Introduction
It is well documented that hypoxia and/or ischemia
can elicit the release of several neurotransmitters (1,2),
including glutamate (3). Such
elevated levels of glutamate, and the subsequent activation of
ionotropic NMDA receptors, can trigger the neuronal damage during
hypoxia and/or ischemia (4,5).
Glutamate homeostasis is therefore crucial to prevent neuronal
death after a hypoxic/ischemic episode. Glutamate transport is the
only mechanism for the removal of glutamate from the extracellular
fluid in the brain (6,7), and it is essential for maintaining
extracellular glutamate below neurotoxic levels in the normal brain
(8). Therefore, glutamate
transporters are considered to play a key role in the process of
increase in extracellular glutamate during hypoxia/ischemia.
To date, 5 distinguishing high-affinity,
Na+-dependent glutamate transporters have been
identified: excitatory amino acid transporter (EAAT)1,
glutamate-aspartate transporter (GLAST), EAAT2, glutamate
transporter-1 (GLT-1), EAAT3, excitatory amino acid carrier 1
(EAAC1), EAAT4 and EAAT5. These transporters are present throughout
the central nervous system (CNS), with GLT-1 being highly abundant
in astroglial cells, whereas GLAST exists at higher levels in
Bergmann glia in the cerebellum (9,10).
GLT-1 plays a critical role in CNS homeostasis, accounting for up
to 70% of glutamate clearance (10,11).
The roles of GLT-1 in hypoxia/ischemia-induced
injury and neuroprotection have attracted extensive attention.
However, the findings are controversial. A pharmacological study
indicated that the GLT-1 blocker reduces the ischemia-induced
glutamate release in rat cortical superfusates (12), revealing that GLT-1 releases
glutamate during ischemia. By contrast, Rao et al (13) reported that the antisense
knockdown of GLT-1 exacerbates ischemia-triggered neuronal damage
in the rat brain, suggesting that GLT-1 takes up glutamate to
protect neurons during ischemia. In addition, ischemic
preconditioning upregulates the GLT-1 protein which may play a role
in the neuroprotective mechanism of preconditioning (14). In neonatal rats, it was shown that
the neuroprotection of ceftriaxone preconditioning against
hypoxia/ischemia-induced neuronal injury is associated with
upregulation of GLT-1 expression (15). On the other hand, an association
between change in GLT-1 expression and hypoxia/ischemia has been
reported by several studies (16,17). Raghavendra et al (16) observed that the expression of
GLT-1 is reduced following transient global ischemia. Conversely,
chronic hypoxia upregulates the expression of EAAC1 and GLT-1, but
not GLAST (17). These findings
support the theory that GLT-1 has a complicated function
(cytoprotective vs. cytotoxic effects) after hypoxic/ischemic
episodes. Thus, it is necessary to explore the roles of GLT-1 in
neuronal injury or the neuroprotective effects in different
hypoxic/ischemic models.
Hydrogen sulfide (H2S), recently
considered a novel neuro-modulator in the CNS, has been shown to
protect astrocytes against H2O2-induced
neural damage by enhancing glutamate uptake (18), suggesting an impact of
H2S.on..lutamate.trans. on glutamate transporters. We
have also demonstrated that H2S protects PC12 cells
against chemical hypoxia-induced injury by inhibiting reactive
oxygen species (ROS) overproduction, extracellular signal-regulated
kinase 1/2 (ERK1/2) and the p38 mitogen-activated protein kinase
(MAPK) signaling pathways (19,20). Since it is reported that ROS and
the activation of the ERK1/2 pathway are involved in the
downregulation of GLT-1 protein expression induced by
H2O2 or amyloid-ß (Aß) in astrocytes
(19,21), we hypothesized that ROS and
ERK1/2-mediated downregulation of GLT-1 might be implicated in
chemical hypoxia-induced neuronal injury and that H2S
might confer neuroprotection by enhancing GLT-1 expression. To test
this hypothesis, PC12 cells, which are derived from chromafin cells
of the adrenal medulla, were exposed to cobalt chloride
(CoCl2), a well-known hypoxia mimetic agent, to
establish a model of chemical hypoxia injury. The effects of
CoCl2 and pretreatment with NaHS (a donor of
H2S) on GLT-1 expression were observed. We found that:
i) CoCl2 significantly inhibits the expression of GLT-1,
ROS and the ERK1/2 pathway contribute to this inhibitory effect;
ii) NaHS pretreatment clearly attenuates the inhibitory effect of
CoCl2 on GLT-1 expression; iii) DHK, a selective
inhibitor of GLT-1, blocks the neuroprotection of H2S
against CoCl2-induced injury in PC12 cells.
