Introduction
Atrial fibrillation (AF) is the most common type of
sustained cardiac arrhythmia, accounting for approximately 1/3 of
hospitalizations for heart rhythm disorders (1). The prevalence of AF is approximately
1% in the general population, and is drastically increasing with
advancing age, rising from approximately 0.1% in individuals
younger than 55 years of age up to almost 10% in those aged 80
years and older (2). The lifetime
risk of developing AF is estimated to be 25% for both men and women
over 40 years of age (3). AF
confers a substantially increased risk of morbidity and mortality.
It increases the risk of stroke by 3- to 5-fold, causing a huge
economic burden on the health care system and an adverse impact on
the quality of life of patients (4). Compared to subjects with sinus
rhythm, patients with AF have a 2-fold increased risk of mortality
(5). Additionally, AF can also
result in other complications, such as adverse hemodynamics,
impaired cognitive function or dementia, congestive heart failure
and tachycardia-induced cardiomyopathy (6). AF is frequently observed as a
complication of various cardiac and systemic conditions, including
hypertension, coronary artery disease, valvular heart disease,
cardiomyopathies and metabolic disorders. Hence, AF is
traditionally regarded as an acquired disorder (1). However, in 30–45% of AF patients, an
underlying cause cannot be identified by routine procedures, and
such AF is defined as ‘idiopathic’ or ‘lone’ (1), of which at least 15% of patients
have a positive family history, so termed familial AF (7). Growing evidence has substantiated
the familial aggregation of AF and an increased susceptibility to
AF in the close relatives of patients with AF, highlighting the
fact that genetic factors play a role in the pathogenesis of AF in
a subset of cases (8–14). Studies on human and medical
genetics have revealed multiple AF-related genes, including
KCNQ1, KCNE2, SCN5A, KCNH2,
KCNJ2, GJA5, KCNA5, KCNE3,
KCNE5, NPPA, NUP155, SCN1B,
SCN2B, SCN3B and GJA1 (15–29). In addition, 4 chromosomal loci
linked to AF have been mapped, although the AF-associated genes
have not yet been discovered (30–33). Nevertheless, AF is genetically
heterogeneous and the genetic determinants for AF in most patients
remain unknown (7).
Recently, there is increasing evidence indicating
the essential role of several transcription factors, including
NKX2-5 and GATA4 in normal cardiogenesis (34–36), and mutations in NKX2–5 and GATA4
have been causally implicated in congenital cardiovascular
abnormalities and AF (37–44).
GATA6 is another member of the GATA family, and its expression and
function overlap with those of GATA4 during cardiac development,
particularly in the regulation of target gene expression
synergistically with NKX2–5 (36,45), which implies the potential
association of functionally impaired GATA6 with AF.
In this study, in order to evaluate the prevalence
of GATA6 mutations in patients with lone AF and characterize
the mechanism by which mutated GATA6 predisposes to AF, the
coding exons and exon/intron boundaries of GATA6 were
sequenced in patients with lone AF in contrast to healthy
individuals, and the functional characteristics of the mutant GATA6
were analyzed in comparison with its wild-type counterpart using a
luciferase reporter assay system.
Materials and methods
Study participants
A total of 140 unrelated patients with lone AF were
identified among the Han Chinese population. The controls were 200
ethnically-matched unrelated healthy individuals. Peripheral venous
blood samples were prepared and clinical data including medical
records, electrocardiogram and echocardiography reports were
collected. The study subjects were clinically classified using a
consistently applied set of definitions (7,43).
Briefly, the diagnosis of AF was made by a standard 12-lead
electrocardiogram demonstrating no P waves and irregular R-R
intervals regardless of the clinical symptoms. Lone AF was defined
as AF occurring in individuals <60 years of age without other
cardiac or systemic diseases by physical examination,
electrocardiogram, transthoracic echocardiogram, and extensive
laboratory tests. Familial AF was defined as the presence of
documented lone AF in 2 or more 1st- or 2nd-degree relatives.
Relatives with AF occurring at any age in the setting of structural
heart disease (hypertensive, ischemic, myocardial or valvular) were
classified as ‘undetermined’ for having an inherited form of AF.
The ‘undetermined’ classification was also used if the
documentation of AF on an electrocardiogram tracing was lacking in
relatives with symptoms consistent with AF (palpitations, dyspnea
and light-headedness), or if a screening electrocardiogram and
echocardiogram were not performed, irrespective of the symptoms.
Relatives were classified as ‘unaffected’ if they were asymptomatic
and had a normal electrocardiogram. Paroxysmal AF was defined as AF
lasting for >30 sec that terminated spontaneously. Persistent AF
was defined as AF lasting for >7 days and requiring either
pharmacological therapy or electrical cardioversion for
termination. AF that was refractory to cardioversion or that was
allowed to continue was classified as permanent. The study protocol
was reviewed and approved by the local institutional ethics
committee and written informed consent was obtained from all
participants prior to investigation.
Genetic studies
Genomic DNA from all participants was extracted from
blood lymphocytes with the Wizard® Genomic DNA
Purification kit (Promega, Madison, WI, USA). Initially, the whole
coding sequence and splice junctions of the GATA6 gene were
screened in 140 unrelated patients with lone AF. Subsequently,
genotyping of GATA6 was performed in the available relatives
of the index patient carrying an identified mutation and 200
ethnically-matched unrelated healthy individuals used as the
controls. The referential genomic DNA sequence of GATA6 was
derived from GenBank (accession no. NT_010966). With the help of
online Primer 3 software (http://frodo.wi.mit.edu), the primer pairs used to
amplify the coding exons (exons 2–7) and intron-exon boundaries of
GATA6 by polymerase chain reaction (PCR) were designed as
shown in Table I. PCR was carried
out using HotStarTaq DNA Polymerase (Qiagen, Hilden, Germany) on a
PE 9700 Thermal Cycler (Applied Biosystems, Foster City, CA, USA)
with standard conditions and concentrations of reagents. Amplified
products were purified with the QIAquick Gel Extraction kit
(Qiagen). Both strands of each PCR product were sequenced with a
BigDye® Terminator v3.1 Cycle Sequencing kit (Applied
Biosystems) under an ABI PRISM 3130XL DNA Analyzer (Applied
Biosystems). The sequencing primers were those designed previously
for specific region amplifications. DNA sequences were viewed and
analyzed with the DNA Sequencing Analysis Software v5.1 (Applied
Biosystems). The variant was validated by resequencing of an
independent PCR-generated amplicon from the subject and met our
quality control threshold with a call rate exceeding 99%.
