Introduction
Status epilepticus (SE) in adult rodents and humans
can cause hippocampal neuronal loss and may result in temporal lobe
epilepsy (TLE) (1–3). Neuronal loss during chronic epilepsy
mainly results from cell apoptosis or necrosis (4–6),
which is triggered by the activation of a number of signal
transduction factors (7). Kainic
acid (KA), an analogue of the excitatory amino acid glutamate, has
been widely used in inducing TLE in animal models (8,9).
KA-induced SE in the amygdaloid complex activates several signal
transduction factors, including B-cell leukemia-2 (Bcl-2),
Bcl-2-associated X protein (Bax), and cysteinyl aspartate-specific
protease-3 (caspase-3) (10,11). In the KA model, deletion or
inhibition of pro-apoptotic genes protects the brain against
seizure-induced neuronal death (12,13). Since currently available
anti-epileptic drugs merely treat symptoms but do not cure the
disease, it is imperative to develop neuroprotective drugs that
prevent apoptosis after SE.
Statins, inhibitors of HMG-CoA reductase, inhibit
cellular synthesis of cholesterol and isoprenoids and are commonly
used to reduce cholesterol levels in humans. In addition to their
lipid-lowering and thus beneficial cardiovascular effects, statins
have also been suggested to exert neuroprotective actions in the
central nervous system. For example, statins play anti-inflammatory
and vasoprotective roles in cultured brain cells and endothelial
cells (14). Statins inhibit a
number of inflammatory processes important to brain damage and
suppress the secretion of cytokines during spinal cord injury and
ischemic stroke (15,16). The neuroprotective effects of
statins have also been reported in various diseases, including
traumatic brain injury, brain ischemia, and Alzheimer’s disease, in
both animal models and clinical studies (17–20). In KA-induced seizure, treatment
with statins provides anti-apoptotic effects (21). However, the mechanisms underlying
the anti-apoptotic role are not clear. In this study, we examined
whether simvastatin regulates apoptosis and exerts its
neuroprotective effects by modulating the expression of Bcl-2 and
caspase-3.
Materials and methods
Ethics statement
Animal care and handling was conducted in compliance
with the Chinese Animal Welfare Act and was approved by the Medical
Ethics Committee of the First Clinical College of Harbin Medical
University (Approval ID, 201001).
Animals
Adult male Wistar rats (n=120), weighing 180–200 g,
were provided by the Animal Center of Jilin University, China. The
rats were housed in individual cages in a controlled environment
(constant 22–25°C; 50–60% humidity; 12/12 h light/dark cycle,
lights on at 7 a.m.) for at least 1 week before being used in the
experiment. The rats had free access to standard laboratory food
and water. In addition, all efforts were made to minimize animal
suffering and to use only the number of animals necessary to
produce reliable scientific data. All the experiments were
conducted in the morning to avoid circadian variations.
Experimental groups and drug
administration
The rats were randomly divided into 4 groups (n=30
per group): a saline group (sham), an epilepsy group, an epilepsy
plus saline group, and an epilepsy plus simvastatin group. In the
sham group, rats received saline intraperitoneally. In the epilepsy
group, KA was dissolved in isotonic saline (pH 7.3) and
administered intraperitoneally to rats at a dose of 10 mg/kg. In
the epilepsy plus saline group, SE were induced in rats with KA
injection followed by oral administration of saline starting at 0.5
h after SE once a day for 3 consecutive days. In the epilepsy plus
simvastatin group, rats were subjected to KA lesions followed by
oral administration of simvastatin (1 mg/kg/day)
(Zocor®; MSD, USA), starting at 0.5 h after SE for 3
consecutive days. The dose was selected according to our previous
study (22,23). The rats were sacrificed at
indicated time points after SE or sham operation.
KA-induced rat seizure model
SE was induced in rats using KA administration.
After KA was administered, the behavior of rats was observed for
3–4 h and documented to determine the duration and severity of
seizure activity using a previously established seizure scoring
scale (24). This method was
widely used to study epilepsy in rodents. The behavior of rats was
divided into 5 stages: stage 1, immobility; stage 2, forelimb
and/or tail extension; stage 3, head bombing, as well as forelimb
clonus with rearing and falling; stage 4, minor clonic seizures;
stage 5, severe tonic-clonic seizures; and stage 6, death. Only
those rats exhibiting at least 2 h of continuous stage 4/5 seizures
were included in this study. Seizure parameters monitored included
latency of convulsions and duration of severe (stage 4/5) seizure
activity.
Histological analysis
Neuronal damage was assessed by histological
examination of brain sections from the dorsal hippocampus of rats
sacrificed at 6, 12, 24, 48, 72 or 96 h after SE or sham operation.
The rats were deeply anesthetized with 4% halothane and then
transcardially perfused with 200 ml of heparinized 0.9% saline
followed by 500 ml of 4% paraformaldehyde (no. 158127;
Sigma-Aldrich, Beijing, China) in 0.1 mol/l of phosphate-buffered
saline (pH 7.4) (P5368; Sigma-Aldrich). After rats were
decapitated, the brains were immersed in 4% paraformaldehyde for 3
days, embedded in paraffin, and sliced on a rotary microtome into
6-mm thick sections. Coronal sections consisting of the dorsal
hippocampus were selected and processed for hematoxylin and eosin
(H&E) staining. The paraffin-embedded sections were dewaxed by
baking at 55–65°C for 45–60 min, cleaned with xylene three times
for 5 min each, and rehydrated with 100% ethanol twice for 5 min
and 70% ethanol once for 5 min. After 5-min washes in distilled
water, the sections were stained with hematoxylin (H9627;
Sigma-Aldrich) for 2 min and then washed twice in running water for
1 min. The sections were dipped in ammonium hydroxide solution and
then washed again using running water for 1 min. After being rinsed
in graded ethanol (80% ethanol for 1 min and 95% ethanol for 1
min), each section was dipped 10 times in eosin-Y (E4009;
Sigma-Aldrich) for 30–45 sec for counterstaining. Following two
additional rinses in 100% ethanol for 1 min each, the sections were
dehydrated in a graded series of alcohol, cleared in xylene, and
coverslipped with Permount.
Semi-quantitative RT-PCR
The rats were anesthetized with 4% halothane and
decapitated at 12 and 72 h after 2 h of SE. The hippocampus was
then immediately isolated and put on an ice-cold glass stage.
Total-RNA was extracted from the hippocampus using an RNA isolation
reagent, TRIzol (TRIzol® Reagent; Invitrogen Life
Technologies, Beijing, China), and reverse transcription was
performed using oligo(dt) priming according to the manufacturer’s
instructions (ThermoScript™ RT-PCR System; Invitrogen Life
Technologies). Primer sequences were as follows: rat caspase-3
(GenBank accession no. NM_012922), F, 5′-CTGGACTGCGGTATTGAG-3′ and
R, 5′-GGAACATCGGATTTGATT-3′; rat Bcl-2 (GenBank accession no.