Materials and methods
Materials
NaHS, CoCl2, N-acetyl-L-cysteine (NAC),
Hoechst 33258, propidium iodide (PI), RNase and Rhodamine 123
(Rh123) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The
cell counter kit-8 (CCK-8) was purchased from Dojindo Laboratories
(Kumamoto, Japan). The DMEM medium and fetal bovine serum (FBS)
were supplied by Gibco-BRL (Grand Island, NY, USA). Anti-GLT-1
antibody was purchased from Abcam (Cambridge, UK). DHK was
purchased from Merck Co. Anti-β-actin antibody, horseradish
peroxidase (HRP)-conjugated secondary antibody and the BCA protein
assay kit were purchased from KangChen Bio-tech, Inc. (Shanghai,
China). Enhanced chemiluminescence (ECL) solution was purchased
from Nanjing KeyGen Biotech Co., Inc. (Nanjing, China).
Cell culture and treatments
The rat pheochromocytoma cell line PC12 cells were
purchased from the Sun Yat-Sen University Experimental Animal
Center, and were grown in DMEM medium supplemented with 10% FBS at
37°C under an atmosphere of 5% CO2 and 95% air.
According to our previous study (20), chemical hypoxia was achieved by
adding CoCl2 at 600 μM into the medium and cells were
incubated in the presence of CoCl2 for the indicated
times. The cytoprotective effects of H2S were observed
by administering 400 μM NaHS (a donor of H2S) for 30 min
prior to exposure to CoCl2 for 24 h. NAC (a scavenger of
ROS) or U0126 (a MEK1/2 inhibitor) was administered 60 or 120 min
prior to exposure of the PC12 cells to 600 μM CoCl2 for
24 h.
Cell viability assay
The CCK-8 assay was employed to investigate the cell
viability of PC12 cells cultured in 96-well plates. After the
indicated treatments, 10 μl CCK-8 solution was added to each well
of the plate and the cells in the plate were incubated for 4 h in
the incubator. The absorbance at 450 nm was measured with a
microplate reader (Molecular Devices, Sunnyvale, CA, USA). Means of
4 well optical density (OD) in the indicated groups were used to
calculate the percentage of cell viability according to the formula
below: Percentage of cell viability (%) = (ODtreatment
group/ODcontrol group) × 100%. The experiment was
repeated 3 times.
Nuclear staining for assessment of
apoptosis with Hoechst 33258
Morphological changes, such as chromosomal
condensation and fragmentation in the nuclei of PC12 cells, were
observed by Hoechst 33258 staining followed by photo-fluorography.
Cells were plated at a density of 1×106 cells/well in 35
mm dishes. Cells were preconditioned with 400 μM NaHS for 30 min,
and subsequently exposed to 600 μM CoCl2 for 48 h. To
test the role of GLT-1 in H2S-induced cytoprotection
against chemical hypoxia-induced apoptosis, cells were treated with
the GLT-1 inhibitor DHK for 30 min prior to preconditioning with
NaHS. At the end of the indicated treatments, cells were harvested
and fixed with 4% paraformaldehyde in 0.1 mol/l phosphate-buffered
saline (PBS, pH 7.4) for 10 min. After rinsing with PBS, the
nuclear DNA was stained with 5 mg/ml Hoechst 33258 solution for 10
min before being rinsed briefly with PBS and then visualized under
a fluorescence microscope (Bx50-FLA; Olympus, Tokyo, Japan). Viable
cells displayed a uniform blue fluorescence throughout the nucleus,
whereas apoptotic cells showed condensed and fragmented nuclei.
