Introduction
Androgens are the main regulators of prostate growth
and differentiation. The proliferation mechanisms activated by
androgens involve transcription factors that operate together to
maintain the balance between inhibition and cell prostate
proliferation (1–3). The androgen receptor (AR) is
expressed in normal prostate epithelial cells, in almost all
primary prostate cancer cells and in most refractory prostate
cancer cells (4). Androgen
concentrations and actions are determinant for prostate enlargement
and are dependent on AR activity (5,6). A
cell line derived from a mouse xenograft prostate carcinoma (PCa),
CWR22R3, maintains a high level of AR expression. These cells were
infected with retroviruses encoding AR-specific short hairpin RNA
(shRNA), and AR downregulation was demonstrated by anti-AR
immunoblotting; also, the cells expressing AR shRNA showed reduced
cell density (6). Finally,
blocking AR activity is a therapeutic approach for benign prostate
hyperplasia (BPH) and PCa (7,8).
While the stimulatory effect of androgens on in
vivo growth and development is well recognized in several
animal models and in human prostate, the in vitro
characterization of these effects is a complex issue. There have
been reports on the effects of androgens on the growth of normal
epithelial prostate cells (9), on
stromal cells of BPH tissue (3)
and on AR expression (7,8,10,11). By contrast, other studies were
not able to show any effect of androgens, at different
concentrations, on the cell proliferation of normal, hyperplasic or
tumoral prostate cells (12–14). The fact that such studies were
carried out on immortalized or transformed cells might be related
to the variation in the results.
Studies using the LNCaP and MOP androgen-dependent
prostate epithelial cancer cell lines have demonstrated a biphasic
effect of androgens on proliferation, in which lower androgen
concentrations have a maximum effect on cell proliferation
(1,15–17). In addition, androgen shutoff genes
(AS1, AS2 and AS3) have been demonstrated in LNCaP cells, and their
gene expression was reported to be induced by higher concentrations
of DHT (10−9 M) in the culture medium (18). We have also recently shown a
stimulatory effect of androgens at low concentrations on cell
proliferation of human non-transformed epithelial prostatic cells
(HNTEP cells). This effect was eradicated by the addition of
hydroxyflutamide (OH-Flutamide), an AR inhibitor (19). Moreover, this biphasic effect on
HNTEP cell proliferation was associated with changes in mRNA levels
of c-myc (19),
c-fos and c-jun (20).
Molecular mechanisms regulating the cell cycle are
key processes in the proliferation and differentiation of a target
tissue. These include both apoptosis and cell progression through
the cell cycle. Apoptosis is a genetically regulated process, which
requires the expression and action of gene products and
coregulators in order to occur (21). Progression through the G1-phase
cell cycle is associated with cyclins and cyclin-dependent kinase
(CDK) complexes. This effect depends on several factors including
the levels of CDK inhibitors (CDKIs) (22). The p21 gene is a CDKI
member of the CIP/KIP family that includes p27 and p57.
p21 prevents DNA synthesis and leads to growth arrest (23,24). The expression of the p21
gene in prostate cells can be activated through growth factors
(25), androgens (26,27) and p53, a suppressor gene
(28).
p53 is a tumor-suppressor gene that regulates
the expression of genes involved in cell cycle inhibition,
apoptosis and genomic stability. The p53 gene is the most
commonly mutated or deleted gene in human cancer (28). Higher levels of mutated p53
were found in prostate intraepithelial neoplasia (PIN), further
implicating p53 mutation or loss as an early event in
prostate tumorigenesis (29). A
recent study showed a decrease in p53 mRNA levels after 8 h
of treatment with dihydrotestosterone in a prostate cell line
(LNCaP) compared with an untreated group. They also found 4
potential AREs in the p53 gene: 1 in the 5′ flanking region,
1 in the first exon and 2 sites in the first intron (30).
Although p21 and p53 appear to be
associated with abnormal growth of prostate epithelial cells under
the influence of androgens (26,27,30,31), their involvement in the
biphasic effect of androgens on prostate cell proliferation has yet
to be clearly defined. Therefore, the aim of the present study was
to determine the effect of androgen at different concentrations on
p21 and p53 gene expression in HNTEP cells.
Materials and methods
Cell culture
Samples of prostate tissue were obtained from
retropubic prostatectomies of 10 patients between 60 and 77 years
of age, diagnosed with BPH. Patients with malignant tumors were
excluded. The local Ethics Committee approved the study protocol.
Informed consent was obtained from all subjects.
HNTEP cells were cultured as previously described
(32). Briefly, after removal of
blood clots, prostate tissue was washed with Hank’s balanced salt
solution (HBSS; Gibco-BRL, Grand Island, NY, USA) plus kanamycin
(0.5 mg/ml) (Sigma Chemical Co., St. Louis, MO, USA), and then
finely minced into 2–3 mm pieces. Tissue fragments were treated
with collagenase type IA (7.5 mg/g of tissue) (Sigma Chemical Co.)
in HBSS. Enzymatic digestion proceeded for 3 h at 37°C with gentle
shaking. The enzymatic reaction was interrupted by the addition of
warm 199 culture medium (Gibco-BRL) plus kanamycin and 10% fetal
bovine serum (FBS) (Gibco-BRL).
Epithelial cells were separated by differential
filtration. Cell suspensions were distributed into 35-mm
tissue-culture dishes (Corning Glassworks, NY, USA),
1x105 cells/dish, or into 24-well tissue-culture plates
(NUNC™, Denmark), 2x104 cells/ml/plate, and maintained
at 37°C in a humidified atmosphere of 95% air/5% CO2
(NuAire, Inc., Plymouth, MN, USA). In addition, since it is
difficult to observe the stimulatory effect of androgen on cell
growth in vitro, a culture medium that was free of growth
factors (such as insulin or EGF) other than those present in FBS
was used. The basal medium consisted of medium 199 containing
kanamycin (0.5 mg/ml) enriched with 5% charcoal-stripped FBS
(cFBS). Cultures were kept in the same medium for the first 2 days,
and then the medium was changed every 2 days.
Cell proliferation
MTT colorimetric assay was used to study cell
proliferation. The MTT assay is based on the reduction of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma Chemical Co.) to a blue formazan product by mitochondrial
dehydrogenase found in viable cells (33). Epithelial cells (2x104
cells/ml) were allowed to adhere overnight in 24-well
tissue-culture plates in basal medium. On the following day,
designated Day 0, the basal medium was removed and the experimental
medium was applied. The experimental medium consisted of basal
medium and treatments with DHT (10−8, 10−10
and 10−13 M) alone or combined with hydroxyflutamide
(OH-FLU) at 10−6 M or ethanol vehicle (control medium).