NM_021850), F, 5′-CTACCCAAGTTAGCATT CC-3′ and R,
5′-CAAAGTCCCTATTTATCCCT-3′; and rat β-actin (GenBank accession no.
NM_031144), F, 5′-AGCCA TGTACGTAGCCATCC-3′ and R, 5′-GCTGTGGTGGTG
AAGCTGTA-3′. The PCR reactions were conducted as follows: 5 min at
94°C; 35 cycles (for caspase-3 and Bcl-2) or 30 cycles (for
β-actin) of 30 sec at 94°C, 30 sec at 55°C, and 30 sec at 72°C; and
final elongation for 10 min at 72°C. The amplified DNA fragments
were 465 bp for caspase-3, 389 bp for Bcl-2, and 222 bp for
β-actin. The PCR products were run on a 2% agarose gel and
visualized by UV light. Band densities were quantified using the
Scion Image software (Scion Corporation Frederick, MD, USA) and
signals from caspase-3 and Bcl-2 were normalized to those from the
housekeeping gene β-actin.
Western blot analysis
Rats were sacrificed at 6, 12, 24, 48, 72 or 96 h
after 2 h of SE or sham operation. Immediately after decapitation,
the hippocampus was quickly dissected and then homogenized using a
Dounce homogenizer and lysed on ice in 400 μl of RIPA buffer
[50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM PMSF, 1 mM EDTA,
1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS]. Proteins,
20 μg per lane as determined using the BCA protein assay kit
(no. 23227; Pierce, Rockford, IL, USA), were separated on 12%
SDS-polyacrylamide gels and transferred to polyvinylidene
difluoride membranes (RPN2020F; GE Healthcare, Beijing, China). The
membranes were first probed with primary antibody against cleaved
caspase-3 (1:1,000; no. 9664; Cell Signaling Technology, Inc.,
Danvers, MA, USA; recognizing the active form p17 of caspase-3),
Bcl-2 (1:50; ab7973; Abcam Inc., Cambridge, MA, USA), or β-actin
(1:200; sc-47778; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA). The specificity of the immunoreactivity for each antibody was
confirmed by preabsorption experiments. After washes, the membranes
were then incubated with horseradish peroxidase-conjugated
secondary antibodies (1:5,000; for caspase-3 and Bcl-2: sc-2004;
and for β-actin: sc-2005; were from Santa Cruz Biotechnology,
Inc.). Subsequently, the protein bands were visualized with ECL
(RPN2109; GE Healthcare) and quantified by densitometry.
Immunohistochemical staining
Rats were anesthetized and perfused as described for
the histological analysis. Coronal sections including the dorsal
hippocampus were selected and processed for immunohistochemical
staining. The paraffin sections were baked for 2 h in an oven,
deparaffinized in xylene, and rehydrated in graded ethanol
solutions. After three 5-min washes in 0.01 M PBS (pH 7.4), the
sections were microwaved in 0.01 M sodium citrate buffer (pH 6.0)
for 10 min, cooled to room temperature naturally, and then washed
three times in PBS, 5 min each. The sections were incubated in 3%
hydrogen peroxide for 10 min, washed using the same procedure as
above, and then blocked in 10% normal goat serum for 1 h at room
temperature. Rabbit monoclonal antibodies against cleaved caspase-3
or Bcl-2 (1:200; no. 2870; Cell Signaling Technology, Inc.) were
diluted in the recommended antibody diluent (no. 8112; Cell
Signaling Technology, Inc.) and incubated with sections overnight
at 4°C. After three rinses in PBS, the sections were incubated with
biotinylated goat anti-rabbit secondary antibody (1:2,000) for 1 h
at room temperature and then with avidin-biotin-peroxidase solution
(ABC reagent) for 30 min at room temperature. Following three
additional washes in PBS, the sections were treated with
3,3′-diaminobenzidine tetrahydrochloride (DAB) for 2 min,
counterstained in hematoxylin, dehydrated, and coverslipped. Normal
goat serum, biotinylated goat anti-rabbit secondary antibody, ABC
reagent, and DAB were all in the ABC staining kit (sc-2018; Santa
Cruz Biotechnology, Inc.). To assess nonspecific immunostaining in
our study, control samples were stained only with secondary
antibody, and no labeling was detected. To count the number of
positive neurons, 10 microscopic fields in the hippocampus, at a
magnification of 50 μm, were randomly chosen from each
section, and the caspase-3- and Bcl-2-positive neurons were
quantified using the Image-Pro Plus software (Media Cybernetics,
Inc., Shanghai, China).
Statistical analysis
Data were expressed as mean ± standard deviation and
analyzed using one-way analysis of variance (ANOVA) and post hoc
Fisher’s PLSD after normality of the distribution was proved.
P<0.05 was considered statistically significant.