Flow cytometric analysis of
apoptosis
Treated PC12 cells were digested with trypsin (2.5
mg/ml), centrifuged at 350 × g for 10 min and the supernatant was
removed. Cells were washed twice with PBS and fixed with 70%
ice-cold ethanol. Cells were then centrifuged at 350 × g for 10
min, washed twice with PBS and adjusted to a concentration of
1×106 cells/ml. Subsequently, 0.5 ml RNase (1 mg/ml in
PBS) was added to a 0.5 ml cell sample. After gentle mixing with PI
(at a terminal concentration of 50 mg/l), mixed cells were filtered
and incubated in the dark at 4°C for 30 min before flow cytometric
analysis (FCM). The PI fluorescence of individual nuclei was
measured by a flow cytometer (Beckman-Coulter, Los Angeles, CA,
USA). Excitation, 488 nm; emission, 615 nm. The research software
matched with FCM was used to analyze all the data of DNA labeling.
In the DNA histogram, the amplitude of the sub-G1 DNA peak, which
is lower than the G1 DNA peak, represents the number of apoptotic
cells. The experiment was repeated 3 times.
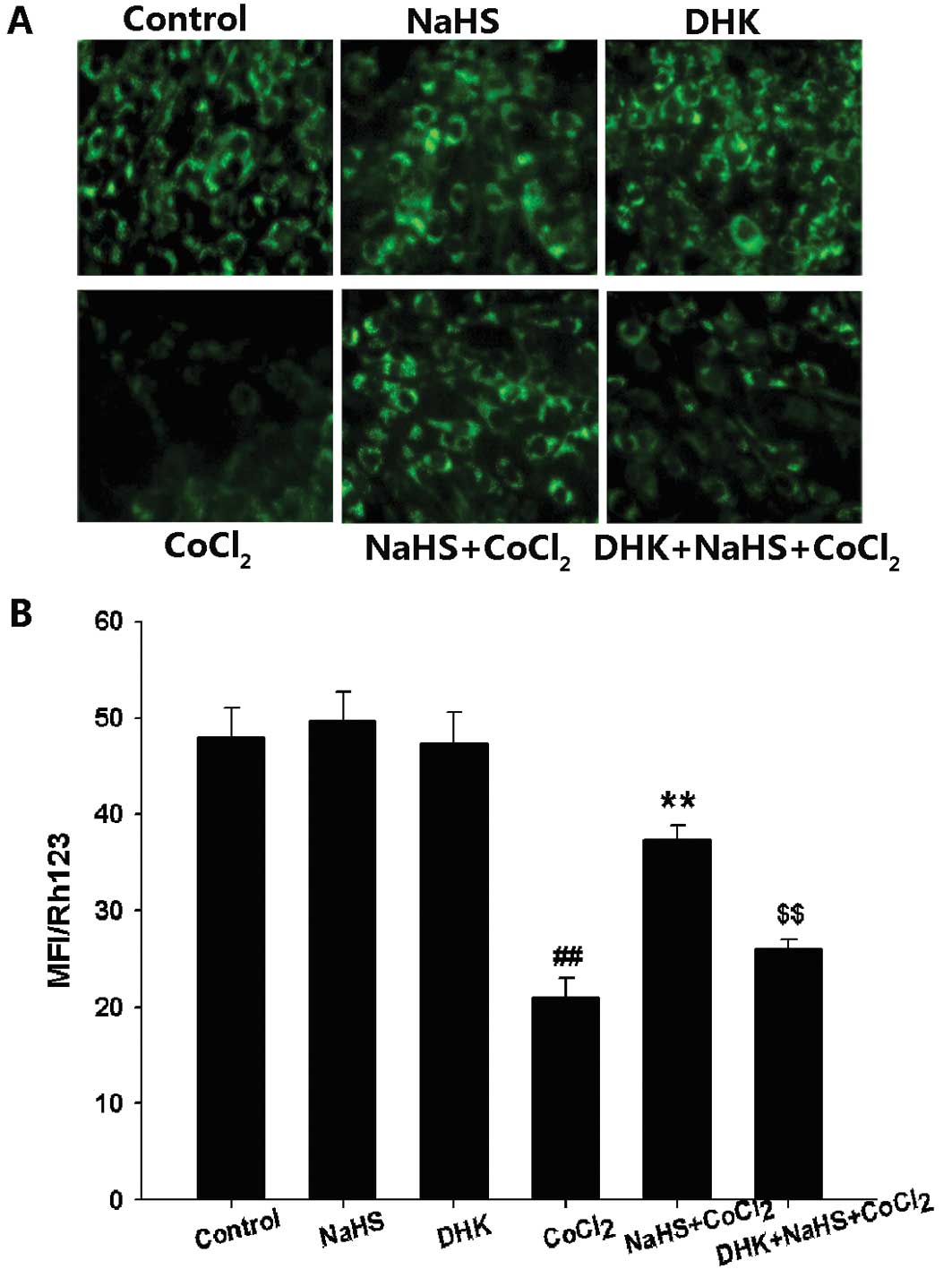
Measurement of MMP
Mitochondrial membrane potential (MMP) was monitored
using the fluorescent dye Rh123, a cell-permeable cationic dye that
preferentially enters into the mitochondria based on the highly
negative MMP. Depolarization of MMP results in loss of Rh123 from
the mitochondria and a decrease in intracellular fluorescence. In
the present study, PC12 cells were cultured in 24-well plates and
treated with 400 μM NaHS for 30 min prior to the administration of
600 μM CoCl2 for 24 h. DHK was administered 30 min prior
to NaHS preconditioning. To evaluate MMP, Rh123 (100 μg/l) was
added to cell cultures for 45 min at 37°C and fluorescence was
measured over the entire field of vision using a fluorescent
microscope connected to an imaging system (BX50-FLA; Olympus). The
mean fluorescence intensity (MFI) of Rh123 from 5 random fields was
