Introduction
Prion diseases or transmissible spongiform
encephalopathies (TSEs) are a group of animal and human brain
diseases that are uniformly fatal and often characterized by a long
incubation period and spongiform degeneration, multifocal
neuropathologic picture of neuronal loss and neuronal death
(1). Prion diseases are peculiar
in that they are caused by an infectious agent, prion, whose main
component is an abnormal isoform (PrPSc) of prion protein (PrP)
(2). A synthetic peptide similar
to sequence 106–126 of human [PrP (106–126)] induces apoptosis in
primary rat hippocampal cultures (3). PrP (106–126), which is composed of
amino acid sequence 106–126 of PrP, has been reported to induce
apoptosis in a variety of cells (4). PrP (106–126) has been used to
explore the neurotoxic mechanisms underlying prion disorders and
has been demonstrated to induce mitochondria dysfunction (3).
Mitochondria fulfill various important roles in
biosynthetic pathways, cellular redox homeostasis, cellular
energetics, cellular differentiation and regulation of programmed
cell death (5). Mitochondrial
dysfunction caused by abnormal regulation of mitochondrial dynamic
proteins may lead to neuropathological changes in prion diseases
(6). In addition, mitochondrial
dysfunction caused by translocation of the bax protein into the
mitochondrial membrane and oxidative stress gives rise to
neurodegeneration in prion disease (7,8).
Mitochondrial inhibitors protect neuronal cells from oxidative
stress-induced cell death (9). An
important effect of mitochondrial dysfunction is reactive oxygen
species (ROS) generation (10).
Oxidative stress results from increased content of
ROS (11). Oxidation of the
mitochondrial permeability transition pores by ROS may contribute
to cytochrome c release due to disruption of the
mitochondrial membrane potential (12). Oxidative stress leads to the
intracellular production of ROS (13). Hence, oxidative stress can cause
irreversible cellular damage since intracellular defense mechanisms
are depleted and therefore cannot protect cells against
ROS-mediated damage (13). In
particular, ROS are recognized as crucial mediators of biological
responses (14), including
neurode-generative disorders by misfolded protein (4). PrP contributes to the neuronal loss
that occurs in prion disorders, through mechanisms involving
modulation of cellular oxidation pathways (4).
Previous reports indicated that insulin-like growth
factor (IGF) signaling can reduce oxidative stress in
neurodegenerative disorders (15). Insulin-like growth factor-1
(IGF-1) is a multifunctional peptide that is structurally similar
to insulin (16) and which is
essential for normal fetal and postnatal growth, development,
metabolism and apoptosis in mammals (16). IGF-1 is a trophic hormone with
multiple neuroprotective actions (17). IGF-1 has salutary effects on
mitochondria. However, the molecular mechanism of IGF-1-mediated
neuronal survival is only beginning to be understood. Moreover, the
pathogenesis of a number of neurodegenerative diseases is
attributed to IGF-1 deficiency (18). IGF-1 has a neuroprotective effect
via regulation of the serine kinase Akt that forms part of the
canonical IGF-I pro-survival signaling pathway (17) and which is altered in
neurodegenerative diseases including Huntington’s disease and
spinocerebellar ataxia (19).
We hypothesized that IGF-1 prevents oxidative stress
and neuronal cell death. In the present study we investigated this
hypothesis and found that IGF-1 treatment prevents prion-mediated
mitochondrial dysfunction and neurotoxicity in neuronal cells. We
tested whether IGF-1 prevents neuronal cell death by PrP (106–126)
and assessed the therapeutic value of IGF-1 in the treatment of
neurodegenerative disorders.
Materials and methods
Cell culture
The SH-SY5Y human neuroblastoma cell line was
obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA). Cells were cultured in Minimum Essential
Medium (MEM; Invitrogen Life Technologies-Gibco-BRL, Grand Island,
NY, USA) that contained 10% fetal bovine serum (FBS; Invitrogen
Life Technologies-Gibco-BRL) and penicillin-streptomycin (both 100
U/ml) in a humidified incubator maintained at 37°C and 5%
CO2.
Reagents
IGF-1 was purchased from Sigma-Aldrich (St. Louis,
MO, USA). Antioxidant agents [glutathione (GSH) and
N-acetylcysteine (NAC)] were purchased from Sigma-Aldrich.
PrP (106–126) treatment
Synthetic PrP (106–126) (sequence,
Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly)
was synthesized by Peptron (Seoul, Korea). The peptide was
dissolved in sterile dimethylsulfoxide (DMSO) at a concentration of
10 mM and stored at −80°C.