Results
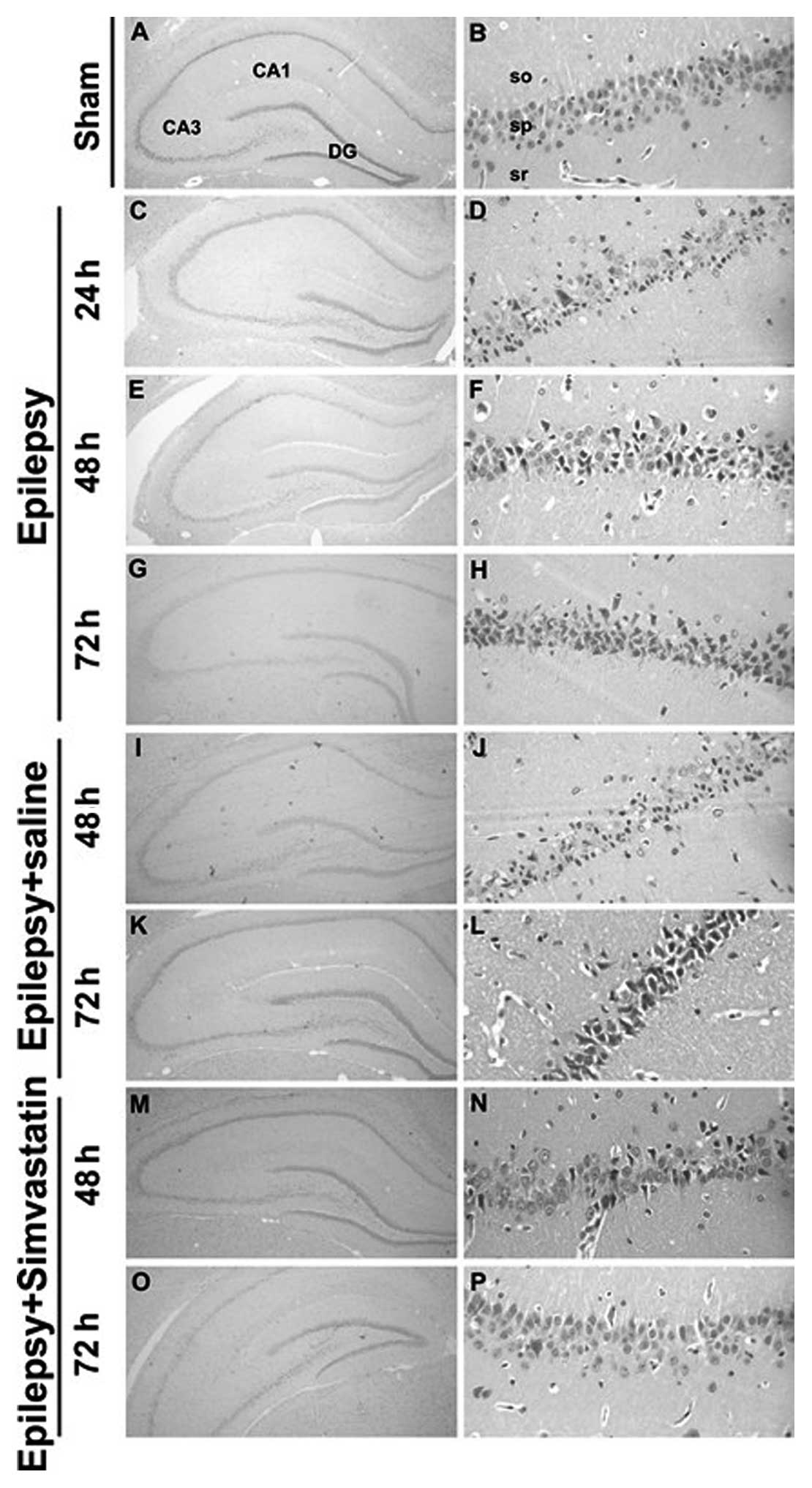
Simvastatin rescues SE-induced neuronal
apoptosis in the hippocampal CA1 region
To examine the effects of SE on the hippocampal CA1
neurons and the role of simvastatin in this process, we conducted
H&E staining of the hippocampal CA1 region of rats. We
demonstrated that 2-h SE induced selective neuronal death. Compared
with hippocampal CA1 sections from the sham group rats (Fig. 1A and B), those from the epilepsy
group showed partial cell death and neuronal loss following the 2-h
SE. Typical morphological characteristics of apoptosis were
observed in CA1 neurons, including cell shrinkage, nuclear
condensation, and fragmentation (Fig.
1C–H). Twenty-four hours after SE, the CA1 cell layer showed a
dramatic loss of neurons (Fig. 1C and
D). The number of apoptotic neurons kept increasing at 48 and
72 h (Fig. 1E–H). Saline intake
did not diminish the effect on neuronal death induced by SE
(Fig. 1I–L). In contrast,
simvastatin administration markedly rescued SE-induced neuronal
apoptosis, especially at 48 and 72 h (Fig. 1M–P).
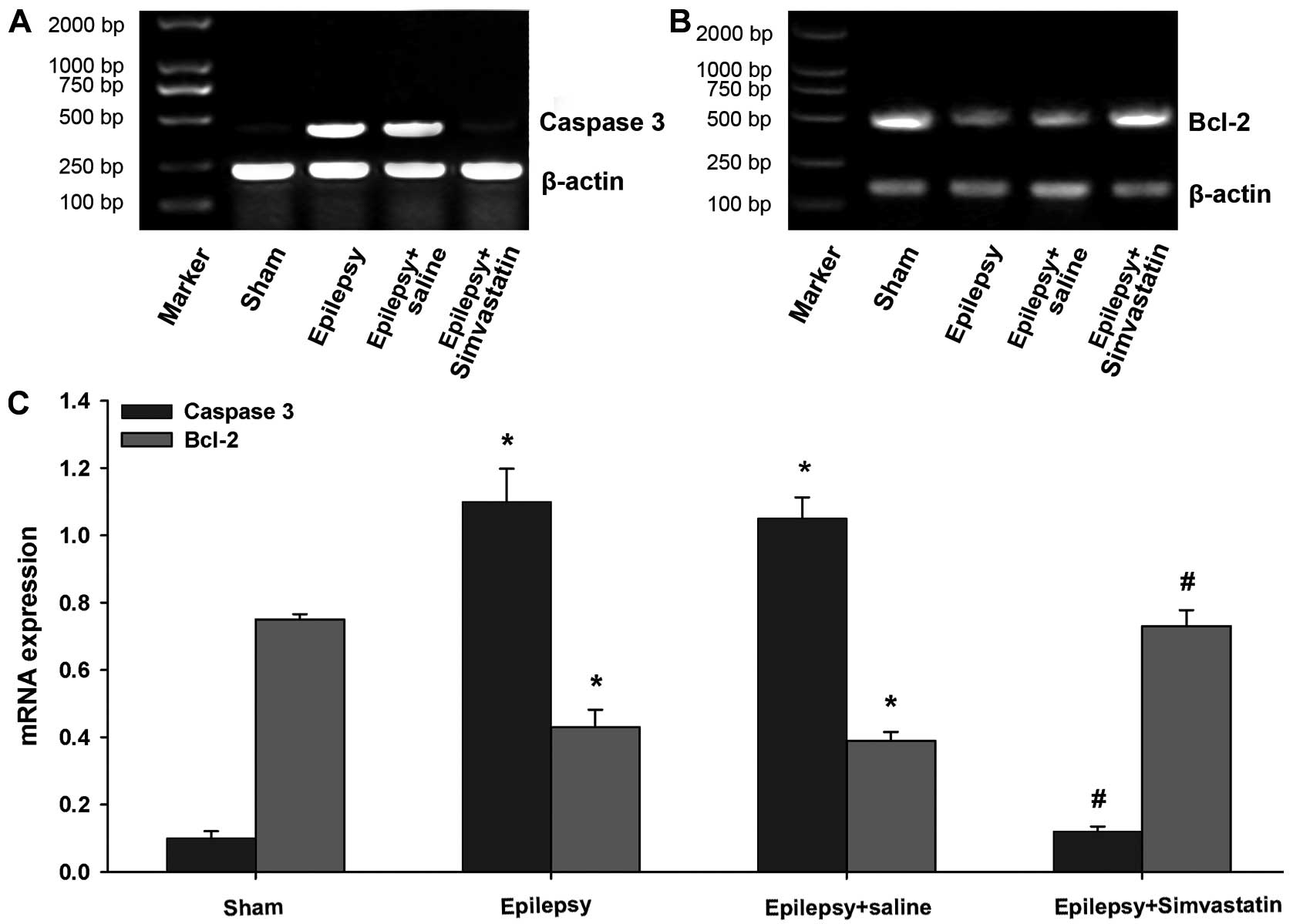
Simvastatin reverses SE-induced changes
in caspase-3 and Bcl-2 mRNA expression in the hippocampus
To analyze the potential mechanisms underlying the
neuroprotective role of simvastatin, we examined the mRNA
expression of the proapoptotic gene, caspase-3, and the
anti-apoptotic gene, Bcl-2, 72 and 12 h following SE, respectively.