analyzed using ImageJ 1.410 software (National Institutes of
Health, Bethesda, MD, USA), and the MFI was taken as an index of
the MMP. The experiment was repeated 3 times.
Western blot assay for protein
expression
After the cells were subjected to the indicated
treatments, they were harvested and lysed with cell lysis solution.
Total protein in the cell lysate was quantified using the BCA
protein assay kit. Sample buffer was added to cytosolic extracts,
and after boiling for 5 min, equal amounts of supernatant from each
sample were fractionated by 10% sodium dodecyl
sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Total
protein in the gel was transferred into polyvinylidene difluoride
(PVDF) membranes. Membranes were blocked for 1.5 h at room
temperature in fresh blocking buffer [0.1% Tween-20 in
Tris-buffered saline (TBS-T) containing 5% fat-free milk] and then
incubated with either anti-GLT-1 (1:2,500 dilution), or
anti-β-actin antibodies (1:5,000 dilution) in freshly prepared
TBS-T with 3% free-fat milk overnight with gentle agitation at 4°C.
After 3 washes with TBS-T, membranes were incubated with
HRP-conjugated goat anti-rabbit secondary antibodies (1:3,000
dilution; KangChen Bio-tech, Inc.) in TBS-T with 3% fat-free milk
for 1.5 h at room temperature. Membranes were washed 3 times with
TBS-T, developed in ECL solution and visualized with X-ray film.
Each experiment was repeated at least 3 times. For quantification,
the film were scanned and analyzed using ImageJ 1.410 software. The
density of specific bands was measured and normalized with the
bands of Action. The experiment was repeated 3 times.
Statistical analysis
Data are representative of experiments performed in
triplicate and are expressed as the mean ± SE. Differences between
groups were analyzed by one-way analysis of variance (ANOVA) using
SPSS 13.0 software, followed by the LSD post hoc comparison test.
P<0.05 was considered to indicate statistically significant
differences.
