuticals (Basel). 15(646)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Prasanna S and Doerksen RJ: Topological
polar surface area: A useful descriptor in 2D-QSAR. Curr Med Chem.
16:21–41. 2009.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Leung DW, Prins KC, Basler CF and
Amarasinghe GK: Ebolavirus VP35 is a multifunctional virulence
factor. Virulence. 1:526–531. 2010.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yuan B, Peng Q, Cheng J, Wang M, Zhong J,
Qi J, Gao GF and Shi Y: Structure of the Ebola virus polymerase
complex. Nature. 610:394–401. 2022.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kalinowsky L, Weber J, Balasupramaniam S,
Baumann K and Proschak E: A diverse benchmark based on 3D matched
molecular pairs for validating scoring functions. ACS Omega.
3:5704–5714. 2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Leon M, Perezhohin Y, Peres F, Popovič A
and Castelli M: Comparing SMILES and SELFIES tokenization for
enhanced chemical language modeling. Sci Rep.
14(25016)2024.PubMed/NCBI View Article : Google Scholar
|
|
55
|
van Tilborg D and Grisoni F: Traversing
chemical space with active deep learning for low-data drug
discovery. Nat Comput Sci. 4:786–796. 2024.PubMed/NCBI View Article : Google Scholar
|