The semiquantitative RT-PCR results showed that the basal
expression levels of caspase-3 and Bcl-2 mRNA were readily
detectable in the hippocampus of the sham rats (Fig. 2). SE significantly increased the
caspase-3 mRNA expression 72 h post-SE (Fig. 2A and C) (P<0.01) and
significantly decreased Bcl-2 mRNA expression 12 h post-SE
(Fig. 2B and C) (P<0.01).
Saline intake did not affect the mRNA expression of caspase-3 and
Bcl-2 following SE (Fig. 2)
(P<0.01 vs. the sham group). Interestingly, simvastatin
treatment completely reversed SE-induced changes in both caspase-3
and Bcl-2 mRNA expression, to levels not significantly different
from that of the sham group (Fig.
2) (P<0.01 vs. the epilepsy group; P>0.05 vs. the sham
group).
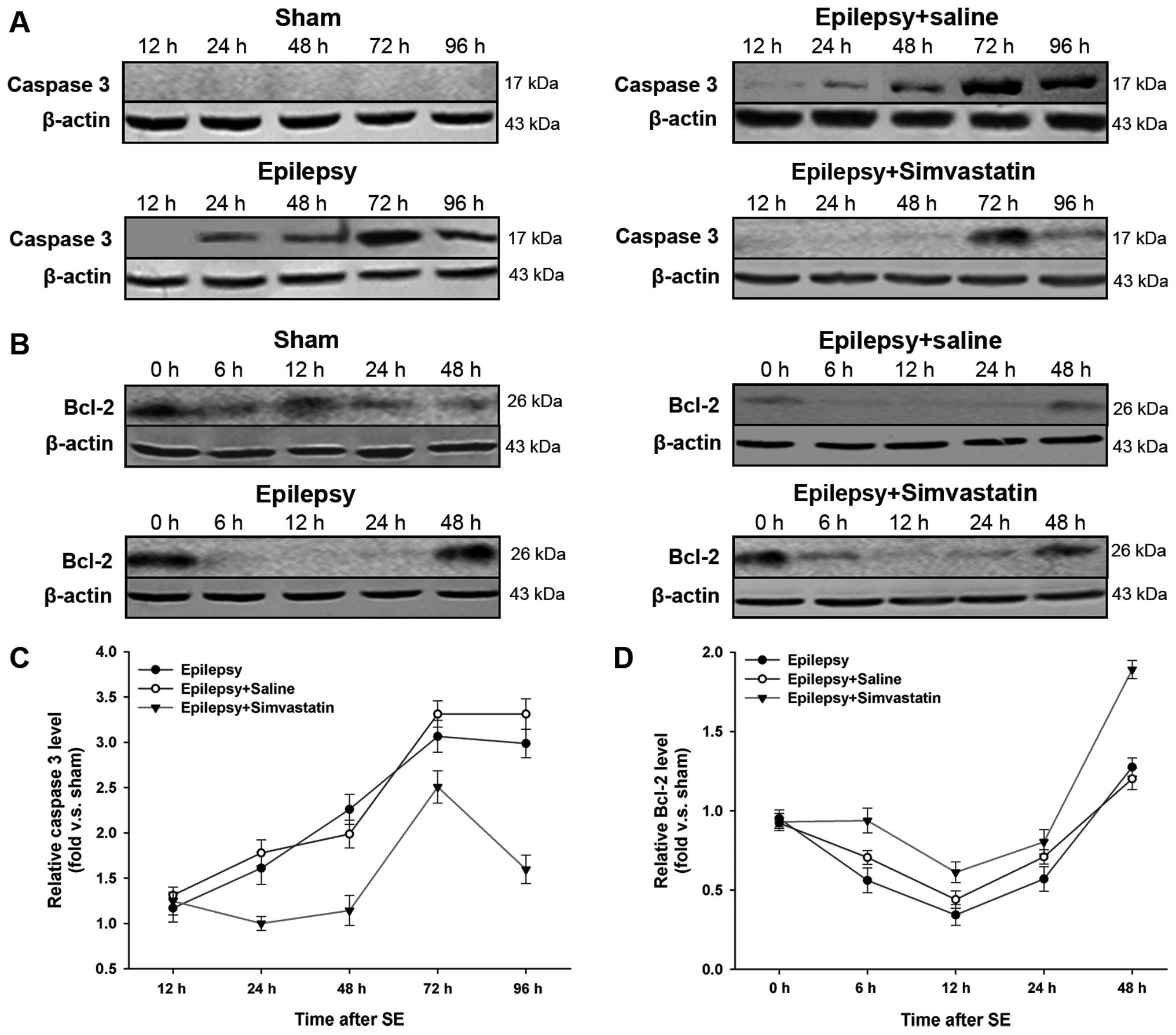
Simvastatin reverses SE-induced changes
in caspase-3 and Bcl-2 protein expression in the hippocampus
(western blot analysis)
We further confirmed the influence on caspase-3 and
Bcl-2 expression by examining protein levels. After SE-induced
activation, full length caspase-3 (32 kDa) is cleaved into 2 mature
subunits, p17 (17 kDa) and p12 (12 kDa). In this study, using an
antibody specific to p17, we compared the levels of activated
caspase-3 in the 4 groups. As expected, p17 staining was not
detected in the sham hippocampus, whereas SE markedly increased the
expression of activated caspase-3. This increase began at 24 h and
peaked at 72 h after SE (Fig. 3A and
C) (P<0.05 vs. the sham group). Treatment with simvastatin,
but not saline, significantly reduced the SE-induced increase of
activated caspase-3 levels (Fig. 3A
and C) (P<0.05 vs. the epilepsy group).
Despite comparable Bcl-2 protein expression in the
sham and epilepsy groups immediately after the 2-h SE (i.e., 0 h),
Bcl-2 levels were significantly decreased from 6 to 24 h (Fig. 3B and D) (P<0.05 vs. the sham
group). Intake of simvastatin, but not saline, significantly
reversed the reduction in Bcl-2 protein levels following SE
(Fig. 3B and D) (P<0.05 vs.
the epilepsy group).