Results
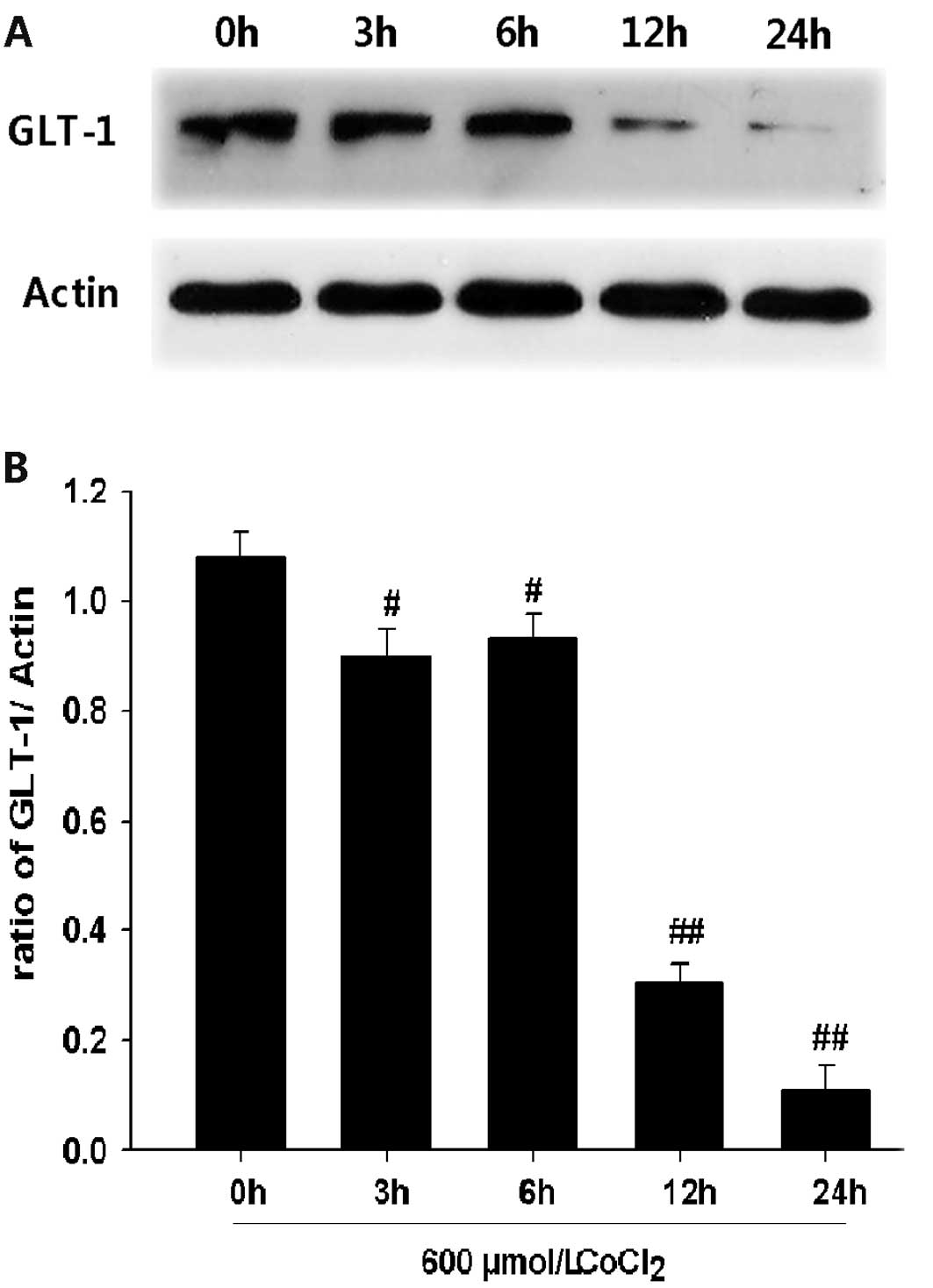
CoCl2 reduces the level of
GLT-1 expression in PC12 cells
In order to explore the effect of CoCl2
on the GLT-1 expression level in PC12 cells, PC12 cells were
exposed to 600 μM CoCl2 for the indicated times (i.e.,
3, 6, 12 and 24 h). Western blot analysis revealed that treatment
with 600 μM CoCl2 caused downregulation of GLT-1
expression in a time-dependent manner (Fig. 1). These data indicate that
chemical hypoxia may reduce GLT-1 protein levels in PC12 cells.