Cells were cultured under routine conditions, and the medium was
changed on Day 2. On Day 6, 50 μl of 5 mg/ml MTT in PBS was
added to each well. After 4 h of incubation at 37°C, the medium was
removed, 100 μl of DMSO was added, and optical density was
measured at 540 nm in an ELISA reader (Benchmark Microplate
Reader). The data represent the mean of 3–6 wells of each
treatment. Experiments were repeated at least three times, using
samples from different patients.
Extraction of RNA and synthesis of
cDNA
Cells were grown in basal medium deprived of serum
for 4 h, and then treated with DHT or ethanol vehicle in different
conditions for 4 h. Subsequently, the mRNA was extracted. The
extraction of RNA and the synthesis of cDNA were carried out as
previously described (19).
Prostatic cells in culture were washed twice with PBS and
homogenized in phenol-guanidine isothiocyanate (TRIzol; Invitrogen,
Carlsbad, CA, USA). Total-RNA was extracted with chloroform and
precipitated with isopropanol by 12,000 x g centrifugation at 4°C.
The RNA pellet was washed twice with 75% ethanol, resuspended in
diethylpyrocarbonate-treated water, and quantified by light
absorbance at 260 nm. First-strand cDNA was synthesized from 1
μg total-RNA, using the SuperScript Preamplification System
(Invitrogen). After denaturing the template RNA and primers at 65°C
for 5 min, reverse transcriptase was added in the presence of 20 mM
Tris-HCl (pH 8.4) plus 50 mM KCl, 2.5 mM MgCl2, 0.5 mM
dNTP mix and 10 mM dithiothreitol, and incubated at 42°C for 50
min. The mixture was heated at 70°C to interrupt the reaction, and
then incubated with E. coli RNase for 20 min at 37°C to
destroy the untranscribed RNA.
Real-time PCR conditions
Amplification and detection were performed with the
MiniOpticon Real-Time PCR detection system (Bio-Rad Life Science
Research, Hercules, CA, USA). Duplicate samples were used. The PCR
mixture contained 1.25 μl SYBR-Green, 2 ng cDNA at 1:50
dilution, 3 mM MgCl2, 20 mM Tris-HCl pH 8.4 plus 50 mM
KCl, 0.2 mM dNTP mix, 1 unit TaqDNA polymerase and 0.4 μM of
each primer in a 25-μl final volume. The reaction conditions
were 94°C for 2 min for hot-start, and 39 cycles of 94°C for 50
sec, 59°C for 30 sec and 72°C for 40 sec for the p53 gene; and 94°C
for 50 sec, 57°C for 30 sec and 72°C for 40 sec for the p21
gene. The sequences of primers employed were: p53 gene,
sense, 5′-CTGAGGTTGGCTCTGACTGTACCA-3′ and antisense,
5′-CTCATTCAGCTCTCGGAACATCTC-3′ (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi);
p21 gene, sense, 5′CTCAG7AGGAGGCGCCATG 3′ and antisense,
5′-GGGCGGATTAGGGCTTCC-3′ (25).
For normalization of the expression levels, the expression of
β2-microglobulin (sense, 5′-ATCCAGCGTACTCCAAAGATTCAG-3′ and
antisense, 5′-AAATTGAAAGTTAACTTATGCACGC-3′ (34) was used as a housekeeping gene.
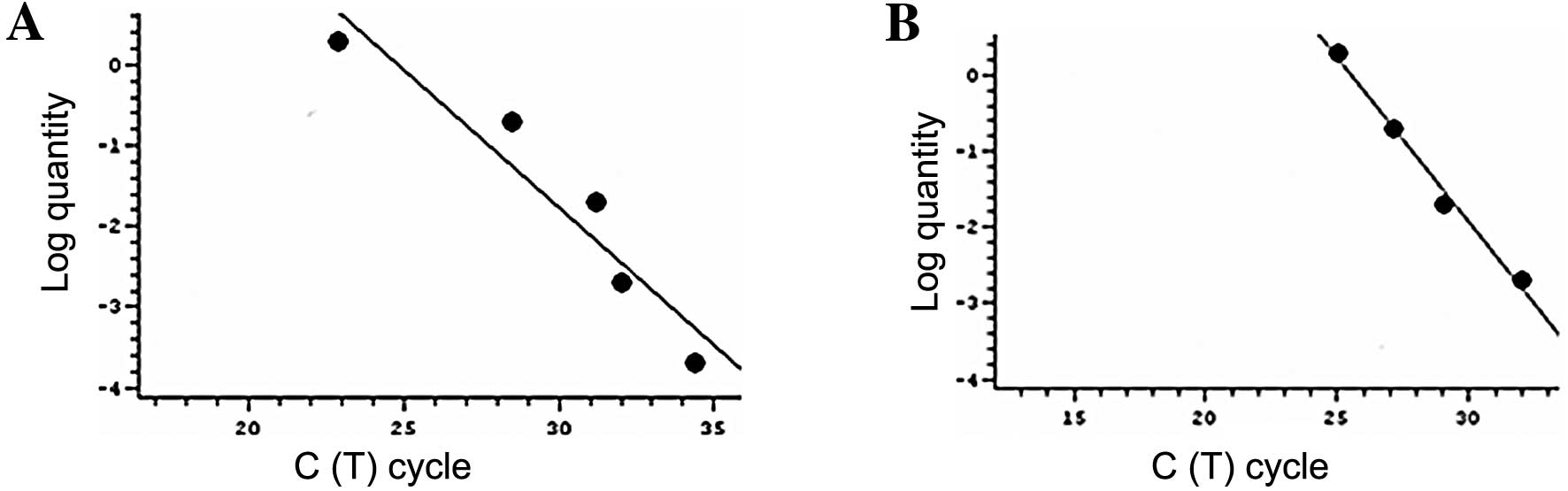
Standard curves and efficiency
All samples were automatically processed for
melting-curve analysis of amplified cDNA. The melting temperature
(Tm) is specific to each amplicon. The melting
temperature for p53 was 89°C, for p21 93°C and for
β2-microglobulin 83°C. Standard curves were constructed by plotting
the CT (cycle threshold) values of the real-time PCR
performed on a dilution series of cDNA standard. The real-time PCR
assay was analyzed in the linear phase, and a linear function was
fitted of the log of relative fluorescence vs. cycle number with a
typical R2 value >0.9 (Fig. 1). The results of the gene
expression corrected by the housekeeping gene were expressed as the
proportional change (1-, 2- or 3-fold) in relation to the control
group.
Western blotting
Protein samples of HNTEP cells in culture were
obtained with TRIzol reagent (Invitrogen) following the
manufacturer’s protocol. The protein concentration was determined
by the Bradford method (35).
Sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis was
carried out using a miniprotein system (Bio-Rad Life Science
Research) with broad-range molecular weight standards. Total
protein of 6.5 μg was loaded onto each lane with a loading
buffer containing 0.375 M Tris (pH 6.8), 50% glycerol, 12% SDS, 0.5
M dithiothreitol and 0.002% bromophenol blue. Samples were heated
at 100°C for 3 min prior to gel loading. Following electrophoresis,
proteins were transferred to nitrocellulose membranes (Schleicher
and Schuell) using an electrophoresis transfer system (mini
Trans-Blot Electrophoretic Transfer Cell) at 110 V for 1–2 h. The
membranes were then washed with TTBS (20 mM Tris-HCl, pH 7.5; 150
mM NaCl; 0.05% Tween-20, pH 7.4) and 8% non-fat dry milk for 90
min. The membranes were incubated overnight at 4°C with the primary
antibody (monoclonal anti-p21 antibody and monoclonal anti-p53)
(Upstate Biotechnology) diluted in TTBS. After washing, the
membranes were incubated for 2 h at room temperature with secondary
antibody (rabbit anti-mouse IgG conjugate 1:3,000 (Bio-Rad Life
Science Research), washed with TBS (20 mM Tris-HCl; 150 mM NaCl, pH
7.5), and developed with the chemoluminescence ECL Western Blotting
system (Amersham) followed by apposition of the membranes to
auto-radiographic film (Kodak X-Omat). The optical density (OD) of
the bands was analyzed using an image-processing system (Image
Master VDS; Pharmacia Biotech, Uppsala, Sweden). Ponceau S staining
was used as a protein loading control for p21 and β-tubulin
(1:10,000) for p53.
Statistical analysis
Data are reported as means and standard error of the
mean (SEM). Differences between groups were assessed by analysis of
variance, followed by Duncan’s test. All analyses were performed
using the Statistical Package for Social Sciences (SPSS, Chicago,
IL, USA). Data were considered to indicate statistically
significant differences at P<0.05.
Results
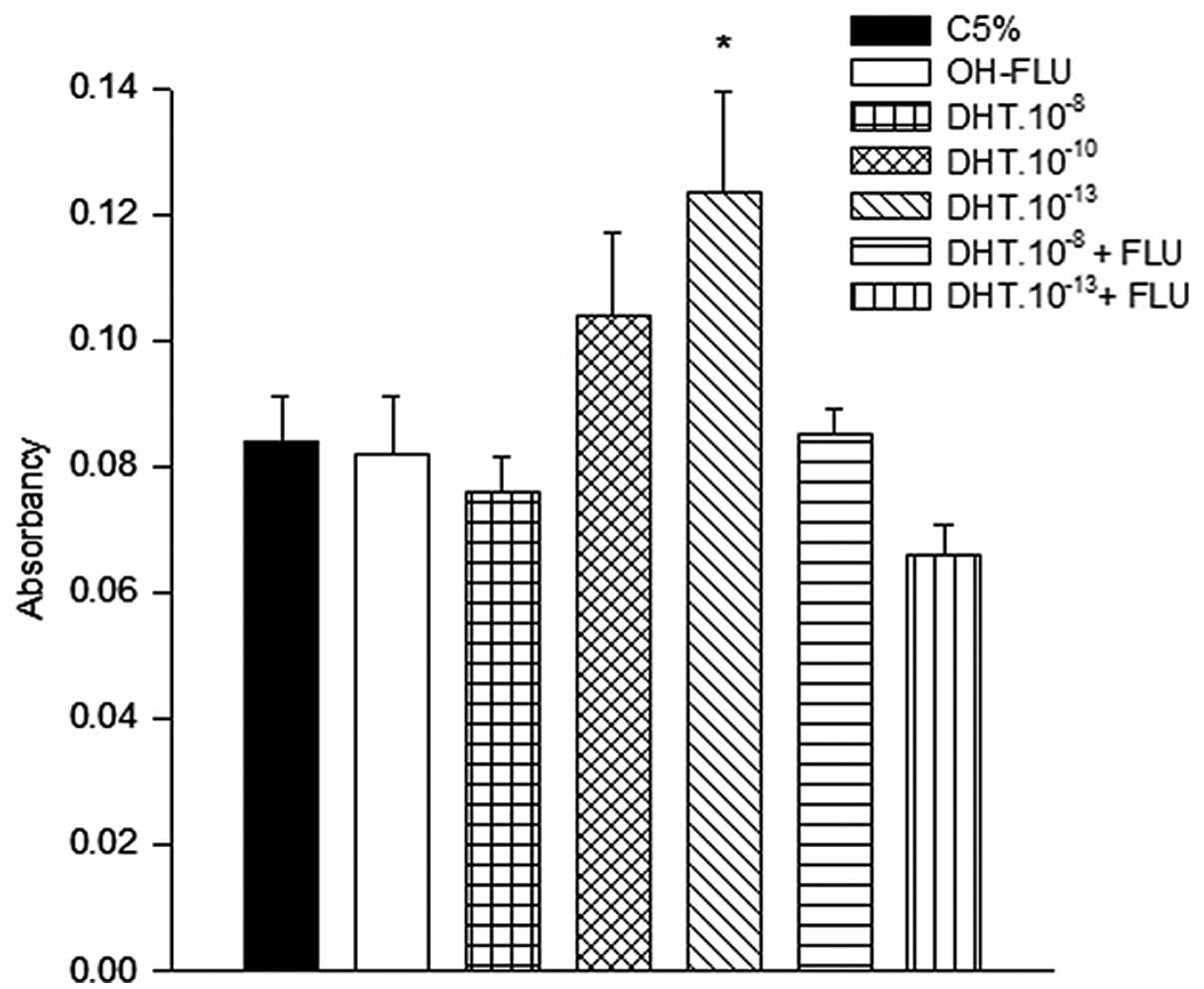
The effect of DHT on the growth of HNTEP cells was
examined on Day 6 of culture. Data were compared to those of cells
treated with control medium. Cells treated with DHT at a
concentration of 10−13 M showed a higher proliferation
rate (P<0.05) compared to cells from the other groups:
DHT.10−8 M, control medium, OH-FLU or
DHT.10−13 M combined with OH-FLU (Fig. 2).
A quantitative analysis of mRNA levels of the
p53 and p21 genes in HNTEP cells was performed after
4 h with different treatment conditions.