Western blot analysis
SH-SY5Y was lysed in a buffer containing 25 mM
HEPES; pH 7.4, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 0.1
mM dithiothreitol (DTT) and protease inhibitor mixture. Proteins
were electrophoretically resolved by 10–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and
immunoblotting was performed as previously described. Equal amounts
of lysate protein were similarly electrophoretically resolved and
electrophoretically transferred to a nitrocellulose membrane.
Immunoreactivity was detected through sequential incubation with
horseradish peroxidase-conjugated secondary antibody and enhanced
chemiluminescence reagents. The antibodies used for immunoblotting
were phospho-c-Jun, N-terminal kinase (JNK; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), Bcl-2 (Santa Cruz
Biotechnology, Inc.) and phospho-AKT (Cell Signaling Technology,
Inc., Cambridge, MA, USA).
Cellular fractionation
SH-SY5Y cells were resuspended in mitochondrial
buffer (210 mM sucrose, 70 mM mannitol, 1 mM EDTA and 10 mM HEPES),
broken by a 26-gauge needle and centrifuged at 700 x g for 10 min.
The postnuclear supernatant was centrifuged at 10,000 x g for 30
min. The pellet was used as the mitochondrial fraction, and the
super-natant was used as the cytosolic fraction. Total proteins
were obtained and subjected to western blotting.
Annexin V assay
Apoptosis was assessed by a commercial Annexin V
assay (Santa Cruz Biotechnology, Inc.), according to the
manufacture’s protocol. Annexin V content was determined by
measuring fluorescence at excitation 488 nm and emission at 525/30
using a Guava easyCyte HT system (Millipore, Billerica, MA,
USA).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
TUNEL analysis was performed to measure the degree
of cellular apoptosis using an in situ ApoBrdU DNA
fragmentation assay kit (BioVision, San Francisco, CA, USA),
following the manufacturer’s instructions.
DCFH-DA assay
SH-SY5Y cells were incubated in MEM (Hyclone
Laboratories, Logan, UT, USA) containing 10 μM
2′,7′-dichlorodihydrofluorescein diacetate (H2-DCFDA) at 37°C for
30 min. Cells were washed with phosphate-buffered saline (PBS) and
lysed in the aforementioned lysis buffer. Cells were transferred to
a clear 96-well plate, and fluorescent emission was measured at 515
nm on bottom read, with an excitation wavelength of 488 nm, using a
SpectraMax M2 instrument (Molecular Devices, Sunnyvale, CA, USA).
SH-SY5Y cells were cultured on cover slips positioned in a 24-well
plate. Cells were incubated in MEM (Hyclone Laboratories)
containing 10 μM H2-DCFDA) at 37°C for 30 min and were then washed
with PBS.
Mitochondrial transmembrane potential
(MTP) assay
The change in MTP was evaluated by the cationic
fluorescent indicator JC-1 (Molecular Probes, Eugene, OR, USA),
which aggregates in intact mitochondria (red fluorescence)
indicating high or normal MTP and low MTP when it remains in
monomeric form in the cytoplasm (green fluorescence). SH-SY5Y cells
were incubated in MEM containing 10 μM JC-1 at 37°C for 30 min,
washed with PBS, and subsequently transferred to a clear 96-well
plate. JC-1 aggregate fluorescent emission was measured at 583 nm
with an excitation wavelength of 526 nm, and JC-1 monomer
fluorescence intensity was measured with an excitation and emission
wavelength of 525 and 530 nm, respectively, using a Guava easyCyte
HT System (Millipore). SH-SY5Y cells were cultured on cover slips
in a 24-well plate, incubated in MEM containing 10 μm JC-1 at 37°C
for 30 min and then washed with PBS. Finally, cells were mounted
with DakoCytomation fluorescent medium and visualized via
fluorescence microscopy.
Statistical analysis
All data are expressed as mean ± standard deviation
(SD), and were compared using the Student’s t-test and the ANOVA
Duncan’s test with the SAS statistical package (SAS, Cary, NC,
USA). The results were considered statistically significant at
*P<0.05 or **P<0.01.
Results
IGF-1 protects against PrP
(106–126)-induced neuronal cell death
IGF-1 is neuroprotective in neurodegenerative
diseases and is involved in Huntington’s disease (20). To examine whether IGF-1 treatment
protects neuronal cells from PrP (106–126)-mediated neurotoxic
effects, SH-SY5Y cells were pretreated with IGF-1 before exposure
to PrP (106–126). The protective effect of IGF-1 was determined by
an Annexin V viability assay. SH-SY5Y cells were pretreated for 12
h with 200 ng/ml IGF-1 and then exposed to 100 μM PrP (106–126) for
24 h. Cells were responsive to PrP (106–126) treatment (43.7%
increase in Annexin V-positive cells) and IGF-1 had no effect on
Annexin V assay results (Fig.
