Introduction
Chromium (Cr) is a common industrial chemical used
in diverse processes including metallurgy, electroplating, leather
tanning, and chroming (1). The
most stable and common oxidation states of Cr are trivalent
chromium [Cr(III)] and hexavalent chromium [Cr(VI)] (2). Cr(III) is required in tracing sugar
amounts and in lipid metabolism (3). The exposure of Cr(III) is considered
less toxic because of its tendency to form insoluble hydrated
complexes which cannot cross cell membranes (4). Cr(VI) enters the body by inhalation,
ingestion, or absorption through skin, and Cr(VI) enters into cells
through an anion transport system (5). Once inside the cells, it can be
reduced to its lower oxidation states, pentavalent chromium [Cr(V)]
and tetravalent chromium [Cr(IV)] (6). The reduction process remains to be
explored and fully understood. Cr(VI) is highly soluble and it
exerts toxic effects in most living organisms. Occupational
exposure to Cr(VI) is associated with several adverse effects of
health, such as contact dermatitis, nasal perforation, and
bronchogenic cancer (7). It has
been reported that Cr(VI) could inhibit DNA, RNA and protein
synthesis in hepatocytes and induce damage to liver structure and
function thus causing toxic hepatitis (8,9).
Reactive oxygen species (ROS) are defined as
oxygen-containing chemical species with reactive chemical
properties including free radicals that contain unpaired electrons,
such as superoxide (O2−), hydroxyl radicals
(HO•), and non-radical molecules like hydrogen peroxide
(H2O2) (10). ROS are formed mainly by the
interaction of oxygen molecules with electrons that escape from the
mitochondrial respiratory chain (MRC) (11). MRC contains five multimeric
protein complexes including reduced nicotinamide adenine
dinucleotide (NADH) dehydrogenase-ubiquinone oxidoreductase (MRCC
I), succinate dehydrogenase-ubiquinone oxidoreductase (MRCC II),
ubiquinone-cytochrome c oxidoreductase (MRCC III), cytochrome c
oxidase (MRCC VI), and ATP synthase (MRCC V) (12). Inhibition of MRCC increases the
electron leakage by blocking the electron transfer, thus enhancing
ROS production (13). ROS can be
scavenged by antioxidative proteins, including catalase, superoxide
dismutase (SOD), and thioredoxin (Trx) (14,15). Although it is believed that ROS
play a key role in the toxic effect of Cr(VI), the precise
mechanisms by which Cr(VI) triggers apoptosis are not fully
understood.
Apoptosis is a process controlled by a specific
signaling pathway and is characterized by cellular shrinkage,
nuclear condensation, and DNA fragmentation (16). Although numerous studies have
suggested that mitochondria stress and caspase activation are the
most typical events required for apoptotic cell death (17), the precise mechanisms by which
Cr(VI) induces apoptosis in hepatocytes have not been elucidated.
In the present study, we demonstrated that Cr(VI) induces
ROS-dependent caspase-3 activation by inhibiting MRCC I activity.
MRCC I is a site for ROS generation as well as Cr(VI) reduction. To
the best of our knowledge, this is the first time that Cr(VI) is
identified as an MRCC I inhibitor. These results present a new
target and mechanism for Cr(VI)-induced cytotoxicity.
Material and methods
Materials
The L-02 hepatocyte line was provided by the China
Center for Type Culture Collection of Wuhan University. The MRCC
substrates (glutamate/malate, succinate, coenzyme Q, vitamin C),
rotenone (ROT) and N-acetylcysteine (NAC) were purchased from
Sigma-Aldrich (St. Louis, MO, USA). RPMI-1640 culture medium, fetal
bovine serum (FBS), and trypsin-EDTA (0.25%) were obtained from
Gibco (Gaithersburg, MD, USA). Potassium dichromate
(K2Cr2O7) was obtained from
Changsha Chemical Reagents Co. (Changsha, China).
Cell culture and chemicals treatment
L-02 hepatocytes were cultured in RPMI-1640 medium
supplemented with 10% (vol/vol) FBS, 2 mM L-glutamine, and
antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) at 37°C
under a humidified atmosphere of 5% CO2. The medium was
changed every other day.
Measurement for cell viability
MTT assay was performed to evaluate cell viability
as previously described (18).
The cells in exponential growth were seeded in 96-well plates with
100 μl of medium containing 104 cells.
K2Cr2O7 solution of indicated final
concentrations (0, 2, 8, 32, 128 and 512 μM) was added. Control
cells and medium controls without cells received DMSO without
K2Cr2O7. After incubation at 37°C
in a 5% CO2 saturated atmosphere for an indicated time period, the
cells were treated with 5 μl 5 mg/ml of MTT solution for an
additional 4 h at 37°C, and then lysed in phosphate-buffered saline
(PBS, pH 7.4) containing 20% sodium dodecyl sulfate (SDS) and 50%
N, N-dimethylformamide (pH 4.5). The absorbance was read on a
multiwell ELISA reader Versamax (Molecular Devices, Sunnyvale, CA,
USA) at 570 nm for each well.
Measurement of ROS production
Intracellular ROS production was determined by
detecting the fluorescent intensity of 2′,7′-dichlorofluorescein
(DCF), the oxidized product of the fluoroprobe 5-(and
6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
(CM-H2DCFDA; Molecular Probes, USA). Briefly, 2×106
cells were collected by centrifugation and were then incubated with
10 μM CM-H2DCFDA in PBS for 40 min at 37°C in the dark. After
incubation, the cells were split into two parts. One part was
checked with a fluorescence microscope equipped with a Leica DC 100
digital camera. The other part was measured with a flow cytometer
with excitation at 488 nm and emission at 535 nm. The amount of ROS
production was considered directly proportional to the fluorescence
intensity.
Western blotting for protein levels
determination
L-02 hepatocytes were lysed using a Mammalian Cell
Lysis kit from Sigma-Aldrich. Western blotting was performed with
the WesternBreeze Chemiluminescent Immunodetection protocol
(Invitrogen, Carlsbad, CA, USA). Proteins were separated by
electrophoresis on 10% sodium dodecyl sulfatepolyacrylamide gels
(SDS-PAGE), and were then transferred to polyvinylidene fluoride
(PVDF) membranes by electroelution. The membranes were incubated
with a primary antibody overnight at 4°C following blocking with 4%
non-fat milk. Membranes were then incubated for 1 h at room
temperature with secondary antibodies, developed with a detection
system and then exposed onto films.