 | Table IThe intronic primers used to amplify
the coding exons and exon-intron boundaries of GATA6. |
Table I
The intronic primers used to amplify
the coding exons and exon-intron boundaries of GATA6.
| Exon | Forward primer (5′
to 3′) | Reverse primer (5′
to 3′) | Amplicon (bp) |
|---|
| 2-a | TTG, TTA, ACC, CGT,
CGA, TCT, CC | GCG, AGG, GTC, TGG,
TAC, ATC, TC | 543 |
| 2-b | TGC, TGT, TCA, CTG,
ACC, TCG, AC | CTG, GGA, GAG, TAG,
GGG, AAG, C | 466 |
| 2-c | CCG, ACA, GCC, CTC,
CAT, ACG | GAA, AAC, AGG, GCC,
CGA, GTG | 539 |
| 3 | GGC, CAA, GGA, GAA,
AAG, CTC, AG | GTT, GGA, ACA, GCC,
GGG, ACA, G | 485 |
| 4 | TCT, TGG, CCC, AGA,
AAA, GTC, AG | TCA, TTT, GCT, GAT,
TCT, TTG, TAA, CTG | 387 |
| 5 and 6 | CTG, GGA, TTA, GAG,
GCG, TGA, GC | TTT, ACT, AGA, GAG,
CAG, CCC, AGT | 473 |
| 7 | ATT, TCT, CCT, GCC,
CTG, GGT, CT | CTG, CAC, AAA, AGC,
AGA, CAC, GA | 382 |
Alignment of multiple GATA6 protein
sequences across species
The multiple GATA6 protein sequences across various
species were aligned using the software ClustalW version 2.0 (an
interactive program at http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Plasmids and site-directed
mutagenesis
The recombinant expression plasmid pcDNA3-hGATA6 was
kindly provided by Dr Angela Edwards-Ghatnekar, from the Division
of Rheumatology and Immunology, Medical University of South
Carolina, Charleston, SC, USA. The atrial natriuretic factor
(ANF)-luciferase reporter gene, which contains the 2600-bp
5′-flanking region of the ANF gene, namely ANF(−2600)-Luc,
was kindly provided by Dr Ichiro Shiojima, from the Department of
Cardiovascular Science and Medicine, Chiba University Graduate
School of Medicine, Chiba, Japan. The identified mutation was
introduced into the wild-type GATA6 using a QuikChange II XL
Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) with
a complementary pair of primers. The mutant was sequenced to
confirm the desired mutation and to exclude any other sequence
variations.
Reporter gene assays
HEK-293 cells were cultured in Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal calf serum. The
ANF(−2600)-Luc reporter construct and an internal control reporter
plasmid [pGL4.75 (hRluc/CMV), Promega] were used in transient
transfection assays to examine the transcriptional activation
function of the GATA6 mutant. HEK-293 cells were transfected
with 0.4 μg of wild-type or mutant pcDNA3-hGATA6 expression
vector, 0.4 μg of ANF(−2600)-Luc reporter construct, and
0.04 μg of pGL4.75 control reporter vector using PolyFect
Transfection Reagent (Qiagen). For co-transfection experiments, 0.2
μg of wild-type pcDNA3-hGATA6, 0.2 μg of mutant
pcDNA3-hGATA6, 0.4 μg of ANF(−2600)-Luc and 0.04 μg
of pGL4.75 were used. Firefly luciferase and Renilla luciferase
activities were measured with the Dual-Glo® luciferase
assay system (Promega) 48 h after transfection. A minimum of 3
independent experiments were performed for wild-type and mutant
GATA6.
Statistical analysis
Data are expressed as the means ± SD. Continuous
variables were examined for normality of distribution and the
Student’s unpaired t-test was used for the comparison of numeric
variables between 2 groups. A comparison of the categorical
variables between 2 groups was performed using Pearson’s
χ2 test or Fisher’s exact test when appropriate. A
two-tailed P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Characteristics of the study
subjects
A total of 140 unrelated patients with lone AF and a
cohort of 200 ethnically-matched unrelated healthy individuals used
as the controls were recruited and clinically evaluated. None of
them had overt traditional risk factors for AF. There were no
significant differences between the patient and control groups in
baseline characteristics including age, gender, body mass index,
blood pressure, fasting blood glucose and serum lipid levels, left
atrial dimension, left ventricular ejection fraction, heart rate at
rest, as well as lifestyle (data not shown). At the time of the
present study, 12 patients were also diagnosed with hypertension in
accordance with the criterion that the average systolic or
diastolic blood pressure (2 readings made after 5 min of rest in
the sitting position) was ≥140 or ≥90 mm Hg, respectively, but at
the time of the initial diagnosis of AF, their blood pressures were
normal. The baseline clinical characteristics of the 140 patients
with lone AF are summarized in Table
II.
| Table IIThe baseline clinical characteristics
of the 140 patients with lone atrial fibrillation. |
Table II
The baseline clinical characteristics
of the 140 patients with lone atrial fibrillation.