1A). As shown in Fig. 1B,
IGF-1 at different concentrations (50, 100, 200 and 400 ng/ml)
significantly attenuated the neurotoxicity induced by 24-h exposure
to 100 μM PrP (106–126). These results were confirmed by
morphological observations of the treated cells using light
microscopy (Fig. 1C). PrP
(106–126)-induced morphological changes were significantly
alleviated by IGF-1. TUNEL assay results revealed that the
concentration of fluorescent-fragmented nuclei increased in the 100
μM PrP (106–126)-treated group compared to the 200 ng/ml
IGF-1-pretreated group and the control group (Fig. 1D). These results suggest that
IGF-1 promotes SH-SY5Y survival by preventing cell death induced by
PrP (106–126).
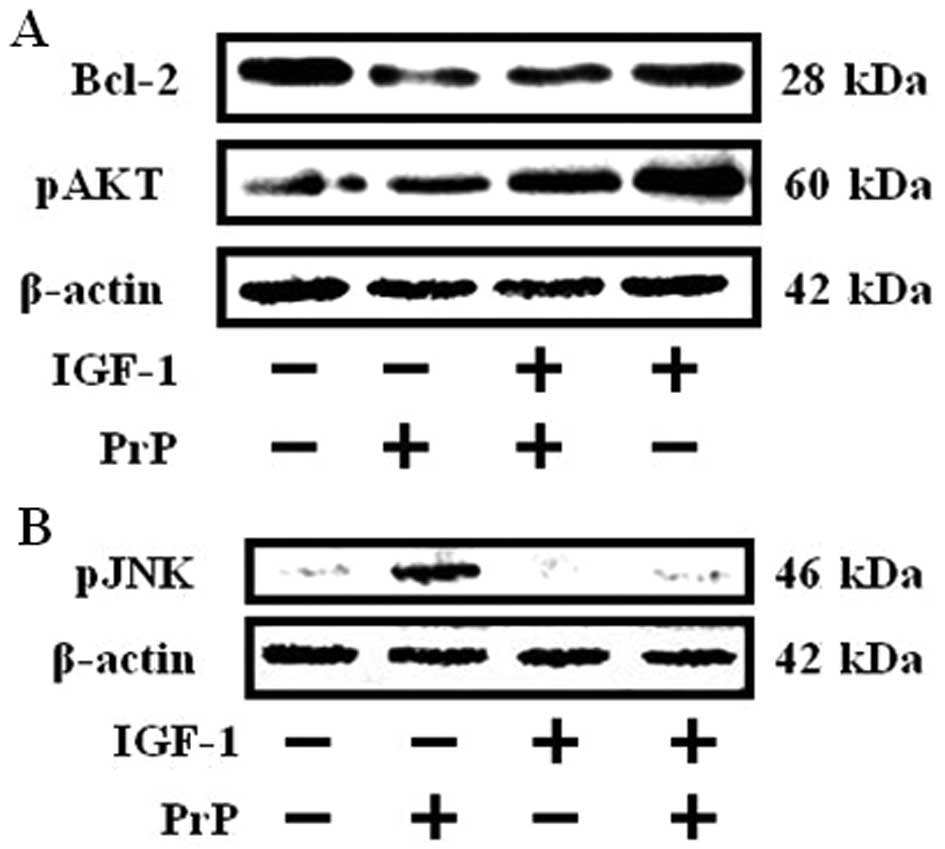
PrP (106–126)-mediated protein expression
is suppressed by IGF-1 treatment
PrP (106–126) impacts the activation of JNK and
expression of the Bcl-2 protein (3), and IGF-1 induces upregulated
expression of the antiapoptotic protein Bcl-2 and expression of a
constitutively active Akt inhibited JNK activation (5,21).
To estimate the effect of IGF-1 on PrP (106–126) affected
activation of JNK and expression of Bcl-2, SH-SY5Y cells were
pretreated for 12 h with 200 ng/ml of IGF-1 and then exposed for 18
h to 100 μM PrP (106–126). Western blot analyses revealed that the
activation of JNK increased and decreased Bcl-2 expression in the
100 μM PrP (106–126)-treated group compared to the IGF-1 (200
ng/ml) pretreated group and the control group (Fig. 2B). However, IGF-1 treatment
inhibited PrP (106–126)-induced activated JNK and inhibited Bcl-2
expression in SH-SY5Y cells (Fig.
2). In addition, IGF-1 enhanced phosphorylation of AKT.
However, PrP (106–126) had no effect on western blotting results
(Fig. 2A). These results suggest
that IGF-1 inhibits PrP (106–126)-induced activation of JNK and
decreases both Bcl-2 expression and AKT activation.