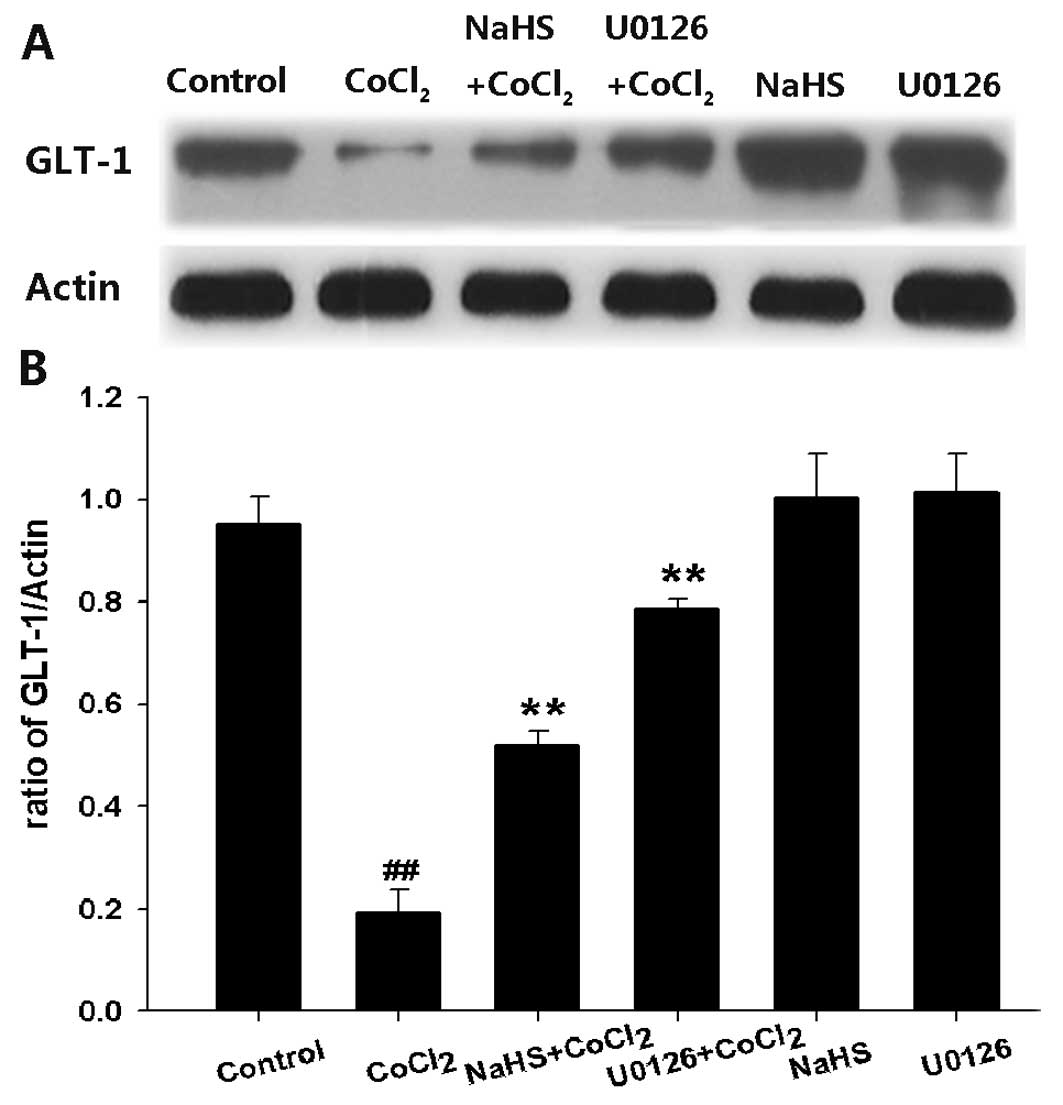
H2S reverses
CoCl2-induced downregulation of GLT-1 expression in PC12
cells
After PC12 cells were exposed to 600 μM
CoCl2 for 24 h, the levels of GLT-1 protein expression
were markedly decreased (Fig. 2).