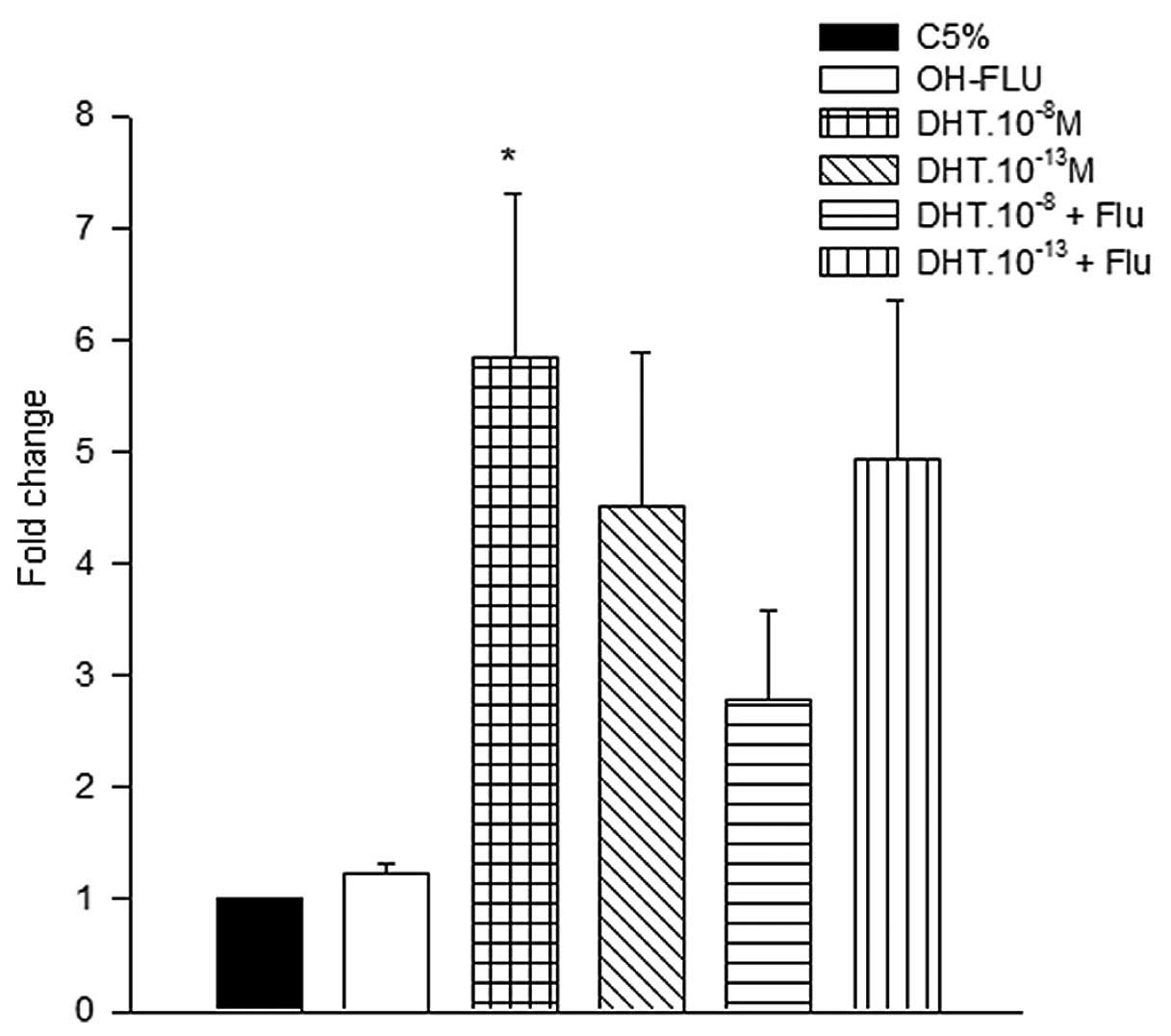
The effect of DHT treatment on p53 and
p21 mRNA levels in HNTEP cells was estimated by quantitative
analysis. The p53 mRNA levels were 6-fold higher in HNTEP
cells treated with DHT.10−8 M compared to the control
group. The p53 mRNA level of this group was also higher
compared with the OH-FLU.10−6, DHT.10−13 and
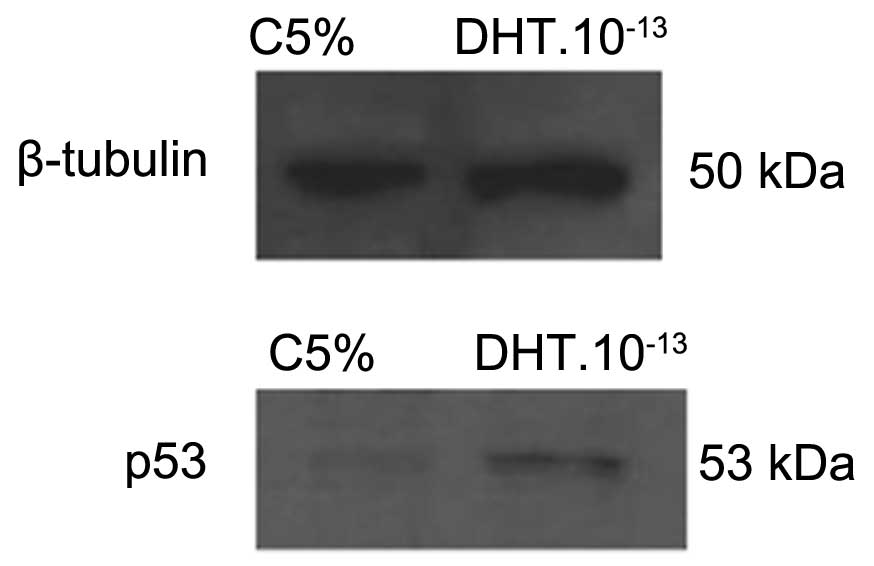
DHT.10−8 + OH-FLU groups (Fig. 3). To confirm the presence of
p53 in HNTEP cells, we evaluated the protein levels of p53
treated for 4 h with DHT at 10−13 M. Western blot
analysis showed an immunoreactivity of p53 corresponding to a 53
kDa protein in HNTEP cells (Fig.
4).