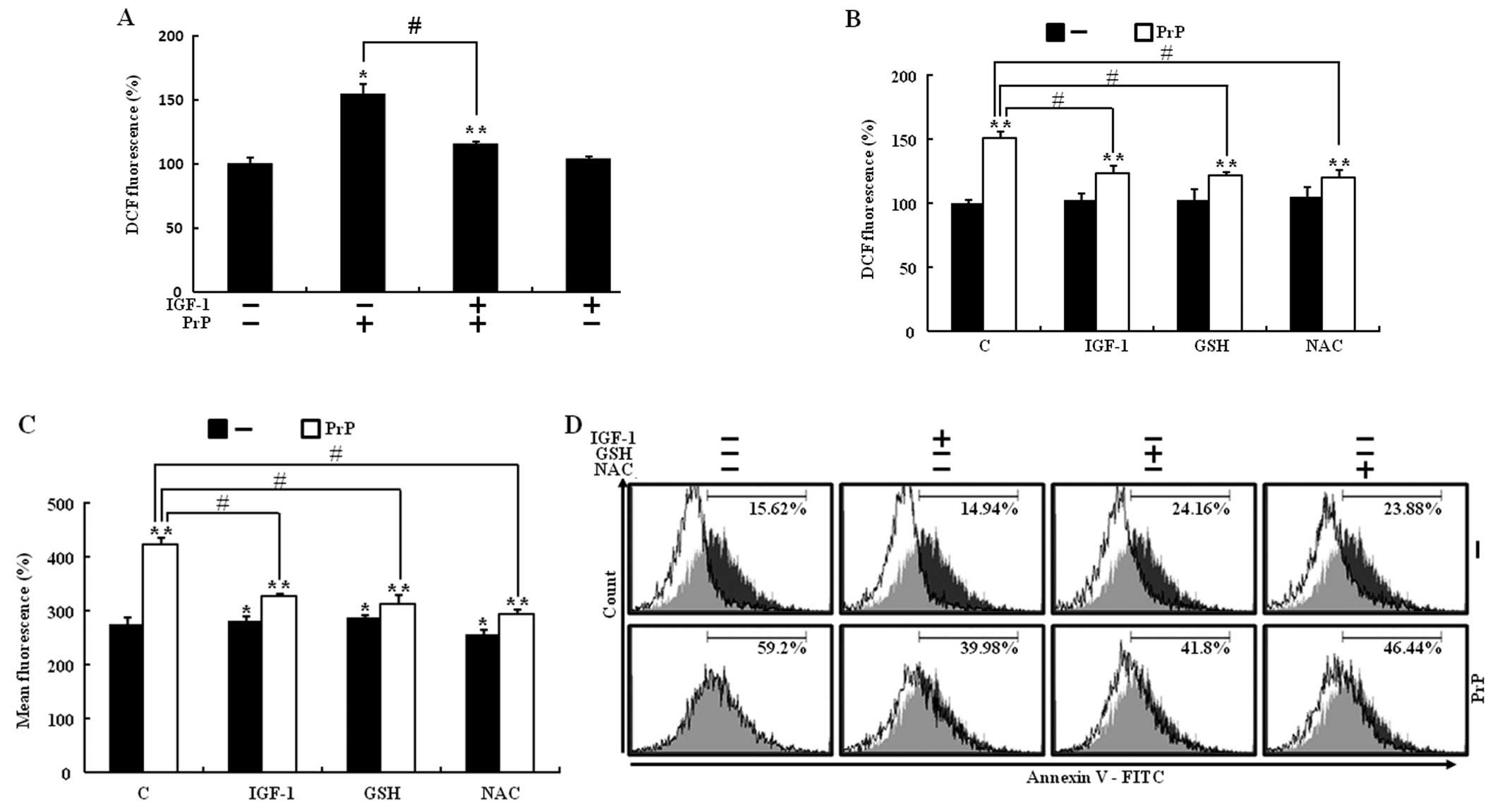
IGF-1 prevents PrP (106–126)-induced ROS
generation
IGF-1 reduces oxidative stress in neuronal cells
(22). Furthermore, PrP (106–126)
induces cell death as a result of its ability to regulate
intracellular ROS production (23). To investigate whether IGF-1
treatment had a neuroprotective effect as a result of inhibited ROS
generation in PrP (106–126)-induced neuronal cell death, SH-SY5Y
cells were pretreated for 12 h with 200 ng/ml of IGF-1 and then
exposed to 100 μM PrP (106–126) for 24 h. A DCFH-DA assay was
carried out to ascertain ROS generation. The addition of IGF-1 did
not change the level of DCFDA intensity, however, IGF-1 inhibited
ROS production in PrP (106–126)-induced increased ROS production
(Fig. 3A). To determine whether
IGF-1 treatment had a neuroprotective effect by decreasing ROS
production in PrP (106–126)-induced neuronal cell death, SH-SY5Y
cells were pretreated with IGF-1, antioxidant agents (GSH and NAC),
and then exposed to PrP (106–126). Following exposure to 100 μM PrP
(106–126), DCF fluorescence intensity in SH-SY5Y cells increased
significantly to 150% of the control value, whereas IGF-1 (200
ng/ml) or antioxidants (800 μM GSH or 4 mM NAC) led to a prominent
decrease in DCF fluorescence intensity (Fig. 3B and C). To investigate whether
decreased ROS production had a protective effect on PrP (106–126)
induced neuronal cell death, an Annexin V assay was used. Treatment
with IGF-1 and both antioxidant agents inhibited PrP
(106–126)-induced neuronal cell death (Fig. 3D). The results suggested that PrP
(106–126)-induced neuronal cell death via increased ROS generation
and IGF-1 treatment had a neuroprotective effect by decreasing ROS
production.