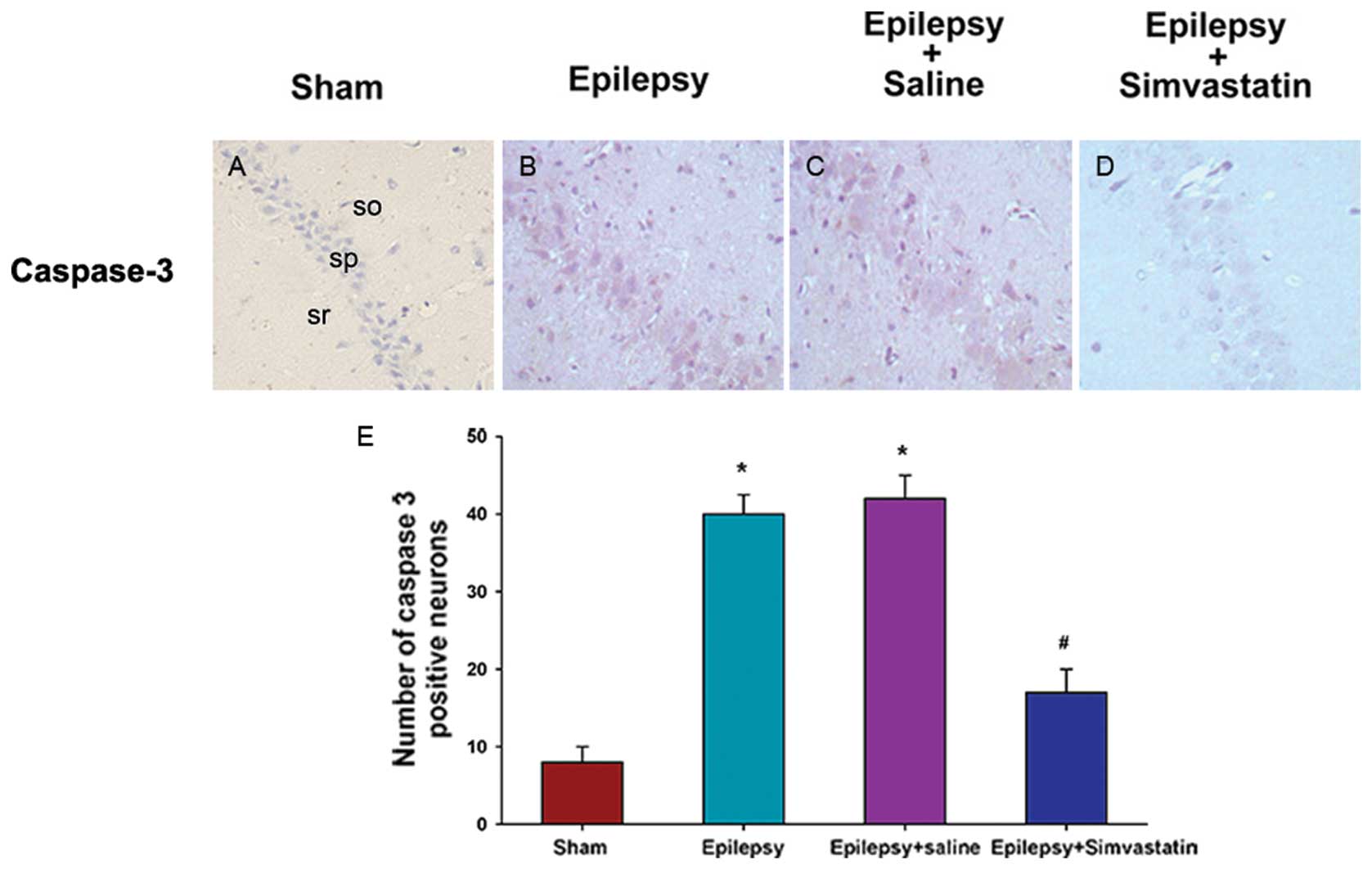
Simvastatin reverses SE-induced changes
in caspase-3 and Bcl-2 protein expression in the hippocampus
(immunohistochemical analysis)
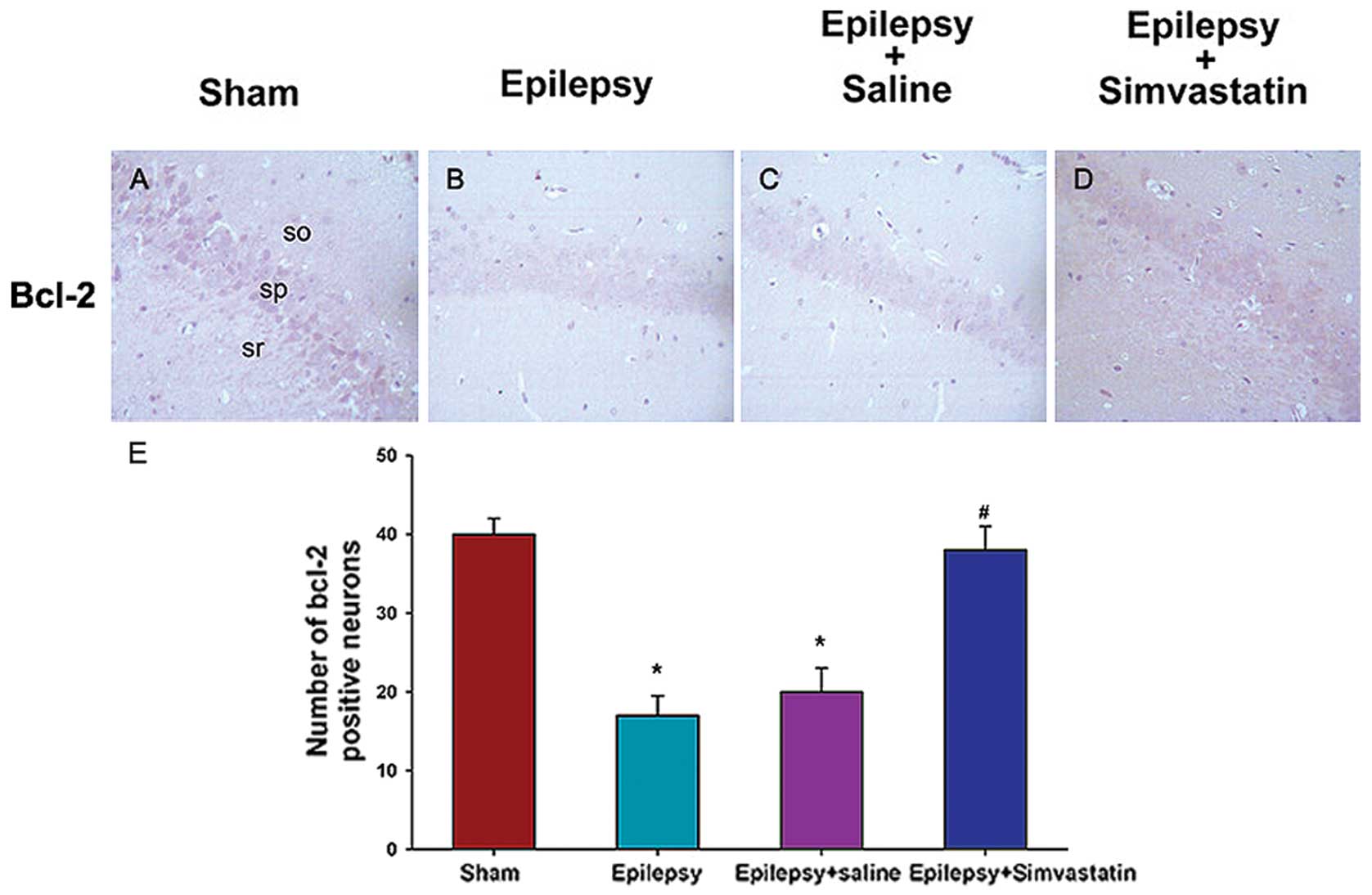
To characterize the cellular distribution of
caspase-3 and Bcl-2 activation after SE, we performed
immunohistochemical staining of caspase-3 in brain sections 72 h
after SE and of Bcl-2 in sections 12 h after SE. Neurons in the
hippocampal CA1 region prepared from sham animals showed some
distribution of caspase-3 (Fig.