| Parameter | No. or
quantity | Percentage or
range |
|---|
| Male | 78 | 56 |
| Age at first
diagnosis of atrial fibrillation (years) | 48.56 | 21–59 |
| Type of atrial
fibrillation at presentation | | |
| Paroxysmal | 92 | 66 |
| Persistent | 35 | 25 |
| Permanent | 13 | 9 |
| Positive family
history of atrial fibrillation | 45 | 32 |
| Underwent
cardioversion | 58 | 41 |
| Cardiac pacemaker
installed | 10 | 7 |
| Resting heart rate
(bpm) | 76.14 | 48–162 |
| Systolic blood
pressure (mmHg) | 128.04 | 92–138 |
| Diastolic blood
pressure (mmHg) | 82.36 | 65–88 |
| Body mass index
(kg/m2) | 22.24 | 20–24 |
| Left atrial
diameter (mm) | 37.90 | 31–46 |
| Left ventricular
ejection fraction (%) | 62 | 50–72 |
| Fasting blood
glucose (mmol/l) | 4.52 | 4–6 |
| Total cholesterol
(mmol/l) | 3.71 | 3–5 |
| Triglycerides
(mmol/l) | 1.24 | 1–2 |
| Medications | | |
| Amiodarone | 96 | 69 |
| Aspirin | 32 | 23 |
| Warfarin | 64 | 46 |
| β-blocker | 28 | 20 |
| Calcium channel
blocker | 17 | 12 |
| Digoxin | 35 | 25 |
GATA6 mutation
Direct sequencing of the coding exons and
exon-intron boundaries of the GATA6 gene was carried out
after PCR amplification of genomic DNA from each of the 140
patients with lone AF. A heterozygous GATA6 mutation was
identified in 1/140 unrelated patients, with a prevalence of
approximately 1.71% for GATA6 mutation. Specifically, A
substitution of T for G in the second nucleotide of codon 469
(c.1406G>T), predicting the transition of glycine (G) into
valine (V) at amino acid 469 (p.G469V) was identified in a patient
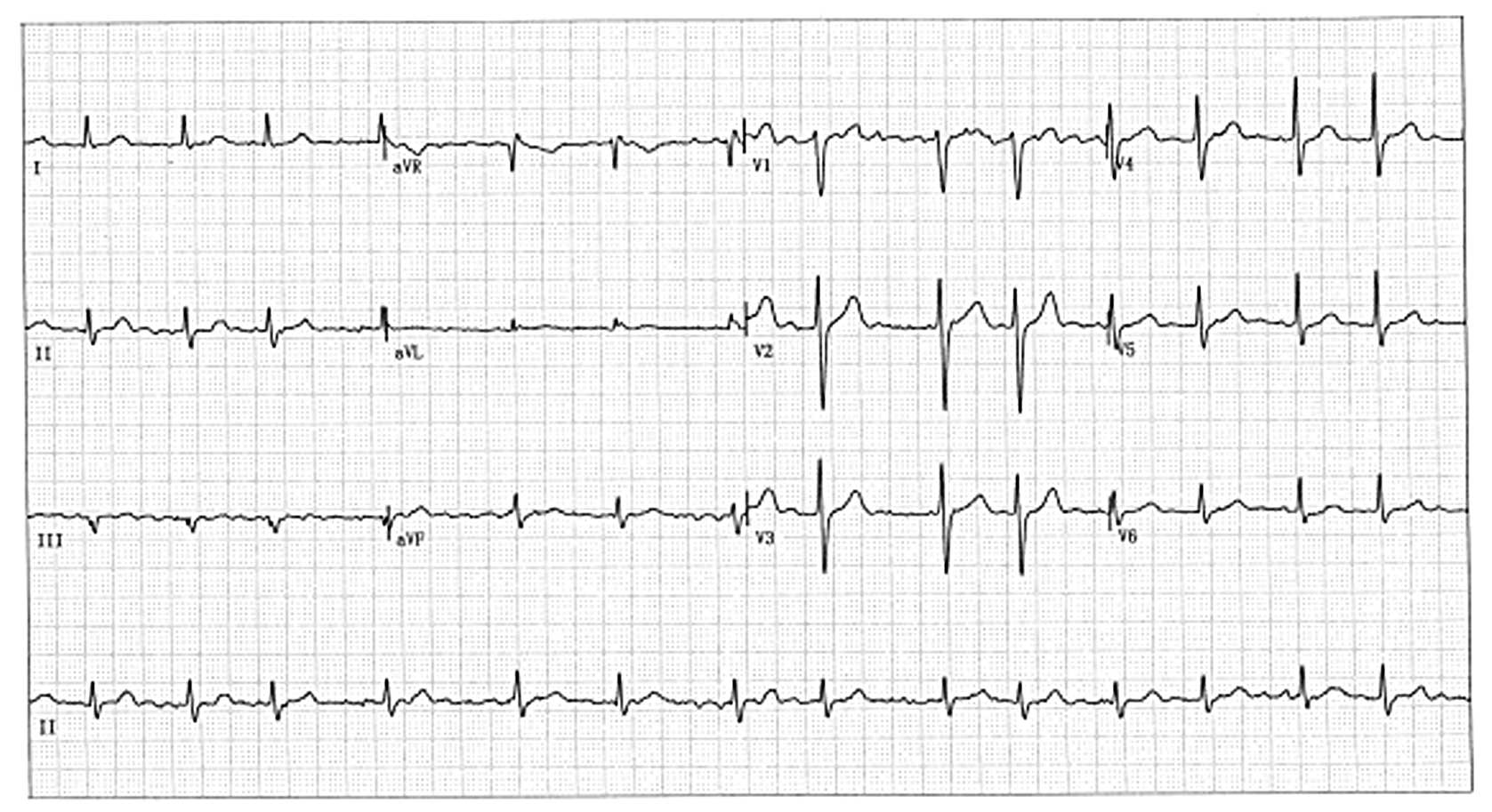
with a positive family history. A representative 12-lead
electrocardiogram of the index patient with AF is recorded in
Fig. 1. The sequence
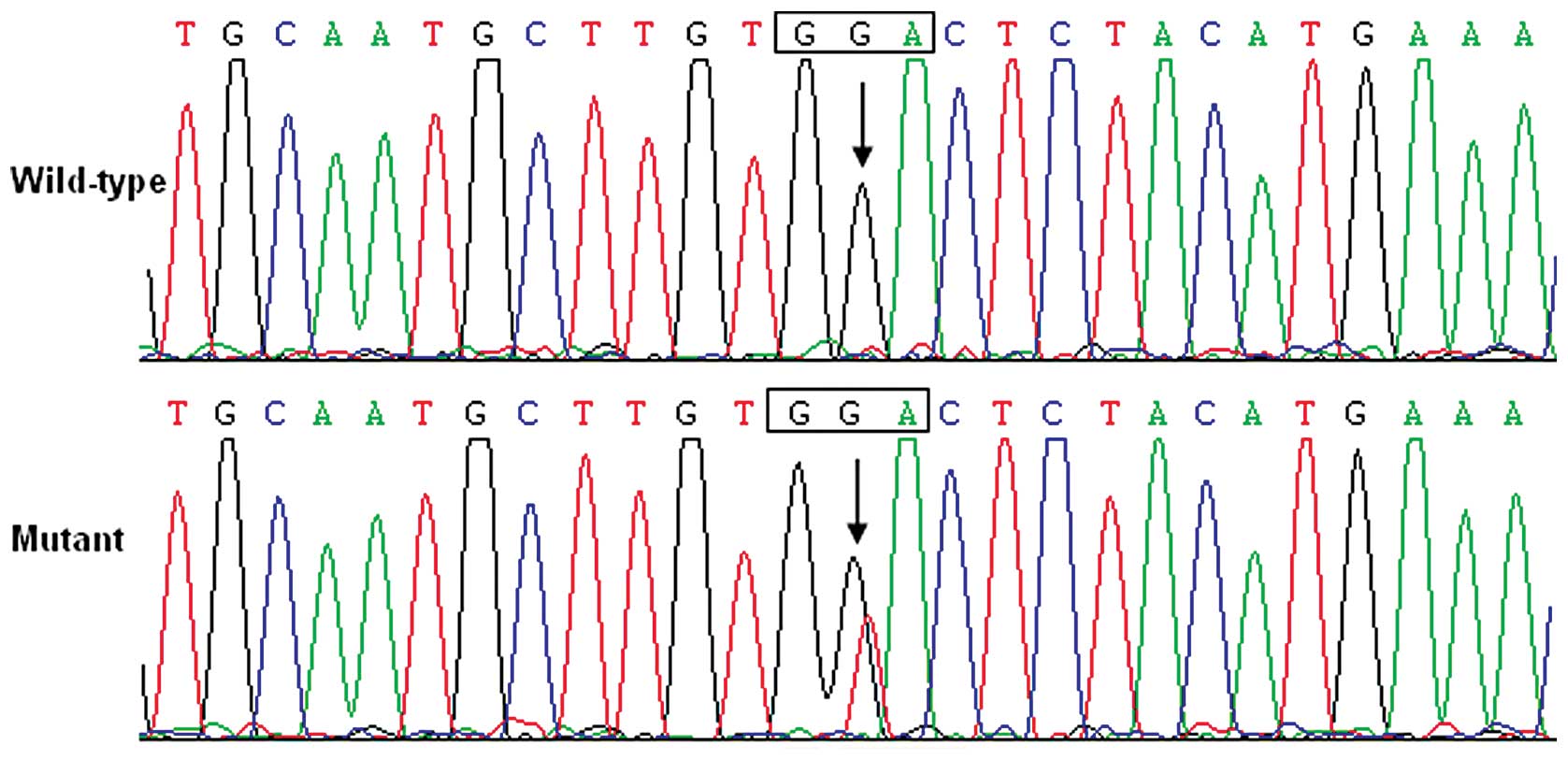
chromatograms showing the detected heterozygous GATA6
sequence mutation of c.406G>T compared with the corresponding
control sequence are shown in Fig.