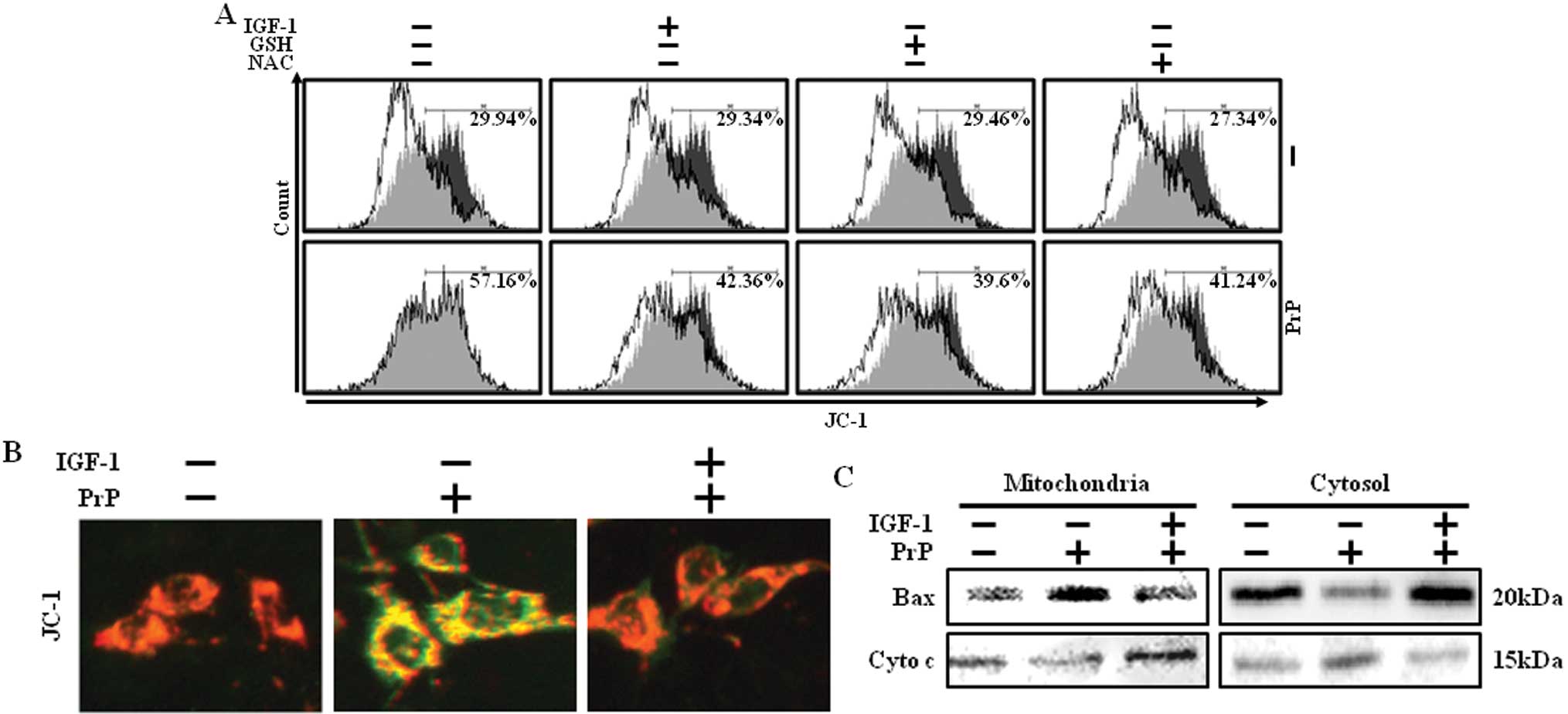
PrP (106–126)-induced mitochondrial
dysfunction can be suppressed by IGF-1
Previous studies have shown that mitochondrial
dysfunction increases oxidative stress and PrP (106–126)-induced
neurotoxicity through induced mitochondrial dysfunction (3,8).