4A) and Bcl-2 (Fig. 5A)
staining in the nucleus or cytoplasm. Under SE treatment, the
caspase-3 expression in the hippocampal CA1 pyramidal cell layer
increased significantly at 72 h (Fig.
4B). In contrast, SE treatment significantly decreased the
expression of Bcl-2, which was predominantly localized in the
cytosol (Fig. 5B). Saline intake
did not affect SE-induced expression patterns of caspase-3
(Fig. 4C) or Bcl-2 (Fig. 5C). In contrast, simvastatin
treatment significantly reduced SE-induced caspase-3 activation
(Fig. 4D) and, rescued SE-induced
reduction in Bcl-2 expression (Fig.
5D) in the hippocampal CA1 region.
To quantitatively analyze caspase-3 and Bcl-2
expression in the hippocampus, the mean intensity was analyzed by
counting the number of positive neurons. Under SE treatment, the
number of caspase-3-positive neurons increased significantly at 72
h (Fig. 4E) (P<0.01 vs. the
sham group), whereas the number of Bcl-2-positive neurons decreased
significantly at 12 h (Fig. 5E)
(P<0.05 vs. the sham group). Treatment with simvastatin, but not
saline, could reverse SE-induced changes in the number of
caspase-3- and Bcl-2-positive neurons (Fig. 4E) (P<0.01 vs. the epilepsy
group) (Fig. 5E) (P<0.05 vs.
the epilepsy group).
Discussion
SE as well as brief and/or repetitive seizures
associated with epilepsy cause neuronal loss in the brain (25). In the KA-induced SE rat model,
pyramidal cells in the CA1 and CA3 regions of the hippocampus
undergo apoptosis and death (26,27). This apoptotic process in
degenerating neurons is characterized by morphological changes,
altered expression of Bcl-2-family proteins, and activation of the
caspase family of cell-death proteases (28–30). Consistent with previous reports,
in this study, we observed that under SE treatment, from 12 to 48
h, a small number of neurons demonstrate the typical
characteristics of apoptosis, including cell shrinkage, nuclear
condensation, and fragmentation, and the number of apoptotic
neurons increases significantly at 72 h.
Apoptosis is triggered by a series of caspase
cascades (31,32). Caspase-3, a crucial apoptotic
regulator that is activated by caspases-9 or -8, is activated
during seizure-induced neuronal death (32,33). Controversially, other studies have
suggested that caspase-3 does not contribute significantly to
SE-induced neuronal necrosis, because caspase-3 activation was not
detected 6 and 24 h after 2-h SE (34,35). In our experiment, caspase-3 mRNA
and protein expression increased in hippocampal neurons 24 h after
the 2-h SE and peaked at 72 h. Our results strongly support the
theory that caspase-3 plays an important role in neuronal cell
death during SE. The sham control rats express only low basal
levels of caspase-3 mRNA and do not exhibit activated caspase-3
protein, and this low level of expression may be interpreted as
mediating normal brain development.
Bcl-2, the founding member of the Bcl-2 family,
suppresses apoptosis primarily via effects on mitochondria and one
of its mechanisms is preventing cytochrome c release (36,37). Under KA-induced SE, Bcl-2 family
members, including Bax and Bcl-2, are activated in the amygdaloid
complex (36,37). In the present study, Bcl-2 mRNA
and protein expression decreased 6 h after the 2-h SE and reached
the trough level at 12 h, suggesting that SE may accelerate
apoptosis by inhibiting Bcl-2 expression. Indeed, Bcl-2
overexpression provides modest protection against hippocampal
seizure damage (38), whereas the
level of SE-induced hippocampal neuronal death in Bcl-w-deficient
mice is twice as severe as that in wild-type mice (39).
Statins are important drugs to ameliorate
neurodegenerative diseases due to its significant neuroprotective
roles as well as the fact that they are well tolerated with
relatively few side effects. Recent clinical trials have reported
the neuroprotective function of statins in Alzheimer’s disease
(40), Parkinson’s disease
(41,42), and multiple sclerosis (43). Although the exact mechanisms still
remain unclear, several important discoveries have been made.
First, statins reduce the circulating cholesterol levels and thus
deplete lipid rafts, which contain many proteins involved in
neurotransmitter systems (44)
and have been linked to the processing of prion protein and the
impairment of dopamine function in Parkinson’s disease (45). Second, statins also exert
cholesterol-independent effects by blocking the production of the
non-sterol isoprenoid products of cholesterol synthesis, such as
farnesylpyrophosphate and geranylgeranylpyrophosphate, which are
active players in protein modification (46). Third, statins can influence the
outcome of neurological insults through peripheral actions,
including reducing oxidative damage, improving vascular function,
and modulating peripheral inflammatory responses. Fourth, statins
activate several neuroprotective and anti-apoptotic signaling
pathways, including the brain-derived neurotrophic factor pathway,
the protein kinase B (PKB/Akt) pathway, and the Ras-ERK signaling
cascade (47–49). In this study, we demonstrated that
treatment with simvastatin significantly attenuates SE-induced
neuronal loss and apoptosis in the hippocampus. Simvastatin
abolishes SE-induced increase in caspase-3 mRNA expression 24 h
after the 2-h SE. A persistent reduction in caspase-3 protein
expression was also observed. In contrast, simvastatin could
significantly reverse SE-induced reduction in Bcl-2 mRNA and
protein expression levels. In conclusion, our results suggest that
the mitochondrial apoptotic pathways participate in the protective
effect of simvastatin against KA-evoked prolonged seizures.
Controversies over the clinical use of statins
include several in vitro studies reporting that statins are
actually neurotoxic and induce cell death in neurons and glia
(50,51). However, these experiments were
usually carried out with very high statin concentrations of 0.1, 1
or even 10 mM. Thus, their physiological significance is unclear
considering that the maximum concentration of pravastatin is only
0.1 mM in the serum of healthy subjects (52). Moreover, statins probably do not
reach this concentration in the central nervous system (53). Different types of statins differ
in their ability to attenuate KA-induced seizures. Simvastatin is
one of the most effective statins against kainate-induced
excitotoxicity (54). Further
studies are required to examine whether simvastatin can influence
apoptotic regulators other than caspase-3 and Bcl-2 and whether the
anti-apoptotic properties of simvastatin depend on the dosages
administered.