The primary antibodies for MRCC including Complex I
subunit NDUFS3 (MS110), Complex II subunit 70 kDa Fp (MS204),
Complex III subunit core 2 (MS304), Complex IV subunit II (MS405),
and ATP synthase subunit α (MS502) were purchased from MitoScience
(Eugene, OR, USA). Antibodies for SOD 1 (#2770), catalase (#8841),
Trx (C63C6) (#2429), caspase-3 (#9662), heat shock protein (HSP)70
(#4872), HSP90 (E289) (#4877), and β-actin (#4967) were purchased
from Cell Signaling Technology (Danvers, MA, USA).
Measurement of activities of respiratory
chain complexes (MRCC) I-IV
The mitochondria were isolated as previously
described with slight modifications (19). Cells were washed twice with cold
PBS, and resuspended with 5 ml buffer A (250 mM sucrose, 20 mM
HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1
mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, pH 7.5).
Cells were homogenized and centrifuged twice at 750 x g for 10 min.
Mitochondria pellets were obtained after centrifugation at 10,000 x
g for 15 min.
The activities of MRCC were determined using
Mitochondrial Respiratory Chain Complexes Activity Assay kits
(Genmed Scientifics, Inc., Shanghai, China). All assays were
performed in a final volume of 1 ml using a UV-9100
spectrophotometer. To establish the optimum conditions for the
release of complexes, the mitochondria were freeze-thawed three
times at 20/−20°C in hypotonic media (25 mM potassium phosphate, 5
mM MgCl2, pH 7.2) before the determination. The activity
of MRCC I [nicotinamide adenine dinucleotide (NADH) coenzyme Q
(CoQ) oxidoreductase, expressed as nmol oxidized NADH/min/mg
protein] was measured following the oxidation of NADH at 340 nm.
The activity of MRCC II (succinate: 2,6-dichlorophenolindophenol
(DCIP) oxireductase, expressed as nmol reduced DCIP/min/mg protein)
was measured following the reduction of DCIP at 600 nm. The
activity of MRCC III (ubiquinol: cytochrome c (Cyt c) reductase,
expressed as nmol reduced Cyt c/min/mg protein) was measured
following the reduction of Cyt c at 550 nm. The activity of MRCC IV
(Cyt c oxidase, expressed as nmol oxidized Cyt c/min/mg protein)
was measured following the oxidation of Cyt c at 550 nm. All
measurements were performed in triplicate.
Measurement of the Cr(VI) reduction rate
in hepatocytes mitochondria
Cr(VI) reduction was determined colorimetrically
with a spectrophotometer using the S-diphenylcarbazide (DPC)
(Nacalai Tesque, Inc., Japan) method (20). The mitochondria isolated from
hepatocytes were pretreated with the MRCC I substrates 10 mM
glutamate/10 mM malate (Glu/Mal), MRCC II substrate 10 mM succinate
(Suc), MRCC III substrate 5 μM CoQ, or MRCC IV substrate 2 mM
vitamin C (Vit C) for 10 min prior to 32 μM Cr(VI) treatment.
Cr(VI) reduction rate was measured at different time points (5, 30
and 60 min). After 3 freeze and thaw cycles, the mitochondria
treatment suspensions (2 ml) were centrifuged for 5 min at 15,000 x
g. The supernatant was added with 20 μl H2SO4
and 20 μl H3PO4, mixed and then added with 80
μl DPC (0.076 x g DPC previously dissolved in 20 ml of 95%
ethanol). Twenty minutes later, the absorbance of the color
produced was measured at 540 nm. Cr(VI) concentration in the sample
was calculated from a standard curve using
K2Cr2O7 as a standard.
Measurement of caspase-3 activity
Caspase-3 activity was detected using a Caspase-3
colorimetric assay kit (Millipore, Billerica, MA, USA). Briefly,
L-02 hepatocytes were harvested and washed twice with ice-cold PBS.
Then cell pellets were incubated with lysis buffer (50 mM Tris-HCl,
1 mM EDTA, and 10 mM ethyleneglycoltetraacetic acid, pH 7.4) for 30
min on ice. After centrifugation at 13,000 x g at 4°C for 5 min,
the supernatants were collected and added with caspase-3 substrate
Ac-DEVD-pNA to the final concentration of 100 μM. The samples were
incubated at 37°C for 1 h and the alternative activity of caspase-3
was described as the cleavage of the colorimetric substrate by
measuring the absorbance at 405 nm.
Flow cytometry analysis for apoptotic
cells
L-02 hepatocytes were harvested by trypsinization
and washed with PBS. Washed cells were treated with FITC-conjugated
Annexin V (0.5 μg/ml final concentration) and propidium iodide (PI,
1 μg/ml final concentration). After incubation for 20 min at room
temperature, the apoptosis was determined by flow cytometry and
analyzed by the CellQuest software. For each measurement, 20,000
cells were analyzed. PI was added to a sample to distinguish early
apoptotic cells (Annexin V-positive, PI-negative) and late
apoptotic cells (positive for both Annexin V and PI).
Statistical analysis
Statistical analysis was performed using SPSS 15.0
one-way analysis of variance (ANOVA) to assess the significance of
differences between groups. The acceptance level of significance
was P<0.05. Results are expressed as mean ± SD.