To determine whether IGF-1 treatment had an antioxidant effect by
prevention of mitochondrial dysfunction in PrP (106–126)-induced
neuronal cell death, an MTP assay was conducted. PrP
(106–126)-treated cells showed increased JC-1 monomers, indicating
low MTP values, while IGF-1 treatment reduced PrP (106–126)-induced
JC-1 monomers, indicating high MTP values (Fig. 4A). Consistent with these results,
fluorescence microscopy also showed that IGF-1 could markedly
reduce the green fluorescence (JC-1 monomer form, gray) of PrP
(106–126)-induced neuronal cell death, and the negative control
cells and IGF-1-treated cells showed red fluorescence (JC-1
aggregate form, white) (Fig. 4B).
Since it has been previously established that the Bax protein is
associated with the mitochondrial apoptotic pathway (24), we examined the effect of IGF-1 on
PrP (106–126)-induced Bax translocation and cytochrome c
release. PrP (106–126)-induced translocation of Bax into
mitochondria and cytochrome c release to the cytosol in
SH-SY5Y cells. By contrast, PrP (106–126)-induced translocation of
Bax and cytochrome c release was blocked when pretreated
with IGF-1 (Fig. 4C).
Collectively, these results indicate that IGF-1 protects against
prion peptide induced-cell death in neuronal cells by blocking Bax
translocation.
Discussion
The synthetic peptide PrP (106–126) is composed of
human prion protein (PrP) amino acid residues 106–126 (25). PrP (106–126) maintains the
neurotoxic properties of the entire pathological PrPSc and is
widely used as a reasonable model to study the mechanism of prion
disorders (25). However, the
mechanism by which this peptide induces cell death in neuronal
cells is not fully understood. Most of these diseases (including
Creutzfeldt-Jakob disease, scrapie and Alzheimer’s disease) are
caused by accumulation of PrPSc (26). Therefore, the accumulation of
PrPSc has been postulated to be a solitary inducer of disease onset
due to neuronal cell death (26).
IGF-1 is a multifunctional peptide that is
structurally similar to insulin and has neuroprotective and
antiapoptotic properties (16).
The IGF-I pro-survival signaling pathway is altered in
neurodegenerative diseases including Huntington’s disease and
spinocerebellar ataxia (19).
Furthermore, scrapie infection affects the expression, binding
affinity and signal transduction mediated by IGF-1R in
neuroblastoma cells (27).
However, the protective effect of IGF-1 on PrP (106–126)-induced
neuronal cells has yet to be fully clarified.
We considered whether IGF-1 could exert a
neuroprotective effect on prion disease, and this prompted our
examination of the possible beneficial effects of IGF-1 on PrP
(106–126)-induced neuronal cell damage. Our results demonstrate
that IGF-1 prevents prion-induced neuronal cell death and
neurotoxicity in SH-SY5Y cells pretreated with IGF-1 for 12 h prior
to 24 h exposure to PrP (106–126) (Fig. 1).
PrP (106–126) induces neuronal cell death by
activating JNK protein phosphorylation and decreasing the level of
the Bcl-2 protein (21,28). JNK, an established mediator of
stress-induced apoptosis, is involved in the neurodegenerative
processes in neurodegenerative disorders (29), and represents a potential
therapeutic target for blockage of apoptosis induced by PrP
(106–126) (11). We demonstrated
the involvement of IGF-1 action on PrP (106–126)-induced
phosphorylated JNK and decreased Bcl-2 protein expression by
western blotting. PrP (106–126) induced the phosphorylation of JNK
and decreased Bcl-2 expression; these actions were inhibited by
IGF-1 (Fig. 2).
Similarly, previous studies have demonstrated that
activation of glycogen synthase kinase 3-β (GSK-3β) precedes the
activation of JNK and that this effect contributes to apoptotic
signaling (16). Markedly, the
activation of GSK-3β is an important mediator of prion
peptide-induced neurodegeneration and neuronal cell survival in
neurodegenerative disorders is mediated by JNK inactivation via
phosphorylated AKT dependent GSK-3β inhibition (16,30). In addition, the protective effects
of IGF-1 are mediated by pAKT expression in neuronal cells
(15). Our results indicate that
the inhibition of PrP (106–126)-induced JNK activation is a
downstream event of AKT/GSK-3β signaling and may add more weight to
GSK-3β blockage in the treatment of prion disorders.
Studies of neurodegenerative diseases have reported
physical disruption of the mitochondria (31). In addition, some recent reports
have shown that PrP (106–126) induces neuronal cell death by
mitochondrial disruption in neuroblastoma cells and mitochondrial
disruption by activated JNK and Bax trans-location (32). Therefore, PrP (106–126)-induced
mitochondrial disruption may possibly be the cause of oxidative
stress in neuronal cells. Oxidative stress results from increased
content of ROS and is the main source of the neuronal cell death;
ROS are particularly high in neurodegenerative disorders. Increased
ROS production by mitochondrial dysfunction ultimately causes cell
death. These results suggest that the increased oxidative stress is
central to the pathogenesis of prion diseases.