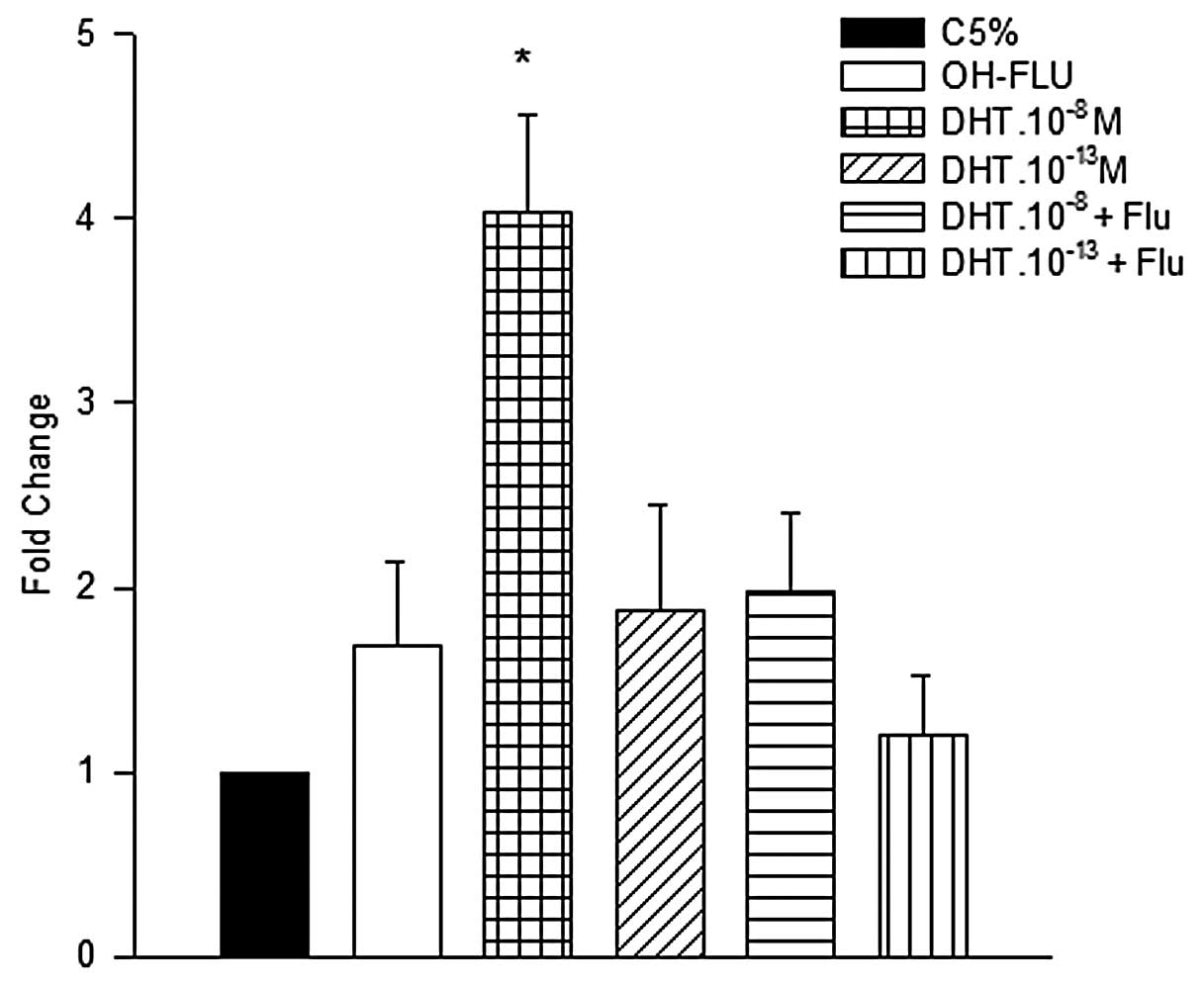
The data presented in Fig. 5 show that HNTEP cells treated with
DHT.10−8 M expressed 4-fold higher p21 mRNA
levels than the control group. This level of expression was also
significantly higher compared with the other groups. The protein
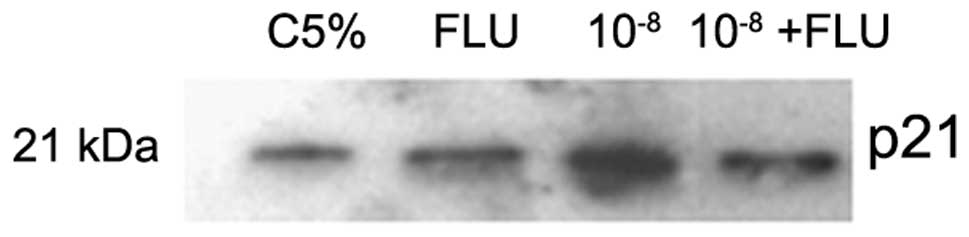
level of p21 (21 kDa) was analyzed after 4 h with androgen
treatment. The data shown in Fig.
6 suggest an increase in the protein level of p21 in the
group treated with DHT.10−8 M compared to the other
groups.
Discussion
The results of the present study showed that low
concentrations of androgens exerted a positive effect on cell
proliferation in HNTEP cells, and high concentrations maintained
proliferation similar to that of the control. There are several
experimental models for the study of prostate growth (1,7,12,36)
and each of these models responds differently to stimulation.
Therefore, these models may not be appropriate for investigating
the effects of androgens on growth and differentiation of prostate
epithelial cells. The epithelial prostate cells (HNTEP) were
established as an in vitro model to study the androgen
dependence of human prostate, and have previously been
characterized both functionally and morphologically (32).
In our study, the effect of the low concentration of
androgen was blocked by adding the anti-androgen OH-FLU to the
culture medium. In a parallel study, we assessed the AR gene
expression in HNTEP cells using the same mRNA employed for the
p21 and p53 analysis in these data. In this case, the
AR gene expression did not change with different treatments of DHT
(data not shown). These data confirmed our previous studies on cell
proliferation (19) and supported
the theory that the mitogenic effect of the low dose of DHT in
HNTEP cells is regulated by its own receptor, the AR. There are few
studies regarding the effects of androgens in non-transformed cell
models. A study by Shao et al (17) confirmed a biphasic effect of
androgens on prostate cancer cells using the prostate cancer cell
line LNCaP. They showed that a concentration higher than 1 nM of a
synthetic androgen, R1881, inhibited cell growth, whereas
concentrations of 0.1 nM and below stimulated proliferation. This
biphasic effect of androgens on cancer cells was previously
demonstrated by other groups (1,15,16), but the effect of androgen on the
growth of the non-transformed cells remains uncertain. The actions
of androgen are controversial and complex, since after binding to
androgen, AR is able to recruit general transcription factors to
its target gene promoters. It has become clear that the
transcriptional activity of AR is regulated by coregulators,
including both coactivators and corepressors, by various mechanisms
(37).
The present study also showed that the expression of
the p53 and p21 genes differs according to the
androgen concentrations added to the culture medium of HNTEP cells.
Thus, cells incubated at low DHT concentrations exhibited lower
p53 and p21 mRNA levels than those treated with DHT
at 10−8 M. The inhibition of these tumor suppressor
genes suggested that under the influence of low concentrations of
androgens, HNTEP cells are induced to progress into the cell
cycle.