Results
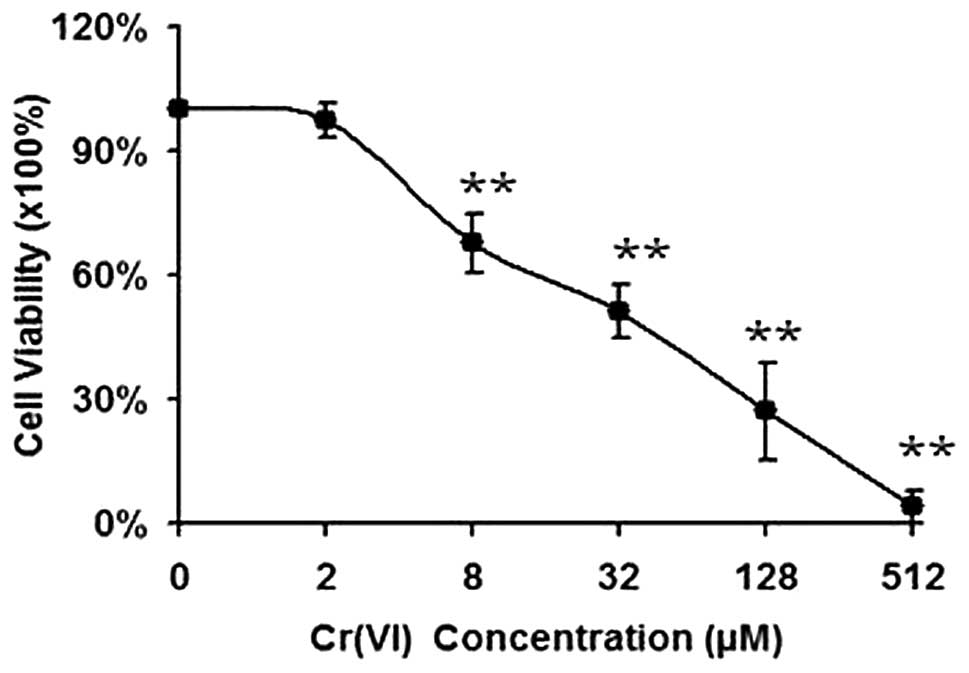
Cr(VI) induces a concentration-dependent
loss of cell viability in L-02 hepatocytes
L-02 hepatocytes exposed to varying doses of Cr(VI)
(2–512 μM) over a 24 h period and a concentration-dependent loss of
cell viability was observed in Fig.
1. The Cr(VI) concentration that required for 50% inhibition of
cell viability (IC50) was 38.97 μM. Therefore, we chose
two concentrations of Cr(VI) (16, 32 μM) for the following
experiments.
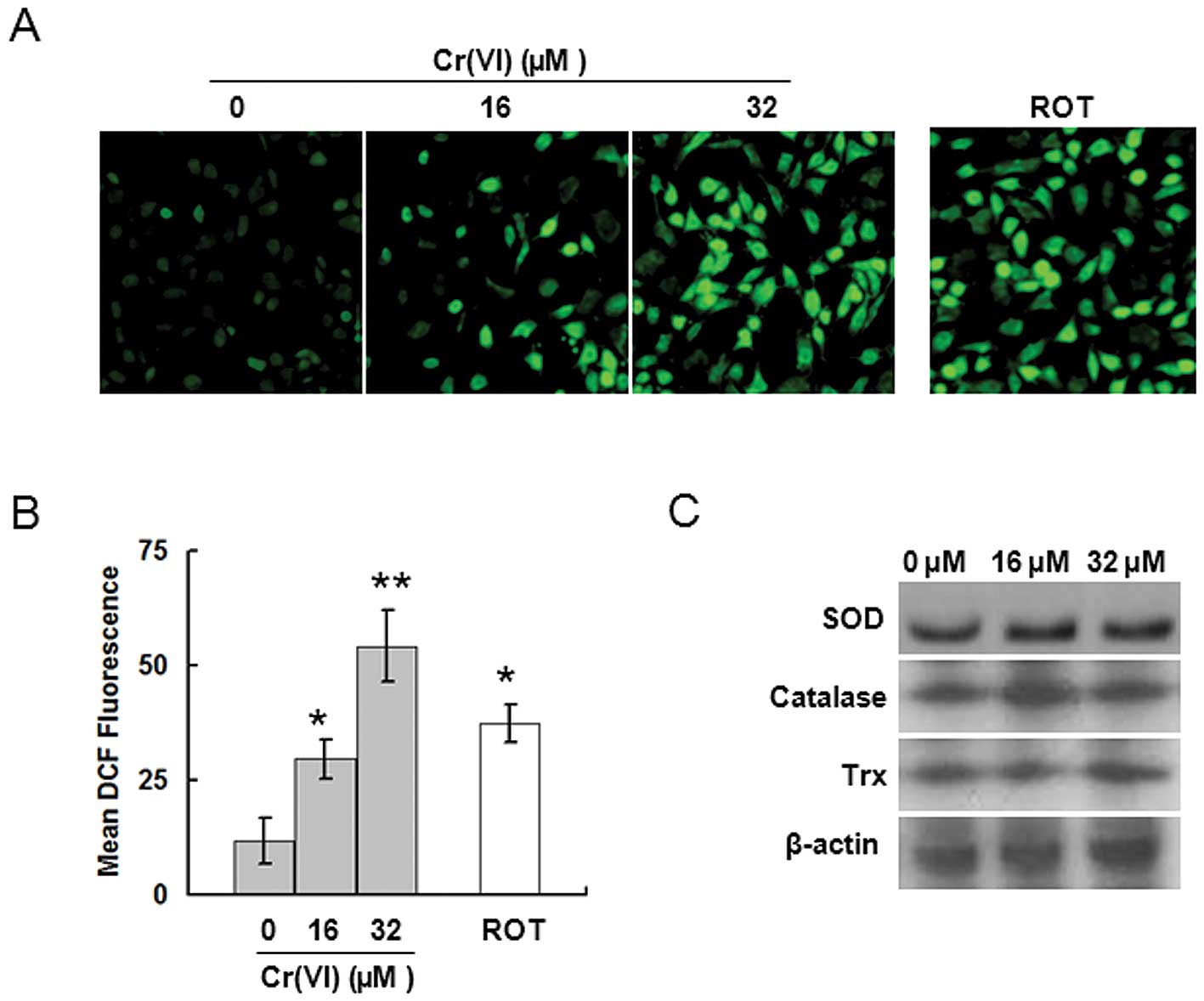
Cr(VI) causes ROS accumulation
ROS play a critical role in mediating the
cytotoxicity induced by Cr(VI), but the targets by which Cr(VI)
induces ROS accumulation are unknown. To identify the targets for
Cr(VI)-induced ROS accumulation, we first measured ROS levels. L-02
hepatocytes exposed to 16 or 32 μM Cr(VI) for 24 h were analyzed
for ROS production utilizing the oxidant-sensitive fluorogenic
probe CM-H2DCFDA. Cr(VI) stimulation induced higher
levels of fluorescence signal in a dose-dependent manner,
indicating the generation of a large amount of intracellular ROS in
the Cr(VI) treatment groups (Fig.