2. The missense mutation was not found in either the control
population or reported in the SNP database (http://www.ncbi.nlm.nih.gov/SNP). Genetic scan of the
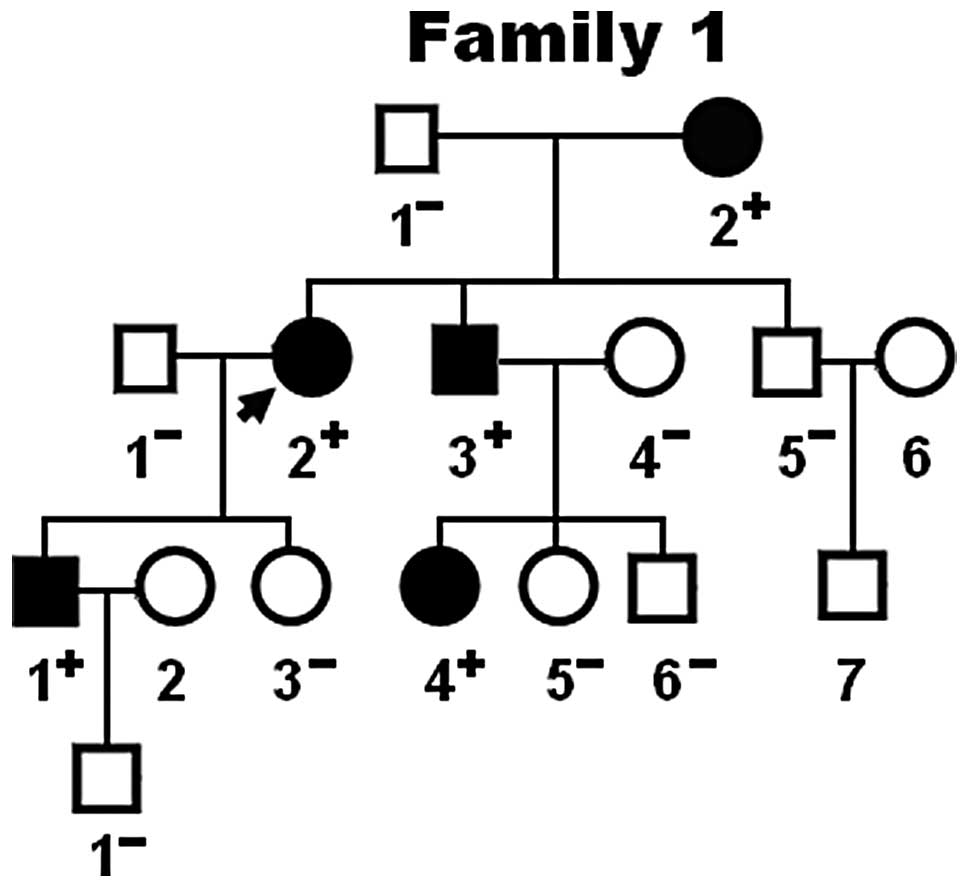
available family members displayed that the mutation was present in
all affected living family members, but absent in unaffected family
members examined. Pedigree analysis demonstrated that the mutation
cosegregated with AF transmitted in an autosomal dominant pattern
in the family with complete penetrance. The pedigree structure of
the family is illustrated in Fig.
3. The phenotypic characteristics and results of genetic
screening of the affected family members are listed in Table III. Congenital atrial septal
defect was confirmed by medical records of previous catheter-based
repairs in 2 AF patients (II-3 and III-4), indicating that AF may
share a common genetic origin with congenital heart disease.
| Table IIIPhenotypic characteristics and status
of GATA6 mutation of the affected pedigree members. |
Table III
Phenotypic characteristics and status
of GATA6 mutation of the affected pedigree members.
Subject information
| Phenotype
| Electrocardiogram
| Echocardiogram
| Genotype
|
|---|
| Identity | Gender | Age at time of
study (years) | Age at diagnosis of
AF (years) | AF
(classification) | Heart rate
(beats/min) | QRS interval
(ms) | QTc | LAD (mm) | LVEF (%) | GATA6 mutation
(G469V) |
|---|
| I-2 | F | 77 | 45 | Permanent | 67 | 90 | 441 | 42 | 65 | ± |
| II-2 | F | 54 | 40 | Persistent | 87 | 114 | 452 | 38 | 60 | ± |
| II-3 | M | 51 | 42 | Paroxysmal | 71 | 100 | 442 | 36 | 58 | ± |
| III-1 | M | 28 | 28 | Paroxysmal | 66 | 88 | 438 | 33 | 67 | ± |
| III-4 | F | 23 | 23 | Paroxysmal | 95 | 116 | 419 | 29 | 65 | ± |
Alignment of multiple GATA6 protein
sequences
A cross-species alignment of GATA6 protein sequences
showed that the altered amino acid was completely conserved, as
presented in Fig. 4, suggesting
that the amino acid is functionally important.