Androgens can stimulate proliferation and
differentiation and suppress apoptosis in the prostate gland
(21). Defects in apoptotic
signaling pathways are common in cancer cells. Inhibition of
apoptosis is important for tumor initiation, since apoptosis is
involved in the process of eliminating cells with different
anomalies that lead to malignant transformation. There are many
apoptosis-modulating proteins, such as Akt and p53 (38,39). Mutations of the p53 gene
are associated with tumors and proliferative disturbance of
prostate tissue (29). In this
study, we worked with the wild-type form of p53,
demonstrating an increase of p53 mRNA levels with the higher
dose of DHT after 4 h of treatment. We showed that the
anti-androgen hydroxyflutamide blocked this effect, indicating that
the p53 gene is a target of androgen modulation in HNTEP cells. In
the present study we also assessed the protein expression of p53 in
HNTEP cells to determine the presence of functional protein. The
results regarding protein levels of p53 are controversial.
Bruckheimer et al (40),
reported an increase in the protein levels of p53 and
Bax after treatment with 1 nM of DHT in a hormone-sensitive
LNCaP-FGC cell line. Moreover, some studies have shown a decrease
in protein levels of the p53 gene in LNCaP after 24 h of
treatment with DHT over a range of 10−11 to
10−7 M (41). These
results could be due to post-transcriptional adjustments which
result in p53 stabilization. Rokhlin et al (30), using the same cell line (LNCaP),
also reported a decrease in the levels of p53 mRNA after 3
days of treatment with 10−11 and 10−9 M of
DHT, and an important role in TNF-α mediated apoptosis in LNCaP
cells (39). There are
androgen-responsive elements in the p53 gene, which may
explain the regulation of this gene expression by androgens
(30). There is little
information on androgen modulation of p53 expression in
non-transformed epithelial prostate cells. Khan et al
(42) reported a higher
expression of the p53 gene in the prostate gland of E6-AP
(E6-associated protein)-null mice, suggesting that E6-AP deletion
attenuates the growth and development of the prostate gland by
interfering with AR function as well as by stimulating
p53-mediated apoptosis.
Previous studies have reported p53 expression
in prostate tumors and immortalized cell lines (43,44). The p53 gene may influence the
expression of other genes; the present study analyzed the
expression of the p21 gene, which has been reported to be a
target of p53 action (23,28,42,45).
The p21 gene induces apoptosis mechanisms and
blocks the progression into the cell cycle (24,45). Although the role of the p21
gene in prostate diseases is not well established, the existence of
an androgen-responsive element in its promotor region suggests that
this gene may be modulated by the androgen receptor (46). Evidence from the LNCaP and HPr-1
cell lines suggests that p21 is involved in the mechanism of
androgen action at different doses (26,27,31,47). Our results showed a decrease in
p21 gene expression in HNTEP cells treated with low androgen
concentrations, and an increase at high doses. These data are
consistent with the results of others who reported an increase in
p21 mRNA levels in LNCaP treated with a high dose
(10−8 M) of synthetic androgen (48). Moreover, a human cell line PC-3
transfected with AR, when treated with 10 nM of DHT, showed
upregulation of p21 levels, and the knockdown of AR
expression also resulted in downregulation of p21 protein
levels (49). A recent study by
semiquantitative RT-PCR analysis showed a high expression of the
p21 gene in DU145-Id4 cells, a lineage of DU145 cells
transfected with Id4 (inhibitor of differentiation 4, a member of
the Id gene family) and PrEC cells (an immortalized lineage of
normal human prostatic epithelial cells) as compared to parental
DU145 cells. In this study a highly significant increase (over
15-fold by real-time PCR) in p53 gene expression was found
in the DU145-Id4 cell line compared to DU145 cells (50). To the best of our knowledge, this
is the first report demonstrating the regulation of suppressor
genes p53 and p21 by androgen in human
non-transformed epithelial prostatic cells in primary culture
(HNTEP).
We also analyzed the protein levels of p21.
Protein expression tended to increase when HNTEP cells were treated
with high concentrations of DHT (10−8 M). The increase
in the p21 protein levels by high doses of androgens has
been demonstrated in other cell-culture models using different
prostate-cell lines (26,27,48). The results of the proliferation
rates of the HNTEP cells submitted to androgen treatment could be
interpreted as indirect evidence favoring the hypothesis of
involvement of these genes in the regulation of the cell cycle,
associated with different doses of androgens. Taken together, these
data may indicate that under the influence of androgens the
expression of both p53 and p21 genes could be
differentially changed in order to allow modulation in cell
proliferation.
Benign prostatic hyperplasia (BPH) is the most
prevalent disease of the prostate. The incidence of BPH coincides
with decreasing circulating testosterone levels in senescence
(51) and is associated with an
imbalance between the rate of cell proliferation and cell death.
The association between low concentrations of androgens and
prostate cell proliferation in our in vitro system of HNTEP
culture cells could be seen as similar to the endocrine events
occurring in male senescence and prostate growth. Thus, changes in
the expression of the p21 and p53 genes might be part
of the mechanisms by which low androgen concentrations are related
to BPH. In this context, studies on suppressor genes continue to be
performed, in order to find molecular markers for states of
uncontrolled growth in target tissues for androgen action (31,43,52–54). In conclusion, the present data
showed that HNTEP cell proliferation could be induced by low
androgen concentrations, and was related to lower p53 and
p21 gene expression at these concentrations. Further studies
are required to assess the intracellular signaling pathway
regulated by p53 and p21 under the influence of
androgen stimulation in human prostate epithelial cells and its
implications for the pathophysiology of benign and neoplastic
transformation of the human adult prostate.
Acknowledgements
The authors thank the Graduate
Research Group (GPPG) at Clinical Hospital de Clínicas de Porto
Alegre for the editorial support provided, and the Urology Services
at Hospital de Clinicas de Porto Alegre, Hospital São Lucas/PUCRS
and Hospital Ernesto Dorneles for the support in obtaining
prostatic tissue. This study was supported in part by Fundação de
Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS) and
Conselho Nacional de Desenvolvimento Científico e Tecnológico
(CNPq).