2A). Quantitative analysis by flow cytometry showed that the
ROS levels were about 3-fold higher in the 16 μM Cr(VI) treatment
group, and were about 5-fold higher in the 32 μM Cr(VI) treatment
group compared with the control group (Fig. 2B). It has been reported that
downregulation of antioxidative proteins results in ROS
accumulation (15). Therefore, we
investigated the effect of Cr(VI) on the expression of
antioxidative proteins such as SOD, catalase and Trx. It was
revealed that Cr(VI) had no significant effect on the three
proteins (Fig. 2C). Therefore, we
reached the conclusion that Cr(VI) does not cause ROS
overproduction by downregulating the antioxidative proteins.
Cr(VI) induces ROS accumulation by
inhibiting the activity of MRCC I
Inhibiting the activity of MRCC promotes ROS
accumulation (13). Thus we
speculated that Cr(VI) may induce ROS accumulation by inhibiting
MRCC activity. We measured the effect of Cr(VI) (16, 32 μM) on
different MRCC activities. Cr(VI) significantly inhibited MRCC I
and slightly inhibited MRCC II at a dose of 32 μM, but had no
effect on MRCC III and IV (Fig.
3A), indicating that MRCC I as well as MRCC II may be targets
of Cr(VI) in mediating ROS accumulation. This was further explored
by measuring the activities of MRCC I and II at different time
points after 32 μM Cr(VI) exposure. MRCC I activity was
significantly inhibited as early as 30 min after Cr(VI) treatment
(Fig. 3B). In contrast, the
activity of MRCC II was not altered until 120 min after Cr(VI)
exposure, indicating that MRCC I is the target of Cr(VI) in
promoting ROS production, and the decrease in MRCC II activity may
be viewed as the result of inhibited MRCC I activity. The data were
further confirmed by western blotting. Cr(VI) significantly
decreased the expression of MRCC I (subunit NDUF S3), but had no
effect on the expression levels of the left three complexes
(Fig. 3C).
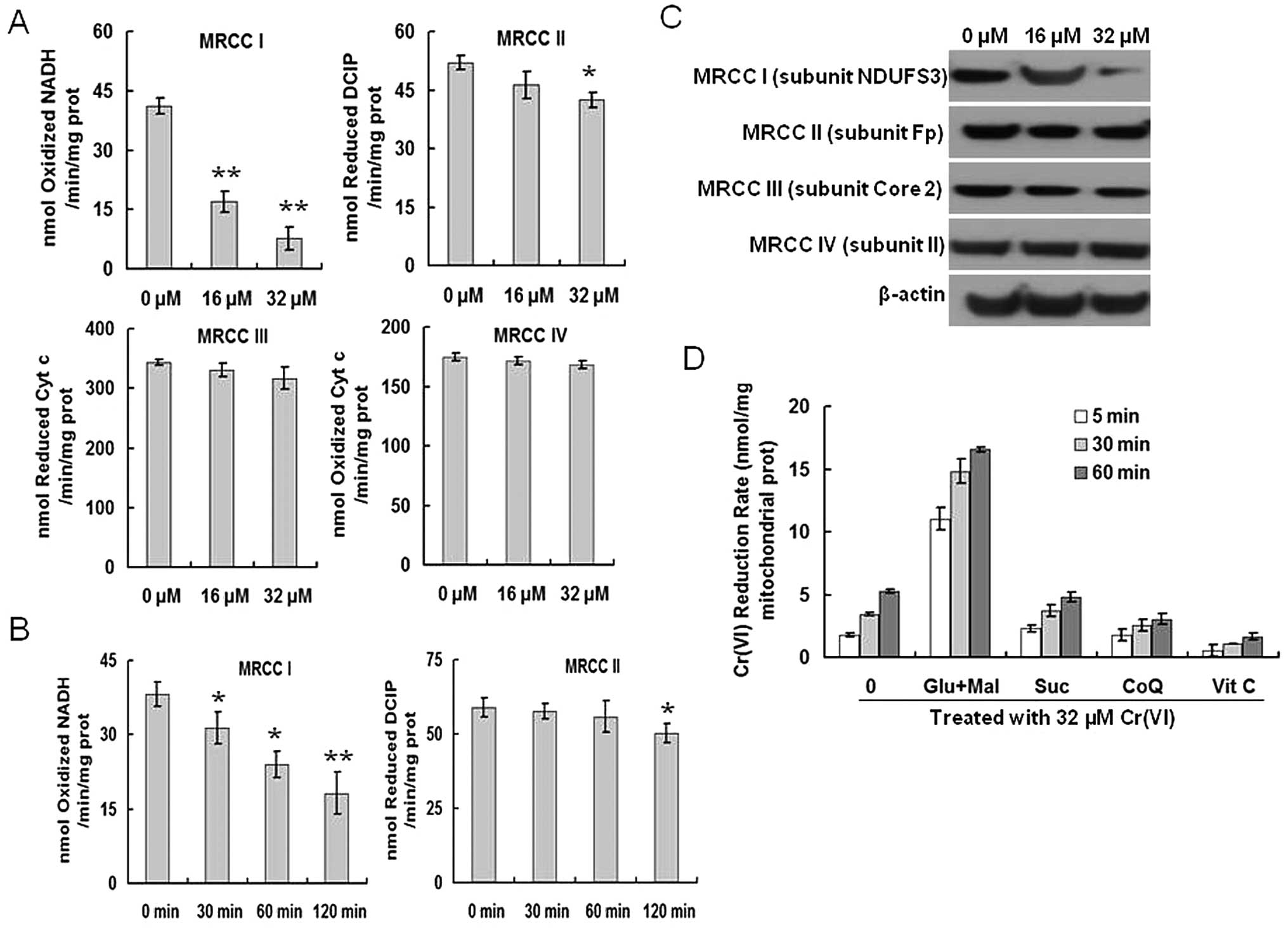 | Figure 3.Cr(VI) targets and inhibits MRCC I.