In conclusion, our study demonstrated that SE
induces apoptosis in the rat hippocampus by increasing the
expression of the pro-apoptotic caspase-3 and reducing the
expression of the anti-apoptotic Bcl-2, in a time-dependent manner.
Treatment with simvastatin rescues SE-induced apoptosis by
reversing SE-produced changes in the expression of caspase-3 and
Bcl-2. This study helps to further our understanding of the
mechanisms of neuronal loss and apoptosis as well as the protective
effects of simvastatin against SE-caused brain injury.
References
|
1.
|
CM DeGiorgioU TomiyasuPS GottDM
TreimanHippocampal pyramidal cell loss in human status
epilepticusEpilepsia332327199210.1111/j.1528-1157.1992.tb02278.x1733757
|
|
2.
|
DG FujikawaHH ItabashiA WuSS ShinmeiStatus
epilepticus-induced neuronal loss in humans without systemic
complications or
epilepsyEpilepsia41981991200010.1111/j.1528-1157.2000.tb00283.x10961625
|
|
3.
|
A PitkanenI KharatishviliH
KarhunenEpileptogenesis in experimental modelsEpilepsia48Suppl
2S13S20200710.1111/j.1528-1167.2007.01063.x
|
|
4.
|
DG FujikawaSS ShinmeiB CaiSeizure-induced
neuronal necrosis: implications for programmed cell death
mechanismsEpilepsia41Suppl
6S9S13200010.1111/j.1528-1157.2000.tb01549.x10999512
|
|
5.
|
JJ RadleyBL JacobsPilocarpine-induced
status epilepticus increases cell proliferation in the dentate
gyrus of adult rats via a 5-HT1A receptor-dependent mechanismBrain
Res966112200310.1016/S0006-8993(02)03989-612646302
|
|
6.
|
D TokuharaS SakumaH HattoriO MatsuokaT
YamanoKainic acid dose affects delayed cell death mechanism after
status epilepticusBrain
Dev2928200710.1016/j.braindev.2006.05.00316790331
|
|
7.
|
J NiquetS AuvinM ArchieStatus epilepticus
triggers caspase-3 activation and necrosis in the immature rat
brainEpilepsia4812031206200710.1111/j.1528-1167.2007.01102.x17441993
|
|
8.
|
Y Ben-AriR CossartKainate, a double agent
that generates seizures: two decades of progressTrends
Neurosci23580587200010.1016/S0166-2236(00)01659-311074268
|
|
9.
|
JP LeiteN Garcia-CairascoEA CavalheiroNew
insights from the use of pilocarpine and kainate modelsEpilepsy
Res5093103200210.1016/S0920-1211(02)00072-412151121
|
|
10.
|
R KovacsS SchuchmannS GabrielO KannJ
KardosU HeinemannFree radical-mediated cell damage after
experimental status epilepticus in hippocampal slice culturesJ
Neurophysiol8829092918200210.1152/jn.00149.200212466417
|
|
11.
|
S NarkilahtiTJ PirttilaK LukasiukJ
TuunanenA PitkanenExpression and activation of caspase 3 following
status epilepticus in the ratEur J
Neurosci1814861496200310.1046/j.1460-9568.2003.02874.x14511328
|
|
12.
|
CT EkdahlP MohapelE ElmerO LindvallCaspase
inhibitors increase short-term survival of progenitor-cell progeny
in the adult rat dentate gyrus following status epilepticusEur J
Neurosci14937945200110.1046/j.0953-816x.2001.01713.x11595032
|
|
13.
|
T EngelBM MurphyS HatazakiReduced
hippocampal damage and epileptic seizures after status epilepticus
in mice lacking proapoptotic PumaFASEB
J24853861201010.1096/fj.09-14587019890018
|
|
14.
|
K PahanFG SheikhAM NamboodiriI
SinghLovastatin and phenylacetate inhibit the induction of nitric
oxide synthase and cytokines in rat primary astrocytes, microglia,
and macrophagesJ Clin Invest10026712679199710.1172/JCI119812
|
|
15.
|
W BalduiniE MazzoniS CarloniProphylactic
but not delayed administration of simvastatin protects against
long-lasting cognitive and morphological consequences of neonatal
hypoxic-ischemic brain injury, reduces interleukin-1beta and tumor
necrosis factor-alpha mRNA induction, and does not affect
endothelial nitric oxide synthase
expressionStroke34200720122003
|
|
16.
|
SF ChenTH HungCC ChenLovastatin improves
histological and functional outcomes and reduces inflammation after
experimental traumatic brain injuryLife
Sci81288298200710.1016/j.lfs.2007.05.02317612572
|
|
17.
|
M EndresU LaufsZ HuangStroke protection by
3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated
by endothelial nitric oxide synthaseProc Natl Acad Sci
USA9588808885199810.1073/pnas.95.15.88809671773
|
|
18.
|
G LiJB ShoferIC RhewAge-varying
association between statin use and incident Alzheimer’s diseaseJ Am
Geriatr Soc5813111317201020533968
|
|
19.
|
D LuA GoussevJ ChenAtorvastatin reduces
neurological deficit and increases synaptogenesis, angiogenesis,
and neuronal survival in rats subjected to traumatic brain injuryJ
Neurotrauma212132200410.1089/089771504772695913
|
|
20.
|
B WolozinJ MangerR BryantJ CordyRC GreenA
McKeeRe-assessing the relationship between cholesterol, statins and
Alzheimer’s diseaseActa Neurol Scand Suppl1856370200616866913
|
|
21.
|
JK LeeJS WonAK SinghI SinghStatin inhibits
kainic acid-induced seizure and associated inflammation and
hippocampal cell deathNeurosci
Lett440260264200810.1016/j.neulet.2008.05.11218583044
|
|
22.
|
A MahmoodA GoussevH KazmiC QuD LuM
ChoppLong-term benefits after treatment of traumatic brain injury
with simvastatin in
ratsNeurosurgery65187192200910.1227/01.NEU.0000343540.24780.D619574841
|
|
23.
|
C XieJ SunW QiaoAdministration of
simvastatin after kainic acid-induced status epilepticus restrains
chronic temporal lobe epilepsyPLoS
One6e24966201110.1371/journal.pone.002496621949812
|
|
24.
|
RJ RacineModification of seizure activity
by electrical stimulation. II Motor seizureElectroencephalogr Clin
Neurophysiol32281294197210.1016/0013-4694(72)90177-04110397
|
|
25.
|
J BengzonP MohapelCT EkdahlO
LindvallNeuronal apoptosis after brief and prolonged seizuresProg
Brain Res135111119200210.1016/S0079-6123(02)35011-812143333
|
|
26.