References
|
1.
|
C SonnenscheinN OleaME PasanenAM
SotoNegative controls of cell proliferation: human prostate cancer
cells and androgensCancer Res493474378119892731169
|
|
2.
|
BJ LongDN GrigoryevIP NnaneY LiuYZ LingAM
BrodieAntiandrogenic effects of novel androgen synthesis inhibitors
on hormone-dependent prostate cancerCancer
Res6066306640200011118046
|
|
3.
|
JM HaynesM FrydenbergH
MajewskiTestosteroneand phorbol ester-stimulated proliferation in
human cultured prostatic stromal cellsCell
Signal13703709200110.1016/S0898-6568(01)00205-411602180
|
|
4.
|
Z CuligA HobischG BartschH KlockerAndrogen
receptor - an update of mechanisms of action in prostate cancerUrol
Res28211219200010.1007/s00240000011111011957
|
|
5.
|
TH LiH ZhaoY PengJ BeliakoffJD BrooksZ
SunA promoting role of androgen receptor in androgen-sensitive and
-insensitive prostate cancer cellsNucleic Acids
Res3527672776200710.1093/nar/gkm19817426117
|
|
6.
|
X YuanT LiH WangT ZhangM BaruaRA BorgesiGJ
BubleyML LuSP BalkAndrogen receptor remains critical for cell-cycle
progression in androgen-independent CWR22 prostate cancer cellsAm J
Pathol169682696200610.2353/ajpath.2006.05104716877366
|
|
7.
|
M BlanchereI BerthautMC PortoisC MestayerI
MowszowiczHormonal regulation of the androgen receptor expression
in human prostatic cells in cultureJ Steroid Biochem Mol
Biol66319326199810.1016/S0960-0760(98)00056-99749837
|
|
8.
|
C AvancesV GeorgetB TerouanneF OrioO
CussenotN MottetP CostaC SultanHuman prostatic cell line PNT1A, a
useful tool for studying androgen receptor transcriptional activity
and its differential subnuclear localization in the presence of
androgens and antiandrogensMol Cell
Endocrinol1841324200110.1016/S0303-7207(01)00669-4
|
|
9.
|
B PlanzQ WangSD KirleyM MarbergerWS
McDougalRegulation of keratinocyte growth factor receptor and
androgen receptor in epithelial cells of the human prostateJ
Urol166678683200110.1016/S0022-5347(05)66042-911458116
|
|
10.
|
K NakanoY FukaboriN ItohW LuM KanWL
McKeehanH YamanakaAndrogen-stimulated human prostate epithelial
growth mediated by stromal-derived fibroblast growth
factor-10Endocr J46405413199910.1507/endocrj.46.40510503993
|
|
11.
|
JT ArnoldX LiuJD AllenH LeKK McFannMR
BlackmanAndrogen receptor or estrogen receptor-beta blockade alters
DHEA-, DHT-, and E(2)-induced proliferation and PSA production in
human prostate cancer
cellsProstate6711521162200710.1002/pros.2058517503469
|
|
12.
|
P BerthonAS WallerJM VilletteL LoridonO
CussenotNJ MaitlandAndrogens are not a direct requirement for the
proliferation of human prostatic epithelium in vitroInt J
Cancer73910916199710.1002/(SICI)1097-0215(19971210)73:6%3C910::AID-IJC25%3E3.0.CO;2-69399675
|
|
13.
|
LE HeislerA EvangelouAM LewJ
TrachtenbergHP ElsholtzTJ BrownAndrogen-dependent cell cycle arrest
and apoptotic death in PC-3 prostatic cell cultures expressing a
full-length human androgen receptorMol Cell
Endocrinol1265973199710.1016/S0303-7207(96)03970-69027364
|
|
14.
|
D KrillJ StonerBR KonetyMJ BecichRH
GetzenbergDifferential effects of vitamin D on normal human
prostate epithelial and stromal cells in primary
cultureUrology54171177199910.1016/S0090-4295(99)00103-X10414747
|
|
15.
|
MO Joly-PharabozMC SoaveB NicolasF
MebarkiM RenaudO FouryY MorelJG AndreAndrogens inhibit the
proliferation of a variant of the human prostate cancer cell line
LNCaPJ Steroid Biochem Mol
Biol556776199510.1016/0960-0760(95)00155-S7577722
|
|
16.
|
MO Joly-PharabozA RuffionA RochL
Michel-CalemardJ AndreJ ChantepieB NicolasG PanayeInhibition of
growth and induction of apoptosis by androgens of a variant of
LNCaP cell lineJ Steroid Biochem Mol
Biol73237249200010.1016/S0960-0760(00)00076-511070352
|
|
17.
|
C ShaoY WangHH YueYT ZhangCH ShiF LiuTY
BaoZY YangJL YuanGX ShaoBiphasic effect of androgens on prostate
cancer cells and its correlation with androgen receptor coactivator
dopa decarboxylaseJ
Androl28804812200710.2164/jandrol.106.00215417581945
|
|
18.
|
P GeckJ SzeleiJ JimenezTM LinC
SonnenscheinA SotoExpression of novel genes linked to the
androgen-induced, proliferative shutoff in prostate cancer cellsJ
Steroid Biochem Mol
Biol63211218199710.1016/S0960-0760(97)00122-29459187
|
|
19.
|
IS SilvaDM MorschL UrnauerPM
SpritzerAndrogen-induced cell growth and c-myc expression in human
non-transformed epithelial prostatic cells in primary cultureEndocr
Res27153169200110.1081/ERC-10010717711428707
|
|
20.
|
IS BrumDM MorschA PozzobonVA BoeriG GeibPM
SpritzerAndrogen-dependent expression of c-jun and c-fos in human
non-transformed epithelial prostatic cells: association with cell
proliferationHormone Res60209214200310.1159/00007403314614224
|
|
21.
|
R ButtyanA ShabsighH PerlmanM
ColombelRegulation of apoptosis in the prostate gland by androgenic
steroidsTrends Endocrinol
Metab104754199910.1016/S1043-2760(98)00104-010322394
|
|
22.
|
MJ SolomonP KaldisRegulation of CDKs by
phosphorylationResults Probl Cell
Differ2279109199810.1007/978-3-540-69686-5_4
|
|
23.
|
S WagaGJ HannonD BeachB StillmanThe p21
inhibitor of cyclin-dependent kinases controls DNA replication by
interaction with PCNANature369574578199410.1038/369574a07911228
|
|
24.
|
CJ SherrJM RobertsCDK inhibitors: positive
and negative regulators of G1-phase progressionGenes
Dev1315011512199910.1101/gad.13.12.150110385618
|
|
25.
|
CN RobsonV GnanapragasamRL ByrneAT
CollinsDE NealTransforming growth factor-beta1 up-regulates p15,
p21 and p27 and blocks cell cycling in G1 in human prostate
epitheliumJ Endocrinol160257266199910.1677/joe.0.16002579924195
|
|
26.
|
S LuSY TsaiMJ TsaiMolecular mechanisms of
androgen-independent growth of human prostate cancer LNCaP-AI
cellsEndocrinology14050545059199910537131
|
|
27.
|
MT LingKW ChanCK ChooAndrogen induces
differentiation of a human papillomavirus 16 E6/E7 immortalized
prostate epithelial cell lineJ
Endocrinol170287296200110.1677/joe.0.170028711431162
|
|
28.
|
KF MacleodN SherryG HannonD BeachT TokinoK
KinzlerB VogelsteinT Jacksp53-dependent and independent expression
of p21 during cell growth, differentiation, and DNA damageGenes
Dev9935944199510.1101/gad.9.8.9357774811
|
|
29.