(A) Cr(VI) significantly inhibits MRCC I activity. L-02 hepatocytes
were treated with Cr(VI) (16, 32 μM) for 24 h. The activities of
MRCC I, II, III and IV were measured with the Mitochondrial
Respiratory Chain Complex Enzyme Activity kits. All columns display
the mean ± SD from three independent experiments.
*P<0.05, **P<0.01, compared to control.
(B) Inhibition of MRCC I activity is involved in mediating ROS
accumulation induced by Cr(VI). L-02 hepatocytes were treated with
32 μM Cr(VI) for the indicated time (30, 60, 120 min). The
activities of MRCC I and II were measured by Mitochondrial
Respiratory Chain Complex Enzyme Activity kit. All columns display
the mean ± SD from three independent experiments.
*P<0.05, **P<0.01, compared to control.
(C) Cr(VI) decreases the protein expression levels of MRCC I. The
hepatocytes were exposed to Cr(VI) (16, 32 μM) for 24 h and then
were processed for western blotting analysis to examine the protein
levels of MRCC I-IV. (D) Cr(VI) reduction occurs at MRCC I. The
mitochondria isolated from hepatocytes were pretreated with MRCC I
substrates 10 mM glutamate/10 mM malate (Glu/Mal), MRCC II
substrate 10 mM succinate (Suc), MRCC III substrate 5 μM coenzyme Q
(CoQ), or MRCC IV substrate 2 mM vitamin C (Vit C) for 10 min prior
to 32 μM Cr(VI) treatment. The Cr(VI) reduction rate was measured
using a spectrophotometer at different time points (5, 30, 60
min). |
Based on the previous results, we inferred that
Cr(VI) accepted the electrons and was reduced at the MRCC I.
Mitochondria can reduce Cr(VI) using MRCC substrates as electron
donors (21). The mitochondria
isolated from hepatocytes were pretreated with the MRCC I
substrates glutamate/malate (Glu/Mal), the MRCC II substrate
succinate (Suc), the MRCC III substrate CoQ, or the MRCC IV
substrate Vit C prior to Cr(VI) treatment. We then measured the
Cr(VI) reduction rate in mitochondria at different time points.
Cr(VI) reduction rate significantly increased in the mitochondria
treated with Glu/Mal, indicating that Cr(VI) reduction occurs at
the MRCC I (Fig. 3D).
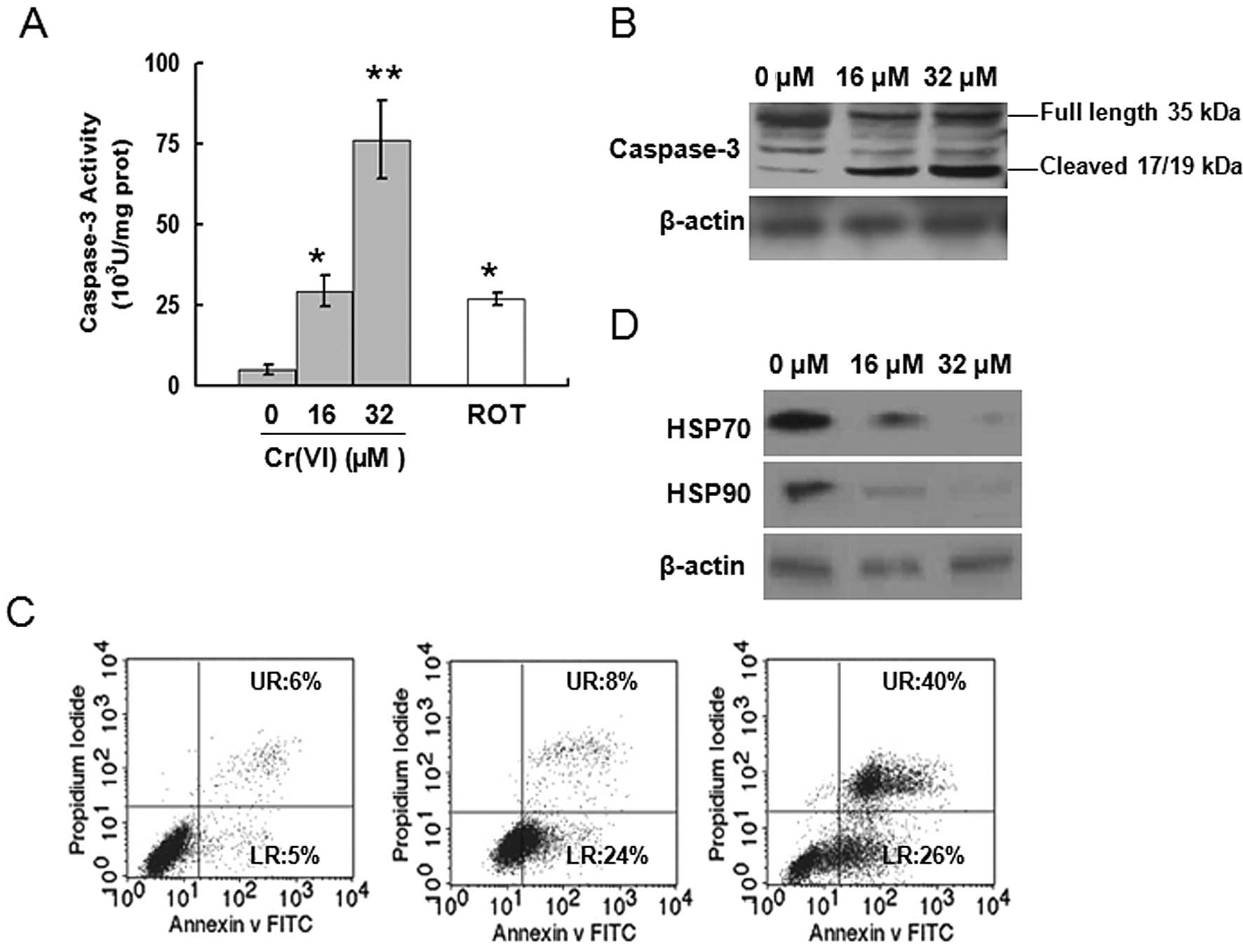
Cr(VI) activates caspase-3
Our results suggested that Cr(VI) acts as an MRCC I
inhibitor. Thus we used the specific MRCC I inhibitor rotenone
(ROT) as a positive control to confirm the functional role of
Cr(VI). ROT at 5 μM for 24 h resulted in the accumulation of
3.6-fold higher ROS compared with control (Fig. 2A and B). Caspase-3 activity was
determined after different doses of Cr(VI) exposure in L-02
hepatocytes. Similar to ROT, Cr(VI) significantly increased
caspase-3 activity in a dose-dependent manner (Fig. 4A), again confirming that Cr(VI)
targets MRCC I to induce the activation of caspase-3. Western
blotting results also revealed that Cr(VI) exposure induced the
increased expression of cleaved caspase-3 (17/19 kDa) and decreased
expressions of full length caspase-3 (35 kDa), thus leading to the
activation of caspase-3 (Fig.