Transcriptional activity of the GATA4
mutants
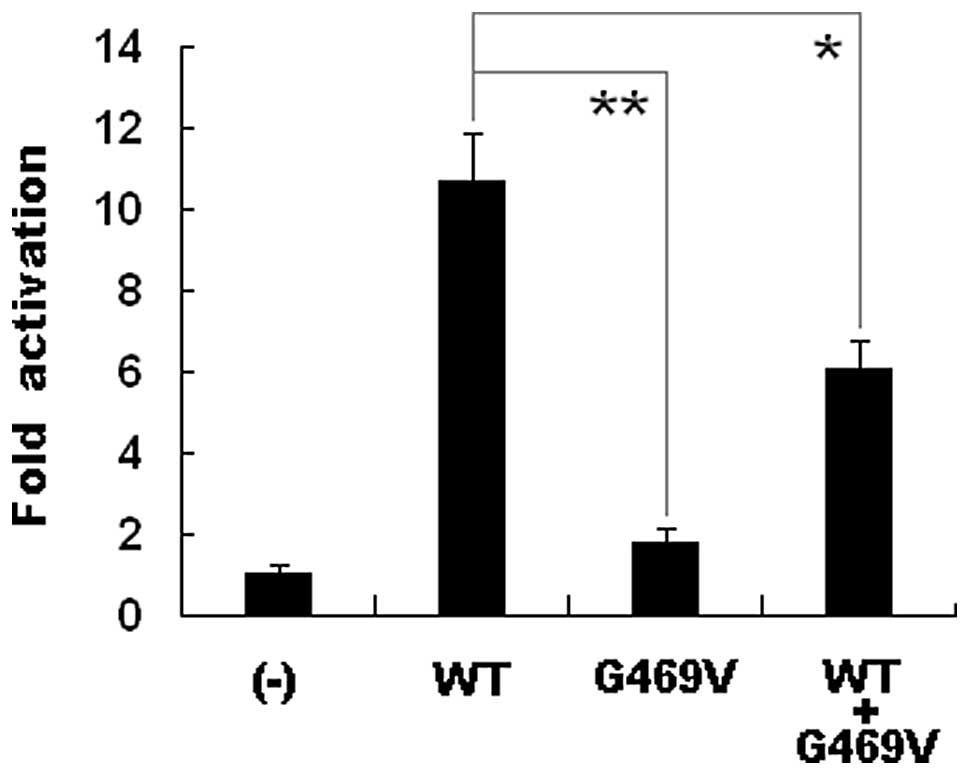
The transcriptional activation characterization of
the mutated GATA6 in HEK-293 cells was examined using one of its
direct cardiac downstream target genes, ANP, as a luciferase
reporter, and the activity of the ANP promoter was presented
as the fold activation of Firefly luciferase activity relative to
Renilla luciferase activity. Equal amounts of wild-type (0.4
μg) and G469V-mutant GATA6 (0.4 μg) activated
the ANP promoter by ∼11- and ∼2-fold, respectively. When the
same amount of wild-type GATA4 (0.2 μg) was
co-transfected with G469V-mutant GATA4 (0.2 μg), the
induced activation of the ANP promoter was ∼6-fold. These
results suggest that the GATA6 mutation has a significantly reduced
activation activity compared with its wild-type counterpart
(Fig. 5).
Discussion
In the present study, a novel heterozygous GTA6
mutation of p.G469V identified in a family with lone AF is
reported. This missense mutation of GATA6 was present in all
the affected family members examined but was absent in the
unaffected family members available and 400 normal chromosomes from
a matched control population. A cross-species alignment of multiple
GATA6 protein sequences exhibited that the altered amino acid was
completely conserved. Functional analysis demonstrated that the
p.G469V mutation of GATA6 was associated with a significantly
decreased transcriptional activity. Therefore, it is highly likely
that functionally impaired GATA6 is involved in the pathogenesis of
AF in this family. To our knowledge, this is the first description
of the correlation between a GATA6 loss-of-function mutation and
susceptibility to familial AF. These results expand the spectrum of
GATA6 mutations linked to AF and provide significant insight into
the molecular basis underlying AF.
GATA transcription factors are a group of DNA
binding proteins characteristic of preferential binding to the
consensus DNA sequence GATA of target gene promoters. The GATA
family comprises 6 members (GATA1–GATA6), of which GATA4, GATA5 and
GATA6 are expressed in various mesoderm and endoderm-derived
tissues, particularly in the embryonic and adult heart (36). GATA6 maps to human chromosome
18q11.1–q11.2 by fluorescence in situ hybridization, which
encodes a predicted 449-amino-acid protein (46). Functionally, GATA6 consists of 2
transcriptional activation domains (TADs), 2 adjacent zinc fingers
(ZFs), and 1 nuclear localization signal (NLS). The 2 TADs are
essential for the transcriptional activity of GATA6. The C-terminal
ZF is required for DNA sequence recognition and binding to the
consensus motif, while the N-terminal ZF is responsible for
stability and sequence specificity of protein-DNA binding as well
as the transcriptional activation by GATA factors. The majority of
the protein-protein interactions of GATA factors are mediated by
its C-terminal ZF. The NLS sequence is associated with the
subcellular trafficking and distribution of GATA6 (36). The GATA6 mutation, p.G469V,
identified in this study is located in the C-terminal ZF;
therefore, it may be expected to exert influence on the
transcriptional activation of GATA6 by interfering with the binding
to the downstream target DNA.
It has been substantiated that GATA6 is an upstream
regulator of multiple genes transcribed during embryogenesis and
cardiac morphogenesis, including the genes that encode the atrial
natriuretic peptide (ANP), brain natriuretic peptide, α-myosin
heavy chain, β-myosin heavy chain and cardiac troponin C and I
(36). Hence, the functional
characteristics of the GATA6 mutations may be explored by
analysis of the transcriptional activity of the ANP promoter
in cells transfected with the GATA6 mutant in contrast to
its wild-type counterpart. In this study, the functional effect of
the novel p.G469V mutation of GATA6 identified in our familial AF
patients was characterized by transcriptional activity assays and
the results demonstrated a significantly decreased transcriptional
activity on a downstream gene, consistent with the loss-of-function
effects of other GATA6 mutations underlying congenital
cardiovascular malformations on the transcriptional activity of the
ANP promoter (47,48). These findings indicate that the
haploinsufficiency or loss-of-function effect resulting from GATA6
mutations is potentially an important pathophysiological mechanism
involved in AF, although the functional roles of the recently
reported AF-related GATA6 mutations remain to be ascertained
(49).