|
JA CollinsCA SchandiKK YoungJ VeselyMC
WillinghamMajor DNA fragmentation is a late event in apoptosisJ
Histochem Cytochem45923934199710.1177/0022155497045007029212818
|
|
27.
|
S WeissO CataltepeAJ ColeAnatomical
studies of DNA fragmentation in rat brain after systemic kainate
administrationNeuroscience74541551199610.1016/0306-4522(96)00148-08865204
|
|
28.
|
J BengzonZ KokaiaE ElmerA NanobashviliM
KokaiaO LindvallApoptosis and proliferation of dentate gyrus
neurons after single and intermittent limbic seizuresProc Natl Acad
Sci USA941043210437199710.1073/pnas.94.19.104329294228
|
|
29.
|
CJ FahertyS XanthoudakisRJ
SmeyneCaspase-3-dependent neuronal death in the hippocampus
following kainic acid treatmentBrain Res Mol Brain
Res70159163199910.1016/S0169-328X(99)00143-610381555
|
|
30.
|
DC HenshallRS ClarkPD AdelsonM ChenSC
WatkinsRP SimonAlterations in bcl-2 and caspase gene family protein
expression in human temporal lobe
epilepsyNeurology55250257200010.1212/WNL.55.2.25010908900
|
|
31.
|
WC EarnshawLM MartinsSH KaufmannMammalian
caspases: structure, activation, substrates, and functions during
apoptosisAnnu Rev
Biochem68383424199910.1146/annurev.biochem.68.1.38310872455
|
|
32.
|
DC HenshallJ ChenRP SimonInvolvement of
caspase-3-like protease in the mechanism of cell death following
focally evoked limbic seizuresJ
Neurochem7412151223200010.1046/j.1471-4159.2000.741215.x10693954
|
|
33.
|
A KondratyevK GaleIntracerebral injection
of caspase-3 inhibitor prevents neuronal apoptosis after kainic
acid-evoked status epilepticusBrain Res Mol Brain
Res75216224200010.1016/S0169-328X(99)00292-210686342
|
|
34.
|
C AnanthS Thameem DheenP
GopalakrishnakoneC KaurDomoic acid-induced neuronal damage in the
rat hippocampus: changes in apoptosis related genes (bcl-2, bax,
caspase-3) and microglial responseJ Neurosci
Res66177190200110.1002/jnr.121011592113
|
|
35.
|
DG FujikawaX KeRB TrinidadSS ShinmeiA
WuCaspase-3 is not activated in seizure-induced neuronal necrosis
with internucleosomal DNA cleavageJ
Neurochem83229240200210.1046/j.1471-4159.2002.01152.x12358747
|
|
36.
|
TC DumasJR McLaughlinDY HoMS LawrenceRM
SapolskyGene therapies that enhance hippocampal neuron survival
after an excitotoxic insult are not equivalent in their ability to
maintain synaptic transmissionExp
Neurol166180189200010.1006/exnr.2000.7500
|
|
37.
|
A GrossJM McDonnellSJ KorsmeyerBCL-2
family members and the mitochondria in apoptosisGenes
Dev1318991911199910.1101/gad.13.15.189910444588
|
|
38.
|
RG PhillipsMS LawrenceDY HoRM
SapolskyLimitations in the neuroprotective potential of gene
therapy with Bcl-2Brain
Res859202206200010.1016/S0006-8993(99)02453-110719065
|
|
39.
|
B MurphyM DunleavyS ShinodaBcl-w protects
hippocampus during experimental status epilepticusAm J
Pathol17112581268200710.2353/ajpath.2007.07026917702891
|
|
40.
|
K HoglundK BlennowEffect of HMG-CoA
reductase inhibitors on beta-amyloid peptide levels: implications
for Alzheimer’s diseaseCNS Drugs214494622007
|
|
41.
|
X HuangH ChenWC MillerLower low-density
lipoprotein cholesterol levels are associated with Parkinson’s
diseaseMov Disord223773812007
|
|
42.
|
AD WahnerJM BronsteinYM BordelonB
RitzStatin use and the risk of Parkinson
diseaseNeurology7014181422200810.1212/01.wnl.0000286942.14552.5118184918
|
|
43.
|
O AktasS WaicziesA SmorodchenkoTreatment
of relapsing paralysis in experimental encephalomyelitis by
targeting Th1 cells through atorvastatinJ Exp
Med197725733200310.1084/jem.2002142512629065
|
|
44.
|
JA AllenRA Halverson-TamboliMM
RasenickLipid raft microdomains and neurotransmitter signallingNat
Rev Neurosci8128140200710.1038/nrn205917195035
|
|
45.
|
EE BenarrochLipid rafts, protein
scaffolds, and neurologic
diseaseNeurology6916351639200710.1212/01.wnl.0000279590.22544.c317938374
|
|
46.
|
J GreenwoodL SteinmanSS ZamvilStatin
therapy and autoimmune disease: from protein prenylation to
immunomodulationNat Rev
Immunol6358370200610.1038/nri183916639429
|
|
47.
|
H WuD LuH JiangSimvastatin-mediated
upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway,
and increase of neurogenesis are associated with therapeutic
improvement after traumatic brain injuryJ
Neurotrauma25130139200810.1089/neu.2007.0369
|
|
48.
|
AM DolgaI GranicIM NijholtPretreatment
with lovastatin prevents N-methyl-D-aspartate-induced
neurodegeneration in the magnocellular nucleus basalis and
behavioral dysfunctionJ Alzheimers Dis17327336200919363269
|
|
49.
|
F ZippS WaicziesO AktasImpact of HMG-CoA
reductase inhibition on brain pathologyTrends Pharmacol
Sci28342349200710.1016/j.tips.2007.05.00117573124
|
|
50.
|
P MarzU OttenAR MiserezStatins induce
differentiation and cell death in neurons and
astrogliaGlia55112200710.1002/glia.2042216998865
|
|
51.
|
Z XiangSA ReevesSimvastatin induces cell
death in a mouse cerebellar slice culture (CSC) model of
developmental myelinationExp
Neurol2154147200910.1016/j.expneurol.2008.09.01018929563
|
|
52.
|
HY PanAP WaclawskiPT FunkeD
WhiganPharmacokinetics of pravastatin in elderly versus young men
and womenAnn Pharmacother271029103319938219432
|
|
53.
|
RE BottiJ TriscariHY PanJ
ZayatConcentrations of pravastatin and lovastatin in cerebrospinal
fluid in healthy subjectsClin
Neuropharmacol14256261199110.1097/00002826-199106000-000101906375
|
|
54.
|
C RamirezI TerceroA PinedaJS
BurgosSimvastatin is the statin that most efficiently protects
against kainate-induced excitotoxicity and memory impairmentJ
Alzheimers Dis24161174201121224519
|