4B). Occurrence of apoptosis was assayed by flow cytometry. The
results of Annexin V-FITC and PI staining showed that Cr(VI)
induced apoptosis in a dose-dependent manner (Fig. 4C). The percentages of both early
apoptotic cells (LR, as reflected in the lower-right-hand quadrant,
Annexin V positive) and late apoptotic cells (UR, depicted in the
upper-right-hand quadrant, positive for both Annexin V and PI) were
significantly increased in the Cr(VI) treatment groups compared
with control (LR, 5%; UR, 6%).
HSP has been identified as caspase-3 negative
regulator and mediates the prevention of apoptosis (22,23). Thus, we determined whether Cr(VI)
also mediated the inhibition of HSP to induce caspase-3 activation.
The levels of HSP70, HSP90 were decreased following Cr(VI)
treatment in a dose-dependent manner (Fig. 4D).
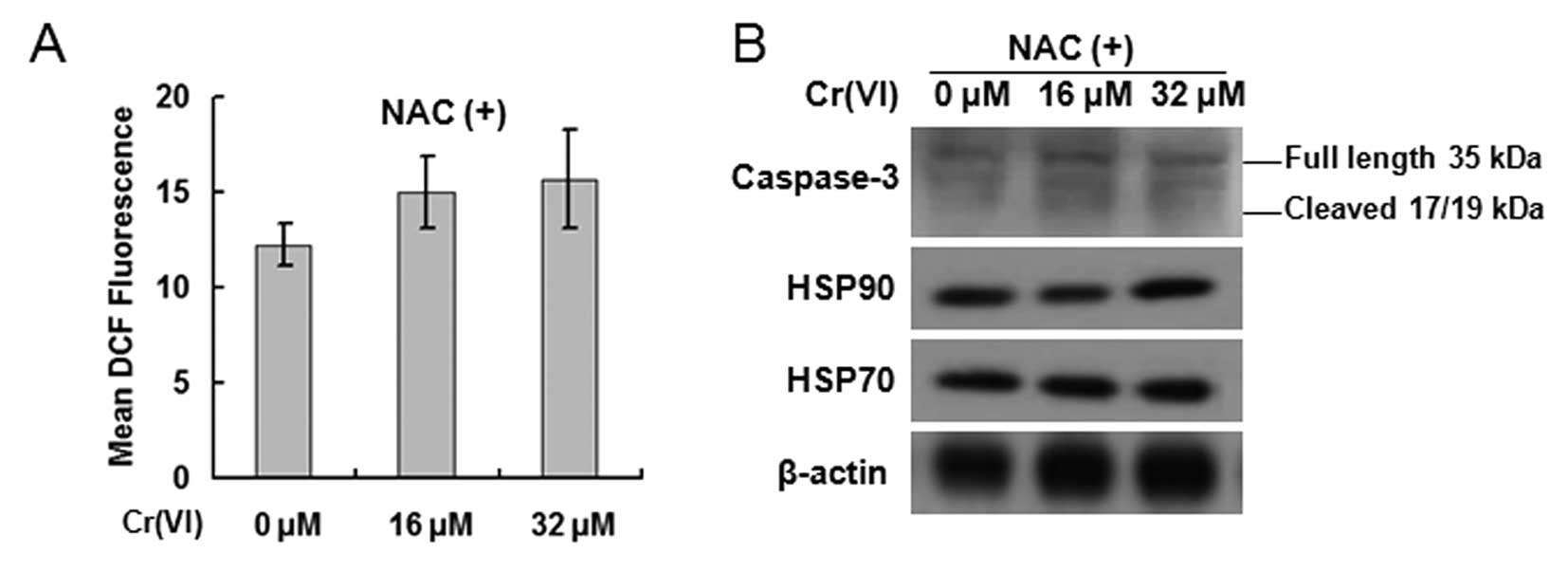
Cr(VI)-induced activation of caspase-3 is
mediated by ROS
In order to confirm that Cr(VI)-induces caspase-3
activation is dependent on ROS function, we used NAC to inhibit
ROS. The hepatocytes were exposed to Cr(VI) (16, 32 μM) in the
presence of 10 mM NAC for 24 h. The production of ROS was blocked
in each group, which confirmed the specificity of NAC (Fig. 5A). Western blot analysis revealed
that NAC blocked the activation of caspase-3 and the inhibition of
HSP70 and HSP90 (Fig. 5B),
suggesting that Cr(VI) activated caspase-3 by inducing
ROS-dependent decrease of HSP70 and HSP90. ROS is essential in
Cr(VI)-induced caspase-3 activation.
Discussion
Cr(VI) displays significant apoptosis-inducing
activity in vivo and in vitro (24,25). Although it is believed that ROS,
DNA damage, and p53 activation played important roles in
Cr(VI)-induced apoptosis, the precise targets and mechanisms remain
to be fully understood.