This study has shown that PrP (106–126)-induced
intracellular ROS production changed by IGF-1 treatment, and was
investigated by antioxidant agents (GSN and NAC) (Fig. 3).
The use of antioxidant agents demonstrated a
protective effect against PrP (106–126)-induced mitochondrial
disruption by inhibiting mitochondrial dysfunction. This protective
effect indicates that IGF-1 treatment may attenuate PrP
(106–126)-induced ROS generation via inhibition of mitochondrial
dysfunction. Furthermore, PrP (106–126) induces mitochondrial
dysfunction by Bax translocation (33). Bax translocation is a
critical event in neuronal apoptosis. Thus, we considered whether
IGF-1 has a neuroprotective effect on prion disease by blocking Bax
translocation. The present results show that IGF-1 blocks PrP
(106–126)-induced Bax translocation (Fig. 4C).
In summary, IGF-1 inhibits PrP (106–126)-induced ROS
production and consequently neuronal cell death by preventing
mitochondria dysfunction and Bax translocation to mitochondria. The
observation that IGF-1 inhibits Bax translocation by preventing
mitochondria dysfunction may have clinical benefits for
neurodegenerative chemotherapy in patients with diseases such as
prion disease.
Acknowledgements
This study was supported by the
Cooperative Research Program for Agriculture Science and Technology
Development (PJ907116) in the Rural Development Administration,
Republic of Korea.
References
|
1.
|
RR NairJK JohnsonPrions and neuro
degenerative diseases (Review)Afr J Biotechnol10236623742011
|
|
2.
|
AR ClarkeGS JacksonJ CollingeThe molecular
biology of prion propagationPhilos Trans R Soc Lond B Biol
Sci356185195200110.1098/rstb.2000.076411260799
|
|
3.
|
CN O’DonovanD TobinTG CotterPrion protein
fragment PrP-(106–126) induces apoptosis via mitochondrial
disruption in human neuronal SH-SY5Y cellsJ Biol
Chem27643516435232001
|
|
4.
|
YH PanYC WangLM ZhangSR DuanProtective
effect of edaravone against PrP106–126-induced PC12 cell deathJ
Biochem Mol Toxicol24235241201020806394
|
|
5.
|
DC LoganThe mitochondrial compartmentJ Exp
Bot5712251243200610.1093/jxb/erj151
|
|
6.
|
HS ChoiJM OhHY SinMitochondrial
dysfunction via differential modulation of mitochondrial
fusion/fission proteins in the brains of scrapie-infected
micePrion41742010
|
|
7.
|
A GrossJM McDonnellSJ KorsmeyerBCL-2
family members and the mitochondria in apoptosisGene
Dev1318991911199910.1101/gad.13.15.189910444588
|
|
8.
|
SI ChoiWK JuEK ChoiMitochondrial
dysfunction induced by oxidative stress in the brains of hamsters
infected with the 263 K scrapie agentActa
Neuropathol96279286199810.1007/s0040100508959754961
|
|
9.
|
Y SagaraK IshigeC TsaiP MaherTyrphostins
protect neuronal cells from oxidative stressJ Biol
Chem2773620436215200210.1074/jbc.M20389520012121989
|
|
10.
|
MS WangS BoddapatiS EmadiMR SierksCurcumin
reduces alpha-synuclein induced cytotoxicity in Parkinson’s disease
cell modelBMC Neurosci1157201020433710
|
|
11.
|
JL EvansID GoldfineBA MadduxGM GrodskyAre
oxidative stress-activated signaling pathways mediators of insulin
resistance and beta-cell
dysfunction?Diabetes5218200310.2337/diabetes.52.1.112502486
|
|
12.
|
HU SimonA Haj-YehiaF Levi-SchafferRole of
reactive oxygen species (ROS) in apoptosis
inductionApoptosis5415418200010.1023/A:100961622830411256882
|
|
13.
|
Y LeshemL SeriA LevineInduction of
phosphatidylinositol 3-kinase-mediated endocytosis by salt stress
leads to intracellular production of reactive oxygen species and
salt tolerancePlant
J51185197200710.1111/j.1365-313X.2007.03134.x17521408
|
|
14.
|
BPS KangS FrencherV ReddyA KesslerA
MalhotraLG MeggsHigh glucose promotes mesangial cell apoptosis by
oxidant-dependent mechanismAm J Physiol Renal
Physiol284F455F466200310.1152/ajprenal.00137.200212419773
|
|
15.