|
S DowningP JacksonP RussellMutations
within the tumor suppressor gene p53 are not confined to a late
event in prostate cancer progression: a review of the evidenceUrol
Oncol6103110200110.1016/S1078-1439(00)00119-811344000
|
|
30.
|
OW RokhlinAF TaghiyevNV GusevaRA GloverPM
ChumakovJE KravchenkoMB CohenAndrogen regulates apoptosis induced
by TNFR family ligands via multiple signaling pathways in
LNCaPOncogene2467736784200510.1038/sj.onc.120883316007156
|
|
31.
|
LG WangL OssowskiAC FerrariOverexpressed
androgen receptor linked to p21WAF1 silencing may be responsible
for androgen independence and resistance to apoptosis of a prostate
cancer cell lineCancer Res6175447551200111606392
|
|
32.
|
PM SpritzerIS SilvaIO OliveiraDM MorschG
CoralCulture of adult human prostatic epithelial cells: A
simplified method for obtaining primary culturesMed Sci
Res233793811995
|
|
33.
|
T MosmannRapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assaysJ Immunol
Methods655563198310.1016/0022-1759(83)90303-46606682
|
|
34.
|
ME TaplinGJ BubleyTD ShusterME FrantzAE
SpoonerGK OgataHN KeerSP BalkMutation of the androgen-receptor gene
in metastatic androgen-independent prostate cancerN Engl J
Med33213931398199510.1056/NEJM1995052533221017723794
|
|
35.
|
M BradfordA rapid and sensitive method for
quantitation of microgram quanties of protein utilizing the
principle of protein-dye bindingAnal
Biochem72248255197610.1016/0003-2697(76)90527-3942051
|
|
36.
|
N OleaK SakabeAM SotoC SonnenscheinThe
proliferative effect of ‘anti-androgens’ on the androgen-sensitive
human prostate tumor cell line
LNCaPEndocrinology126145714631990
|
|
37.
|
B HeJ ChenZ BasrawalaH XinD ChoubeyThe
FXXLF motif mediates androgen receptor-specific interactions with
coregulatorsJ Biol
Chem2771022610235200210.1074/jbc.M11197520011779876
|
|
38.
|
TO ChanSE RittenhousePN TsichlisAKT/PKB
and other D3 phosphoinositide-regulated kinases: kinase activation
by phosphoinositide-dependent phosphorylationAnnu Rev
Biochem689651014199910.1146/annurev.biochem.68.1.965
|
|
39.
|
OW RokhlinAV GudkovS KwekRA GloverAS
GewiesMB Cohenp53 is involved in tumor necrosis
factor-alpha-induced apoptosis in the human prostatic carcinoma
cell line
LNCaPOncogene1919591968200010.1038/sj.onc.120345310773886
|
|
40.
|
EM BruckheimerK SpurgersNL WeigelC
LogothetisTJ McDonnellRegulation of Bcl-2 expression by
dihydrotestosterone in hormone sensitive LNCaP-FGC prostate cancer
cellsJ
Urol16915531557200310.1097/01.ju.0000055140.91204.c712629413
|
|
41.
|
PV NantermetJ XuY YuP HodorD HolderS
AdamskiMA GentileDB KimmelS HaradaD GerholdIdentification of
genetic pathways activated by the androgen receptor during the
induction of proliferation in the ventral prostate glandJ Biol
Chem27913101322200410.1074/jbc.M31020620014576152
|
|
42.
|
OY KhanG FuA IsmailS SrinivasanX CaoY TuS
LuZ NawazMultifunction steroid receptor coactivator, E6-associated
protein, is involved in development of the prostate glandMol
Endocrinol20544559200610.1210/me.2005-011016254014
|
|
43.
|
JR ZhouL YuLF ZerbiniTA LibermannGL
BlackburnProgression to androgen-independent LNCaP human prostate
tumors: cellular and molecular alterationsInt J
Cancer110800806200410.1002/ijc.2020615170660
|
|
44.
|
SR SchwarzeY ShiVX FuPA WatsonDF
JarrardRole of cyclin-dependent kinase inhibitors in the growth
arrest at senescence in human prostate epithelial and uroepithelial
cellsOncogene2081848192200110.1038/sj.onc.120504911781834
|
|
45.
|
R LiS WagaGJ HannonD BeachB
StillmanDifferential effects by the p21 CDK inhibitor on
PCNA-dependent DNA replication and
repairNature371534537199410.1038/371534a07935768
|
|
46.
|
S LuG JensterDE EpnerAndrogen induction of
cyclin-dependent kinase inhibitor p21 gene: role of androgen
receptor and transcription factor Sp1 complexMol
Endocrinol14753760200010.1210/mend.14.5.046110809237
|
|
47.
|
S YehYC HuM RahmanHK LinCL HsuHJ TingHY
KangC ChangIncrease of androgen-induced cell death and androgen
receptor transactivation by BRCA1 in prostate cancer cellsProc Natl
Acad Sci USA971125611261200010.1073/pnas.19035389711016951
|
|
48.
|
S LuM LiuDE EpnerSY TsaiMJ TsaiAndrogen
regulation of the cyclin-dependent kinase inhibitor p21 gene
through an androgen response element in the proximal promoterMol
Endocrinol13376384199910.1210/mend.13.3.025410076995
|
|
49.
|
F AlimirahJ ChenZ BasrawalaH XinD
ChoubeyDU-145 and PC-3 human prostate cancer cell lines express
androgen receptor: implications for the androgen receptor functions
and regulationFEBS
Lett58022942300200610.1016/j.febslet.2006.03.04116580667
|
|
50.
|
JP CareyAJ AsirvathamO GalmTA GhogomuJ
ChaudharyInhibitor of differentiation 4 (Id4) is a potential tumor
suppressor in prostate cancerBMC
Cancer9173200910.1186/1471-2407-9-17319500415
|
|
51.
|
JD McConnellProstatic growth: new insights
into hormonal regulationBr J Urol7651019957544215
|
|
52.
|
A GotohT ShirakawaY WadaM FujisawaH OkadaS
KamidonoK HamadaThe growth inhibitory effect of p21 adenovirus on
androgen-dependent and -independent human prostate cancer cellsBJU
Int92314318200310.1046/j.1464-410X.2003.04318.x12887490
|
|
53.
|
H MiyakeA TolcherME GleaveAntisense Bcl-2
oligodeoxynucleotides inhibit progression to androgen-independence
after castration in the Shionogi tumor modelCancer
Res5940304034199910463603
|
|
54.
|
J ConcatoD JainE UchioH RischWW LiCK
WellsMolecular markers and death from prostate cancerAnn Intern
Med150595603200910.7326/0003-4819-150-9-200905050-0000519414838
|