However, pretreatment of PC12 cells with 400 μM NaHS for 30 min
before exposure to CoCl2 reversed this effect,
suggesting that NaHS preconditioning may enhance GLT-1 protein
expression level in CoCl2-treated PC12 cells.
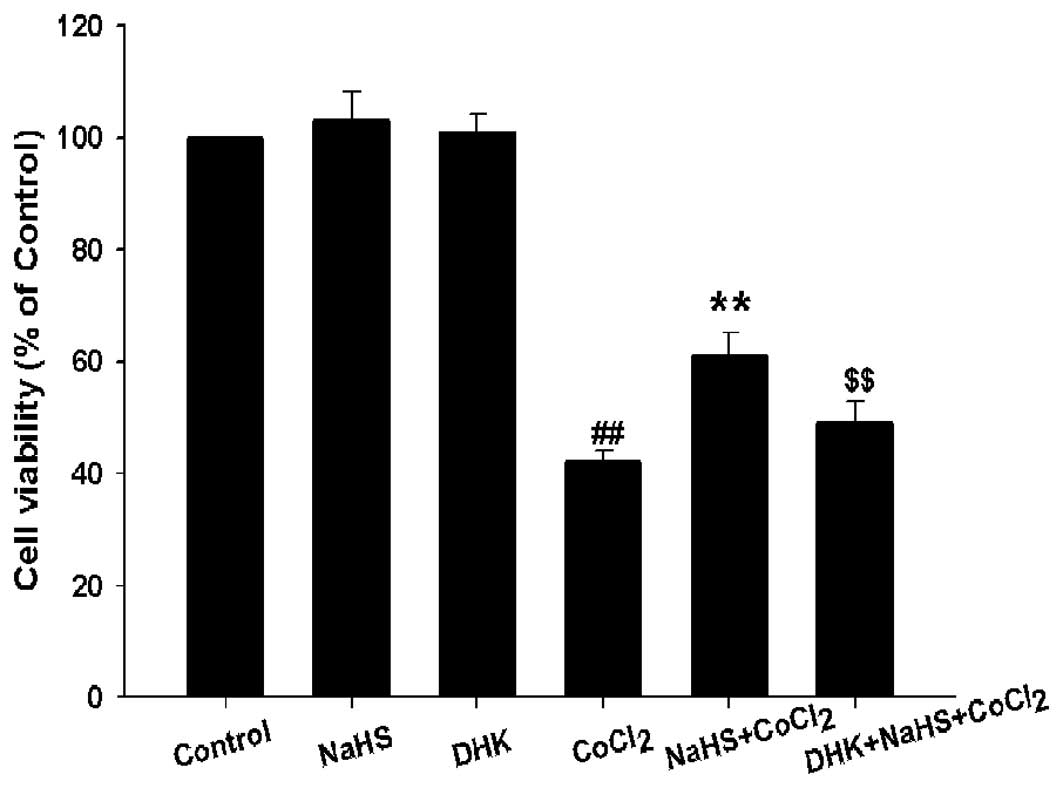
GLT-1 is involved in the cytoprotection
of H2S against CoCl2-induced injury
To explore whether GLT-1 is involved in the
cytoprotection of H2S against CoCl2-induced
injuries, PC12 cells were pretreated with DHK (a inhibitor of
GLT-1) at 400 μM for 30 min prior to NaHS preconditioning followed
by exposure to 600 μM CoCl2 for 24 h. As shown in
Fig. 3, DHK pretreatment
significantly blocked the protection of NaHS preconditioning