The findings that functionally compromised GATA6
confers susceptibility to AF may be partially ascribed to the
abnormally developed pulmonary vein myocardium (50–52). The pulmonary venous vessels are
ensheathed by a layer of myocardium termed pulmonary myocardial
sleeve, which has been demonstrated to be responsible for the
initiation and perpetuation of AF by several potential
arrhythmogenic mechanisms, including intrinsic pacemaker activity
and properties facilitating re-entrance (50–52). Genetic labeling lineage-tracing
studies have shown that NKX2-5 is expressed in the atrial and
pulmonary myocardium and is essential for the localized formation
of the sinoatrial node during embryonic development. NKX2-5 can act
as a suppressor of the sinoatrial node lineage gene program, which
limits pacemaker activity to the sinus and atrioventricular node.
When the NKX2-5 protein level was lowered in a hypomorphic model,
the pulmonary cardiomyocytes switched to connexin40-negative,
HCN4-positive cells, a nodal-like phenotype with pacemaker activity
(51). In NKX2-5-deficient
mouse embryos, HCN4 was activated in the entire embryonic
heart tube, whereas connexin40 expression was lost, and the
ectopic expression of pacemaker cells was observed throughout the
heart tube (53). In humans, AF
has been reported as an isolated phenotype or a part of compound
phenotypes in patients harboring NKX2-5 mutations (40,54,55). Therefore, as a transcriptionally
cooperative partner of NKX2-5, GATA6, when a
loss-of-function mutation occurs, may conduce to the formation of
the pulmonary myocardium sleeve and the shift of the pulmonary
myocardium to a sinoatrial node-like phenotype by reducing the
expression of NKX2-5, hence producing an atrial
electrophysiological substrate favoring AF.
There are a large number of downstream genes
transactivated by GATA6, and mutations in several target genes have
been associated with AF, including β-myosin heavy chain, ANP and
connexin40 (24,25,56–58). Therefore, it is highly likely for
mutated GATA6 to predispose to AF by decreasing the expression of
target genes.
Of note, congenital atrial septal defect was
documented in 2 AF patients carrying the p.G469V mutation of GATA6.
Similar to our current findings, congenital cardiac defects were
previously confirmed in 3 AF patients carrying heterozygous GATA6
mutations, including atrial septal defect in 1 missense mutation
(p.Q206P) carrier and ventricular septal defect in 2 truncation
mutation (p.Y265X) carriers (49). These observations suggest that AF
may share a common genetic origin with congenital heart
disease.
In conclusion, the results from the present study
link a novel mutation in the cardiac transcription factor, GATA6,
to familial AF and provides novel insight into the molecular
mechanism involved in AF, as well as insight into potential
therapies for the prevention and treatment of this common type of
arrhythmia.
Acknowledgements
We are indebted to participants for
their dedication to the study. This study was supported in part by
grants from the National Natural Science Fund of China (81070153
and 30570768), the National Basic Research Program of China
(2010CB912604), the Personnel Development Foundation of Shanghai,
China (2010019), the Key Program of Basic Research of Shanghai,
China (10JC1414000, 10JC1414001 and 10JC1414002) and the Natural
Science Fund of Shanghai, China (10ZR1428000).
References
|
1.
|
V FusterLE RydénDS CannomHJ CrijnsAB
CurtisKA EllenbogenJL HalperinGN KayJY Le HuezeyJE LoweAmerican
College of Cardiology Foundation/American Heart Association Task
Force2011 ACCF/AHA/HRS focused updates incorporated into the
ACC/AHA/ESC 2006 guidelines for the management of patients with
atrial fibrillation: a report of the American College of Cardiology
Foundation/American Heart Association Task Force on practice
guidelinesCirculation123e269e3672011
|
|
2.
|
AS GoEM HylekKA PhillipsY ChangLE
HenaultJV SelbyDE SingerPrevalence of diagnosed atrial fibrillation
in adults: national implications for rhythm management and stroke
prevention: the AnTicoagulation and Risk Factors in Atrial
Fibrillation (ATRIA)
StudyJAMA28523702375200110.1001/jama.285.18.2370
|
|
3.
|
DM Lloyd-JonesTJ WangEP LeipMG LarsonD
LevyRS VasanRB D’AgostinoJM MassaroA BeiserPA WolfEJ
BenjaminLifetime risk for development of atrial fibrillation: the
Framingham Heart
StudyCirculation11010421046200410.1161/01.CIR.0000140263.20897.4215313941
|
|
4.
|
PA WolfRD AbbottWB KannelAtrial
fibrillation as an independent risk factor for stroke: the
Framingham
StudyStroke22983988199110.1161/01.STR.22.8.9831866765
|
|
5.
|
EJ BenjaminPA WolfRB D’AgostinoH
SilbershatzWB KannelD LevyImpact of atrial fibrillation on the risk
of death: the Framingham Heart
StudyCirculation98946952199810.1161/01.CIR.98.10.9469737513
|
|
6.
|
JW MagnaniM RienstraH LinMF SinnerSA
LubitzDD McManusJ DupuisPT EllinorEJ BenjaminAtrial fibrillation:
current knowledge and future directions in epidemiology and
genomicsCirculation12419821993201110.1161/CIRCULATIONAHA.111.03967722042927
|
|
7.
|
D DarbarKJ HerronJD BallewA JahangirBJ
GershWK ShenSC HammillDL PackerTM OlsonFamilial atrial fibrillation
is a genetically heterogeneous disorderJ Am Coll
Cardiol4121852192200310.1016/S0735-1097(03)00465-012821245
|
|
8.
|
PT EllinorDM YoergerJN RuskinCA
MacRaeFamilial aggregation in lone atrial fibrillationHum
Genet118179184200510.1007/s00439-005-0034-816133178
|
|
9.
|
DO ArnarS ThorvaldssonTA ManolioG
ThorgeirssonK KristjanssonH HakonarsonK StefanssonFamilial
aggregation of atrial fibrillation in IcelandEur Heart
J27708712200610.1093/eurheartj/ehi72716428254
|
|
10.
|
MJ JunttilaMJ RaatikainenJS PerkiömäkiK
HongR BrugadaHV HuikuriFamilial clustering of lone atrial
fibrillation in patients with saddleback-type ST-segment elevation
in right precordial leadsEur Heart
J28463468200710.1093/eurheartj/ehl47417242012
|
|
11.
|
IE ChristophersenLS RavnE
Budtz-JoergensenA SkyttheS HaunsoeJH SvendsenK ChristensenFamilial
aggregation of atrial fibrillation: a study in Danish twinsCirc
Arrhythm
Electrophysiol2378383200910.1161/CIRCEP.108.78666519808493
|
|
12.