|
D DavilaI Torres-AlemanNeuronal death by
oxidative stress involves activation of FOXO3 through a two-arm
pathway that activates stress kinases and attenuates insulin-like
growth factor I signalingMol Biol
Cell1920142025200810.1091/mbc.E07-08-081118287535
|
|
16.
|
L WangHJ YangYY XiaZW FengInsulin-like
growth factor 1 protects human neuroblastoma cells SH-EP1 against
MPP+-induced apoptosis by AKT/GSK-3β/JNK
signalingApoptosis1514701479201010.1007/s10495-010-0547-z20963499
|
|
17.
|
JL TrejoE CarroE Garcia-GallowayI
Torres-AlemanRole of insulin-like growth factor I signaling in
eurodegenerative diseasesJ Mol Med
(Berl)82156162200410.1007/s00109-003-0499-714647921
|
|
18.
|
J ZhongWH LeeHydrogen peroxide attenuates
insulin-like growth factor-1 neuroprotective effect, prevented by
minocyclineNeurochem
Int51398404200710.1016/j.neuint.2007.04.00517531350
|
|
19.
|
L LaviolaA NatalicchioS PerriniF
GiorginoAbnormalities of IGF-I signaling in the pathogenesis of
diseases of the bone, brain, and fetoplacental unit in humansAm J
Physiol Endocrinol
Metab295E991E999200810.1152/ajpendo.90452.200818713961
|
|
20.
|
N SalehS MoutereauA DurrNeuroendocrine
disturbances in Huntington’s diseasePLoS One4e49622009
|
|
21.
|
S PugazhenthiA NesterovaC SableAkt/protein
kinase B up-regulates Bcl-2 expression through cAMP-response
element-binding proteinJ Biol
Chem2751076110766200010.1074/jbc.275.15.1076110753867
|
|
22.
|
H GustafssonT SoderdahlG JonssonJO
BrattengA ForsbyInsulin-like growth factor type 1 prevents
hyperglycemia-induced uncoupling protein 3 down-regulation and
oxidative stressJ Neurosci
Res77285291200410.1002/jnr.2014215211595
|
|
23.
|
M PietriA CapriniS
Mouillet-RichardOverstimulation of PrPC signaling pathways by prion
peptide 106–126 causes oxidative injury of bioaminergic neuronal
cellsJ Biol Chem2812847028479200616864581
|
|
24.
|
A NechushtanCL SmithI LamensdorfSH YoonRJ
YouleBax and Bak coalesce into novel mitochondria-associated
clusters during apoptosisJ Cell
Biol15312651276200110.1083/jcb.153.6.126511402069
|
|
25.
|
JS SeoJW SeolMH MoonJK JeongYJ LeeSY
ParkHypoxia protects neuronal cells from human prion protein
fragment-induced apoptosisJ
Neurochem112715722201010.1111/j.1471-4159.2009.06496.x19919574
|
|
26.
|
A SakudoK IkutaPrion Protein functions and
dysfunction in prion diseasesCurr Med
Chem16380389200910.2174/09298670978700267319149584
|
|
27.
|
P OstlundH LindegrenC PetterssonK
BedecsUp-regulation of functionally impaired insulin-like growth
factor-1 receptor in scrapie-infected neuroblastoma cellsJ Biol
Chem2763611036115200110.1074/jbc.M10571020011461928
|
|
28.
|
J CarimaloS CronierG PetitActivation of
the JNK-c-Jun pathway during the early phase of neuronal apoptosis
induced by PrP106–126 and prion infectionEur J
Neurosci2123112319200515932590
|
|
29.
|
S HunotM VilaP TeismannJNK-mediated
induction of cyclooxygenase 2 is required for neurodegeneration in
a mouse model of Parkinson’s diseaseProc Natl Acad Sci
USA101665670200414704277
|
|
30.
|
M PerezAI RojoF WandosellJ Diaz-NidoJ
AvilaPrion peptide induces neuronal cell death through a pathway
involving glycogen synthase kinase 3Biochem
J372129136200310.1042/BJ2002159612578563
|
|
31.
|
E SzegezdiSE LogueAM GormanA
SamaliMediators of endoplasmic reticulum stress-induced
apoptosisEMBO Rep7880885200610.1038/sj.embor.740077916953201
|
|
32.
|
A CorsaroS ThellungV VillaPrion protein
fragment 106–126 induces a p38 MAP kinase-dependent apoptosis in
SH-SY5Y neuroblastoma cells independently from the amyloid fibril
formationAnn NY Acad Sci10106106222003
|
|
33.
|
JK JeongMH MoonYJ LeeJW SeolSY
ParkMelatonin-induced autophagy protects against human prion
protein-mediated neurotoxicityJ Pineal ResJan302012(Epub ahead of
print).
|