ROS are defined as oxygen-containing chemical
species with reactive chemical properties. MRC is the most
important source of ROS within most cells, and ROS produced from
the inhibition of MRCC are of pathological importance in a wide
variety of degenerative diseases and cancer (26). SOD, catalase and Trx are main
antioxidative proteins involved in ROS clearance. It is reported
that some chemotherapeutic agents cause ROS-dependent cytotoxicity
by downregulating the expression of the antioxidative proteins to
facilitate ROS overproduction (27,28). However, in the present study we
observed that Cr(VI) did not affect the expression levels of the
antioxidative proteins, indicating that the antioxidative system
was not involved in Cr(VI)-induced ROS accumulation. ROT is a
cytotoxic agent that has been shown to induce ROS-dependent
cytotoxicity by specifically targeting MRCC I (29). We found that after Cr(VI)
exposure, MRCC I and II were inhibited, especially the former. By
measuring the activities of MRCC I and II at different time points
after 32 μM Cr(VI) exposure, we found that the inhibition of MRCC I
activity occurred at least 90 min earlier than the inhibition of
MRCC II, indicating that MRCC I is the target of Cr(VI) in
promoting ROS production, and the decrease in MRCC II activity may
be viewed as the result of inhibited MRCC I activity. By comparing
with ROT which can also induce the activation of caspase-3, we
identified Cr(VI) as a novel MRCC I inhibitor. However, whether
Cr(VI) can directly inhibit MRCC I activity required further study.
Isolated rat liver mitochondria are also capable of reducing Cr(VI)
(20), and the reduction of
Cr(VI) has been suggested to occur at the expense of MRCC I,
interfering with the electron flow and inducing the generation of
hydroxyl radicals (HO•) via the Fenton-mechanism
(30). In the present study, by
applying different MRCC substrates as electron donors, we also
confirmed that Cr(VI) could accept the electrons leaked from MRCC I
and that the reduction occurs at MRCC I.
Caspase-3 is an executioner caspase that has
virtually no activity until it is cleaved after apoptotic signaling
events have occurred (31). It
has been suggested that ROS-dependent caspase-3 activation is
achieved by Cyt c, which can be released after the disturbance of
MRC (32). p53 can also activate
caspase-3 (33). The caspase-3
activation that was observed in this study was not p53-dependent,
as Cr(VI) can still activate caspase-3 when p53 was blocked by
Pifithrin-α (PFT-α) (data not shown). HSPs are a class of
functionally related proteins, the expression of which will be
upregulated when the cells are exposed to elevated temperatures or
other factors (34). HSP has been
shown to antagonize apoptosis-inducing factors, such as caspases-9
and -3 (35). The present study
provided the evidence that Cr(VI) can activate caspase-3 by
inducing ROS-dependent decrease of HSP70 and HSP90. In order to
confirm our hypothesis that ROS play a key role in Cr(VI)-mediated
cytotoxicity, we used NAC to inhibit the accumulation of ROS. NAC
successfully blocked the activation of caspase-3 and the inhibition
of HSP70 and HSP90, suggesting that ROS is essential in
Cr(VI)-induced caspase-3 activation.
In conclusion, we demonstrated that Cr(VI) targets
MRCC I and induces ROS accumulation, and the accumulated ROS act as
the key intermediate that downregulates HSP70, HSP90 to induces
caspase-3 activation. Therefore, in the present study, MRCC I has
been identified as a new target and a new mechanism for the
apoptosis-inducing activity displayed by Cr(VI).
Acknowledgements
We thank all of our laboratory members
for their ideas and suggestions. This study was supported by the
National Science Foundation of China (no. 30972511).
References
|
1.
|
J BarnhartOccurrences, uses, and
properties of chromiumRegul Toxicol
Pharmacol26S3S7199710.1006/rtph.1997.11329380835
|
|
2.
|
BD KergerDJ PaustenbachGE CorbettBL
FinleyAbsorption and elimination of trivalent and hexavalent
chromium in humans following ingestion of a bolus dose in drinking
waterToxicol Appl
Pharmacol141145158199610.1016/S0041-008X(96)80020-28917687
|
|
3.
|
J YeX ShiGene expression profile in
response to chromium-induced cell stress in A549 cellsMol Cell
Biochem222189197200110.1023/A:101797441505211678601
|
|
4.
|
K ChoureyMR ThompsonJ Morrell-FalveyGlobal
molecular and morphological effects of 24-hour chromium(VI)
exposure on Shewanella oneidensis MR-1Appl Environ
Microbiol7263316344200610.1128/AEM.00813-0616957260
|
|
5.
|
PH ConnettKE WetterhahnMetabolism of the
carcinogen chromate by cellular constituentsStructures and Bonding
Inorganic Elements in
BiochemistrySpringer-VerlagBerlin93124198310.1007/BFb0111319
|
|
6.
|
X ShiA ChiuCT ChenReduction of chromium
(VI) and its relationship to carcinogenesisJ Toxicol Environ Health
B Crit Rev287104199910.1080/10937409928124110081526
|
|
7.
|
P VenierA MontaldiF MajoneV BianchiAG
LevisCytotoxic, mutagenic and clastogenic effects of industrial
chromium
compoundsCarcinogenesis313311338198210.1093/carcin/3.11.13316758977
|
|
8.
|
M GunaratnamMH GrantCr (VI) inhibits DNA,
RNA and protein syntheses in hepatocytes: involvement of
glutathione reductase, reduced glutathione and DT-diaphoraseToxicol
In Vitro22879886200810.1016/j.tiv.2008.01.00518321676
|
|
9.
|
AI RafaelA AlmeidaP SantosA role for
transforming growth factor-beta apoptotic signaling pathway in
liver injury induced by ingestion of water contaminated with high
levels of Cr(VI)Toxicol Appl
Pharmacol224163173200710.1016/j.taap.2007.07.00417692352
|
|
10.
|
Z UngvariBF KrasnikovA CsiszarTesting
hypotheses of aging in long-lived mice of the genus Peromyscus:
association between longevity and mitochondrial stress resistance,
ROS detoxification pathways, and DNA repair efficiencyAge
(Dordr)30121133200810.1007/s11357-008-9059-y
|
|
11.
|
RO PoytonKA BallPR CastelloMitochondrial
generation of free radicals and hypoxic signalingTrends Endocrinol
Metab20332340200910.1016/j.tem.2009.04.00119733481
|
|
12.