|
YQ YangXL ZhangXH WangHW TanHF ShiWY FangX
LiuFamilial aggregation of lone atrial fibrillation in the Chinese
populationIntern
Med4923852391201010.2169/internalmedicine.49.413021088338
|
|
13.
|
SA LubitzX YinJD FontesJW MagnaniM
RienstraM PaiML VillalonRS VasanMJ PencinaD LevyAssociation between
familial atrial fibrillation and risk of new-onset atrial
fibrillationJAMA30422632269201010.1001/jama.2010.169021076174
|
|
14.
|
CS FoxH PariseRB D’Agostino SrDM
Lloyd-JonesRS VasanTJ WangD LevyPA WolfEJ BenjaminParental atrial
fibrillation as a risk factor for atrial fibrillation in
offspringJAMA29128512855200410.1001/jama.291.23.285115199036
|
|
15.
|
YH ChenSJ XuS BendahhouXL WangY WangWY
XuHW JinH SunXY SuQN ZhuangKCNQ1 gain-of-function mutation in
familial atrial
fibrillationScience299251254200310.1126/science.107777112522251
|
|
16.
|
Y YangM XiaQ JinS BendahhouJ ShiY ChenB
LiangJ LinY LiuB LiuIdentification of a KCNE2 gain-of-function
mutation in patients with familial atrial fibrillationAm J Hum
Genet75899905200410.1086/42534215368194
|
|
17.
|
TM OlsonVV MichelsJD BallewSP ReynaML
KarstKJ HerronSC HortonRJ RodehefferJL AndersonSodium channel
mutations and susceptibility to heart failure and atrial
fibrillationJAMA293447454200510.1001/jama.293.4.44715671429
|
|
18.
|
K HongP BjerregaardI GussakR BrugadaShort
QT syndrome and atrial fibrillation caused by mutation in KCNH2J
Cardiovasc
Electrophysiol16394396200510.1046/j.1540-8167.2005.40621.x15828882
|
|
19.
|
M XiaQ JinS BendahhouY HeMM LarroqueY
ChenQ ZhouY YangY LiuB LiuA Kir2.1 gain-of-function mutation
underlies familial atrial fibrillationBiochem Biophys Res
Commun33210121019200510.1016/j.bbrc.2005.05.05415922306
|
|
20.
|
MH GollobDL JonesAD KrahnL DanisXQ GongQ
ShaoX LiuJP VeinotAS TangAF StewartSomatic mutations in the
connexin 40 gene (GJA5) in atrial fibrillationN Engl J
Med35426772688200610.1056/NEJMoa05280016790700
|
|
21.
|
TM OlsonAE AlekseevXK LiuS ParkLV ZingmanM
BienengraeberS SattirajuJD BallewA JahangirA TerzicKv1.5
channelopathy due to KCNA5 loss-of-function mutation causes human
atrial fibrillationHum Mol
Genet1521852191200610.1093/hmg/ddl14316772329
|
|
22.
|
A LundbyLS RavnJH SvendsenS HaunsSP
OlesenN SchmittKCNE3 mutation V17M identified in a patient with
lone atrial fibrillationCell Physiol
Biochem214754200810.1159/00011374618209471
|
|
23.
|
LS RavnY AizawaGD PollevickJ Hofman-BangJM
CordeiroU DixenG JensenY WuE BurashnikovS HaunsoGain of function in
IKs secondary to a mutation in KCNE5 associated with atrial
fibrillationHeart
Rhythm5427435200810.1016/j.hrthm.2007.12.01918313602
|
|
24.
|
DM Hodgson-ZingmanML KarstLV ZingmanDM
HeubleinD DarbarKJ HerronJD BallewM de AndradeJC Burnett JrTM
OlsonAtrial natriuretic peptide frameshift mutation in familial
atrial fibrillationN Engl J
Med359158165200810.1056/NEJMoa070630018614783
|
|
25.
|
X RenC XuC ZhanY YangL ShiF WangC WangY
XiaB YangG WuIdentification of NPPA variants associated with atrial
fibrillation in a Chinese GeneID populationClin Chim
Acta411481485201010.1016/j.cca.2009.12.01920064500
|
|
26.
|
X ZhangS ChenS YooS ChakrabartiT ZhangT
KeC ObertiSL YongF FangL LiMutation in nuclear pore component
NUP155 leads to atrial fibrillation and early sudden cardiac
deathCell13510171027200810.1016/j.cell.2008.10.02219070573
|
|
27.
|
H WatanabeD DarbarDW KaiserK
JiramongkolchaiS ChopraBS DonahuePJ KannankerilDM RodenMutations in
sodium channel beta1- and beta2-subunits associated with atrial
fibrillationCirc Arrhythm
Electrophysiol2268275200910.1161/CIRCEP.108.77918119808477
|
|
28.
|
P WangQ YangX WuY YangL ShiC WangG WuY
XiaB YangR ZhangFunctional dominant-negative mutation of sodium
channel subunit gene SCN3B associated with atrial fibrillation in a
Chinese GeneID populationBiochem Biophys Res
Commun39898104201010.1016/j.bbrc.2010.06.04220558140
|
|
29.
|
IL ThibodeauJ XuQ LiG LiuK LamJP VeinotDH
BirnieDL JonesAD KrahnR LemeryParadigm of genetic mosaicism and
lone atrial fibrillation: physiological characterization of a
connexin 43-deletion mutant identified from atrial
tissueCirculation122236244201010.1161/CIRCULATIONAHA.110.961227
|
|
30.
|
R BrugadaT TapscottGZ CzernuszewiczAJ
MarianA IglesiasL MontJ BrugadaJ GironaA DomingoLL BachinskiR
RobertsIdentification of a genetic locus for familial atrial
fibrillationN Engl J
Med336905911199710.1056/NEJM1997032733613029070470
|
|
31.
|
PT EllinorJT ShinRK MooreDM YoergerCA
MacRaeLocus for atrial fibrillation maps to chromosome
6q14–16Circulation10728802883200312782570
|
|
32.
|
C ObertiL WangL LiJ DongS RaoW DuQ
WangGenome-wide linkage scan identifies a novel genetic locus on
chromosome 5p13 for neonatal atrial fibrillation associated with
sudden death and variable
cardiomyopathyCirculation11037533759200410.1161/01.CIR.0000150333.87176.C7
|
|
33.
|
PG VoldersQ ZhuC TimmermansPM EurlingsX
SuYH ArensL LiRJ JongbloedM XiaLM RodriguezYH ChenMapping a novel
locus for familial atrial fibrillation on chromosome10p11–q21Heart
Rhythm44694752007
|
|
34.
|
DL HuFK ChenYQ LiuYH ShengR YangXQ KongKJ
CaoHT GuLM QianGATA-4 promotes the differentiation of P19 cells
into cardiac myocytesInt J Mol Med26365372201020664952
|
|
35.
|
BG BruneauThe developmental genetics of
congenital heart
diseaseNature451943948200810.1038/nature0680118288184
|
|
36.
|
S PikkarainenH TokolaR KerkeläH
RuskoahoGATA transcription factors in the developing and adult
heartCardiovasc
Res63196207200410.1016/j.cardiores.2004.03.02515249177
|
|
37.
|
J WangM FangXY LiuYF XinZM LiuXZ ChenXZ
WangWY FangX LiuYQ YangA novel GATA4 mutation responsible for
congenital ventricular septal defectsInt J Mol
Med28557564201121637914
|
|
38.
|
XY LiuJ WangJH ZhengK BaiZM LiuXZ WangX
LiuWY FangYQ YangInvolvement of a novel GATA4 mutation in atrial
septal defectsInt J Mol Med281723201121373748
|
|
39.
|
J WangYF XinXY LiuZM LiuXZ WangYQ YangA
novel NKX2-5 mutation in familial ventricular septal defectInt J
Mol Med27369375201121165553
|
|
40.
|
I Gutierrez-RoelensL De RoyC OvaertT
SluysmansK DevriendtHG BrunnerM VikkulaA novel CSX/NKX2-5 mutation
causes autosomal-dominant AV block: are atrial fibrillation and
syncopes part of the phenotype?Eur J Hum
Genet1413131316200610.1038/sj.ejhg.520170216896344
|
|
41.
|
LH BoldtMG PoschA PerrotM PolotzkiS RolfAS
ParwaniM HuemerA WutzlerC OzcelikW HaverkampMutational analysis of
the PITX2 and NKX2–5 genes in patients with idiopathic atrial
fibrillationInt J Cardiol145316317201020022124
|
|
42.
|
MG PoschLH BoldtM PolotzkiS RichterS RolfA
PerrotR DietzC OzcelikW HaverkampMutations in the cardiac
transcription factor GATA4 in patients with lone atrial
fibrillationEur J Med
Genet53201203201010.1016/j.ejmg.2010.03.00820363377
|
|
43.
|
YQ YangMY WangXL ZhangHW TanHF ShiWF
JiangXH WangWY FangX LiuGATA4 loss-of-function mutations in
familial atrial fibrillationClin Chim
Acta41218251830201110.1016/j.cca.2011.06.01721708142
|
|
44.
|
JQ JiangFF ShenWY FangX LiuYQ YangNovel
GATA4 mutations in lone atrial fibrillationInt J Mol
Med2810251032201121874226
|
|
45.
|
Y ZhangN RathS HannenhalliZ WangT CappolaS
KimuraE Atochina-VassermanMM LuMF BeersEE MorriseyGATA and Nkx
factors synergistically regulate tissue-specific gene expression
and development in
vivoDevelopment134189198200710.1242/dev.0272017164424
|
|
46.
|
E SuzukiT EvansJ LowryL TruongDW BellJR
TestaK WalshThe human GATA-6 gene: structure, chromosomal location,
and regulation of expression by tissue-specific and
mitogen-responsive
signalsGenomics38283290199610.1006/geno.1996.06308975704
|
|
47.
|
X LinZ HuoX LiuY ZhangL LiH ZhaoB YanY
LiuY YangYH ChenA novel GATA6 mutation in patients with tetralogy
of Fallot or atrial septal defectJ Hum
Genet55662667201010.1038/jhg.2010.84
|
|
48.
|
GF ZhengD WeiH ZhaoN ZhouYQ YangXY LiuA
novel GATA6 mutation associated with congenital ventricular septal
defectInt J Mol Med2910651071201222407241
|
|
49.
|
YQ YangXH WangHW TanWF JiangWY FangX
LiuPrevalence and spectrum of GATA6 mutations associated with
familial atrial fibrillationInt J
Cardiol155494496201210.1016/j.ijcard.2011.12.09122257684
|
|
50.
|
M HaïssaguerreP JaïsDC ShahA TakahashiM
HociniG QuiniouS GarrigueA Le MourouxP Le MétayerJ
ClémentySpontaneous initiation of atrial fibrillation by ectopic
beats originating in the pulmonary veinsN Engl J
Med33965966619989725923
|
|
51.
|
MT MommersteegNA BrownOW PrallC de Gier-de
VriesRP HarveyAF MoormanVM ChristoffelsPitx2c and Nkx2–5 are
required for the formation and identity of the pulmonary
myocardiumCirc Res1019029092007
|
|
52.
|
MT MommersteegVM ChristoffelsRH AndersonAF
MoormanAtrial fibrillation: a developmental point of viewHeart
Rhythm618181824200910.1016/j.hrthm.2009.07.01119726237
|
|
53.
|
MT MommersteegWM HoogaarsOW PrallC de
Gier-de VriesC WieseDE CloutVE PapaioannouNA BrownRP HarveyAF
MoormanVM ChristoffelsMolecular pathway for the localized formation
of the sinoatrial nodeCirc
Res100354362200710.1161/01.RES.0000258019.74591.b317234970
|
|
54.
|
Y WatanabeDW BensonS YanoT AkagiM
YoshinoJC MurrayTwo novel frameshift mutations in NKX2.5 result in
novel features including visceral inversus and sinus venosus type
ASDJ Med Genet39807811200210.1136/jmg.39.11.80712414819
|
|
55.
|
S PabstB WollnikE RohmannY HintzK GlänzerH
VetterG NickenigC GrohéA novel stop mutation truncating critical
regions of the cardiac transcription factor NKX2-5 in a large
family with autosomal-dominant inherited congenital heart
diseaseClin Res Cardiol973942200810.1007/s00392-007-0574-0
|
|
56.
|
EJ GruverD FatkinGA DoddsJ KissloBJ
MaronJG SeidmanCE SeidmanFamilial hypertrophic cardiomyopathy and
atrial fibrillation caused by Arg663His beta-cardiac myosin heavy
chain mutationAm J
Cardiol8313H18H199910.1016/S0002-9149(99)00251-910750581
|
|
57.
|
YQ YangX LiuXL ZhangXH WangHW TanHF ShiWF
JiangWY FangNovel connexin40 missense mutations in patients with
familial atrial
fibrillationEuropace1214211427201010.1093/europace/euq27420650941
|
|
58.
|
YQ YangXL ZhangXH WangHW TanHF ShiWF
JiangWY FangX LiuConnexin40 nonsense mutation in familial atrial
fibrillationInt J Mol Med26605610201020818502
|