|
SK YonallyRA CapaldiThe F(1)F(0) ATP
synthase and mitochondrial respiratory chain complexes are present
on the plasma membrane of an osteosarcoma cell line: An
immunocytochemical
studyMitochondrion6305314200610.1016/j.mito.2006.10.001
|
|
13.
|
N DiasC BaillyDrugs targeting
mitochondrial functions to control tumor cell growthBiochem
Pharmacol70112200510.1016/j.bcp.2005.03.02115907809
|
|
14.
|
W XuL NgoG PerezM DokmanovicPA
MarksIntrinsic apoptotic and thioredoxin pathways in human prostate
cancer cell response to histone deacetylase inhibitorProc Natl Acad
Sci USA1031554015545200610.1073/pnas.060751810317030815
|
|
15.
|
M InoueEF SatoM NishikawaMitochondrial
generation of reactive oxygen species and its role in aerobic
lifeCurr Med Chem1024952505200310.2174/092986703345647714529465
|
|
16.
|
MP WaalkesDA FoxSR PatiernoMJ McCabeMetals
and disorders of cell accumulation: modulation of apoptosis and
cell proliferationToxicol
Sci56255261200010.1093/toxsci/56.2.25510910982
|
|
17.
|
A GhelliC ZannaAM PorcelliLeber’s
hereditary optic neuropathy (LHON) pathogenic mutations induce
mitochondrial-dependent apoptotic death in transmitochondrial cells
incubated with galactose mediumJ Biol Chem278414541502003
|
|
18.
|
J Cinatl JrJ CinatlPH DrieverSodium
valproate inhibits in vivo growth of human neuroblastoma
cellsAnticancer
Drugs8958963199710.1097/00001813-199711000-000079436639
|
|
19.
|
N BrustovetskyT BrustovetskyR JemmersonJM
DubinskyCalcium-induced Cytochrome c release from CNS mitochondria
is associated with the permeability transition and rupture of the
outer membraneJ
Neurochem80207218200210.1046/j.0022-3042.2001.00671.x11902111
|
|
20.
|
D RybergJ AlexanderInhibitory action of
hexavalent chromium (Cr(VI)) on the mitochondrial respiration and a
possible coupling to the reduction of Cr(VI)Biochem
Pharmacol3324612466198410.1016/0006-2952(84)90718-46466363
|
|
21.
|
A ArilloF MelodiaR FracheReduction of
hexavalent chromium by mitochondria: methodological implications
and possible mechanismsEcotoxicol Environ
Saf14164177198710.1016/0147-6513(87)90059-53691371
|
|
22.
|
MC KamradtF ChenVL CrynsThe small heat
shock protein alpha B-crystallin negatively regulates cytochrome
cand caspase-8-dependent activation of caspase-3 by inhibiting its
autoproteolytic maturationJ Biol
Chem2761605916063200110.1074/jbc.C100107200
|
|
23.
|
JA RibeilY ZermatiJ VandekerckhoveHsp70
regulates erythropoiesis by preventing caspase-3-mediated cleavage
of GATA-1Nature445102105200710.1038/nature0537817167422
|
|
24.
|
DL CarlisleDE PritchardJ SinghSR
PatiernoChromium (VI) induces p53-dependent apoptosis in diploid
human lung and mouse dermal fibroblastsMol
Carcinog28111118200010.1002/1098-2744(200006)28:2%3C111::AID-MC7%3E3.0.CO;2-Y10900468
|
|
25.
|
XF WangML XingY ShenX ZhuLH XuOral
administration of Cr (VI) induced oxidative stress, DNA damage and
apoptotic cell death in
miceToxicology2281623200610.1016/j.tox.2006.08.00516979809
|
|
26.
|
DC WallaceMitochondrial diseases in man
and
mouseScience28314821488199910.1126/science.283.5407.148210066162
|
|
27.
|
KN IslamY KayanokiH KanetoTGF-beta1
triggers oxidative modifications and enhances apoptosis in HIT
cells through accumulation of reactive oxygen species by
suppression of catalase and glutathione peroxidaseFree Radic Biol
Med2210071017199710.1016/S0891-5849(96)00493-5
|
|
28.
|
W WangM AdachiR
KawamuraParthenolide-induced apoptosis in multiple myeloma cells
involves reactive oxygen species generation and cell sensitivity
depends on catalase
activityApoptosis1122252235200610.1007/s10495-006-0287-2
|
|
29.
|
N LiK RaghebG LawlerMitochondrial complex
I inhibitor rotenone induces apoptosis through enhancing
mitochondrial reactive oxygen species productionJ Biol
Chem27885168525200310.1074/jbc.M210432200
|
|
30.
|
XL ShiNS DalalEvidence for a Fenton-type
mechanism for the generation of .OH radicals in the reduction of
Cr(VI) in cellular mediaArch Biochem
Biophys2819095199010.1016/0003-9861(90)90417-W2166480
|
|
31.
|
J WaltersC PopFL ScottA constitutively
active and uninhibitable caspase-3 zymogen efficiently induces
apoptosisBiochem J424335345200910.1042/BJ2009082519788411
|
|
32.
|
AW Abu-QareMB Abou-DoniaBiomarkers of
apoptosis: release of cytochrome c, activation of caspase-3,
induction of 8-hydroxy-2′-deoxyguanosine, increased
3-nitrotyrosine, and alteration of p53 geneJ Toxicol Environ Health
B Crit Rev4313332200111503418
|
|
33.
|
E PaitelR FahraeusF CheclerCellular prion
protein sensitizes neurons to apoptotic stimuli through
Mdm2-regulated and p53-dependent caspase-3-like activationJ Biol
Chem2781006110066200310.1074/jbc.M21158020012529324
|
|
34.
|
A De MaioHeat shock proteins: facts,
thoughts, and dreamsShock111121999
|
|
35.
|
HM BeereBB WolfK CainHeat-shock protein 70
inhibits apoptosis by preventing recruitment of procaspase-9 to the
Apaf-1 apoptosomeNat Cell
Biol2469475200010.1038/3501950110934466
|