against CoCl2-induced cytotoxicity, the cell viability
was considerably decreased, from 62±2.3% to 50±2.1% (P<0.01)
(Fig. 3). Moreover, pretreatment
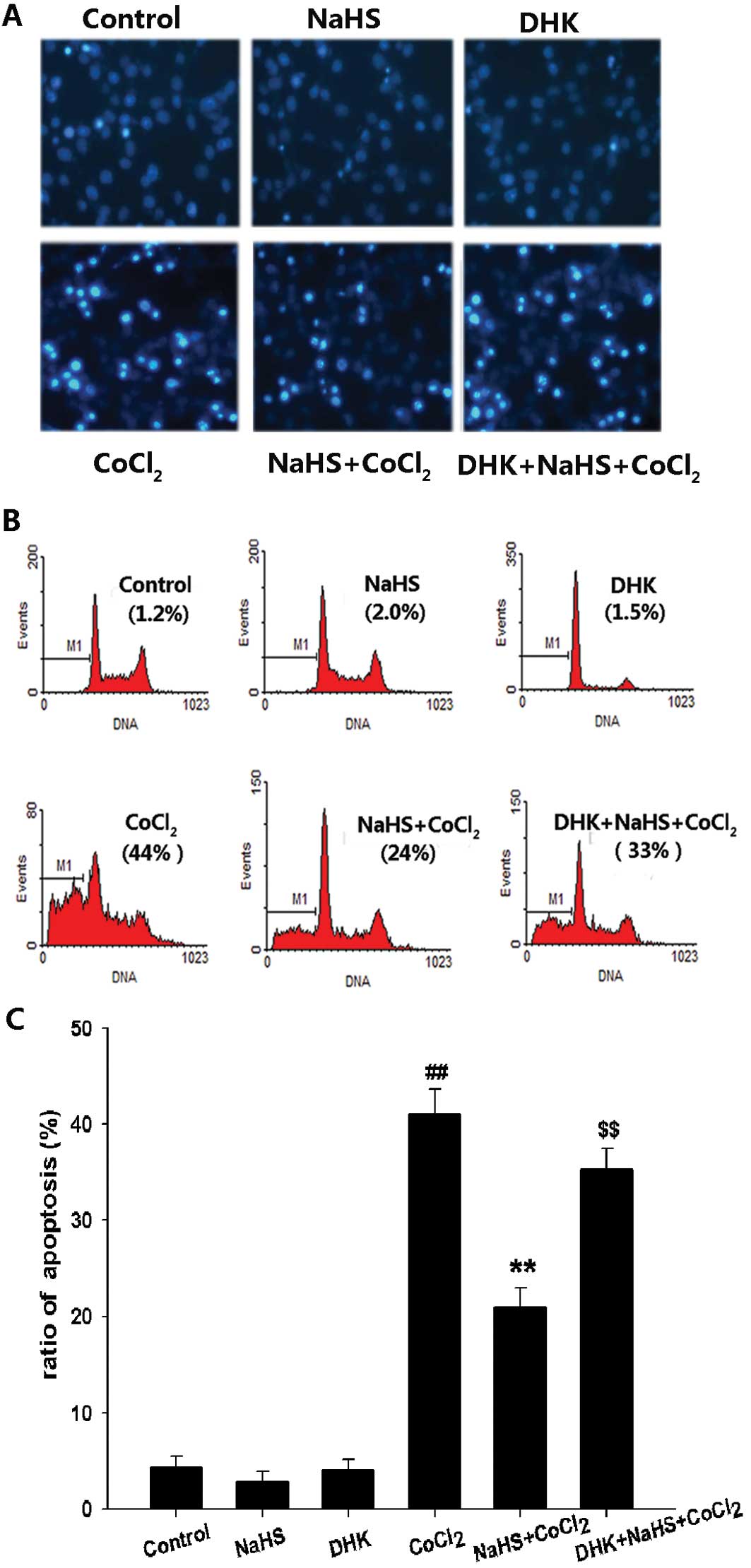
with 400 μM DHK also markedly inhibited H2S-induced
anti- apoptotic effects, increasing the number of apoptotic cells
with nuclear condensation and fragmentation (Fig. 4A) as well as the apoptotic
percentage of PC12 cells compared with the NaHS pretreatment +
CoCl2 group (P<0.01) (Fig. 4B). Additionally, pretreatment of
PC12 cells with DHK for 30 min before 400 μM NaHS preconditioning
clearly inhibited H2S-induced preservation of MMP
(Fig. 5). These findings suggest
that GLT-1 contributes to the cytoprotection of H2S
against CoCl2-induced injuries.
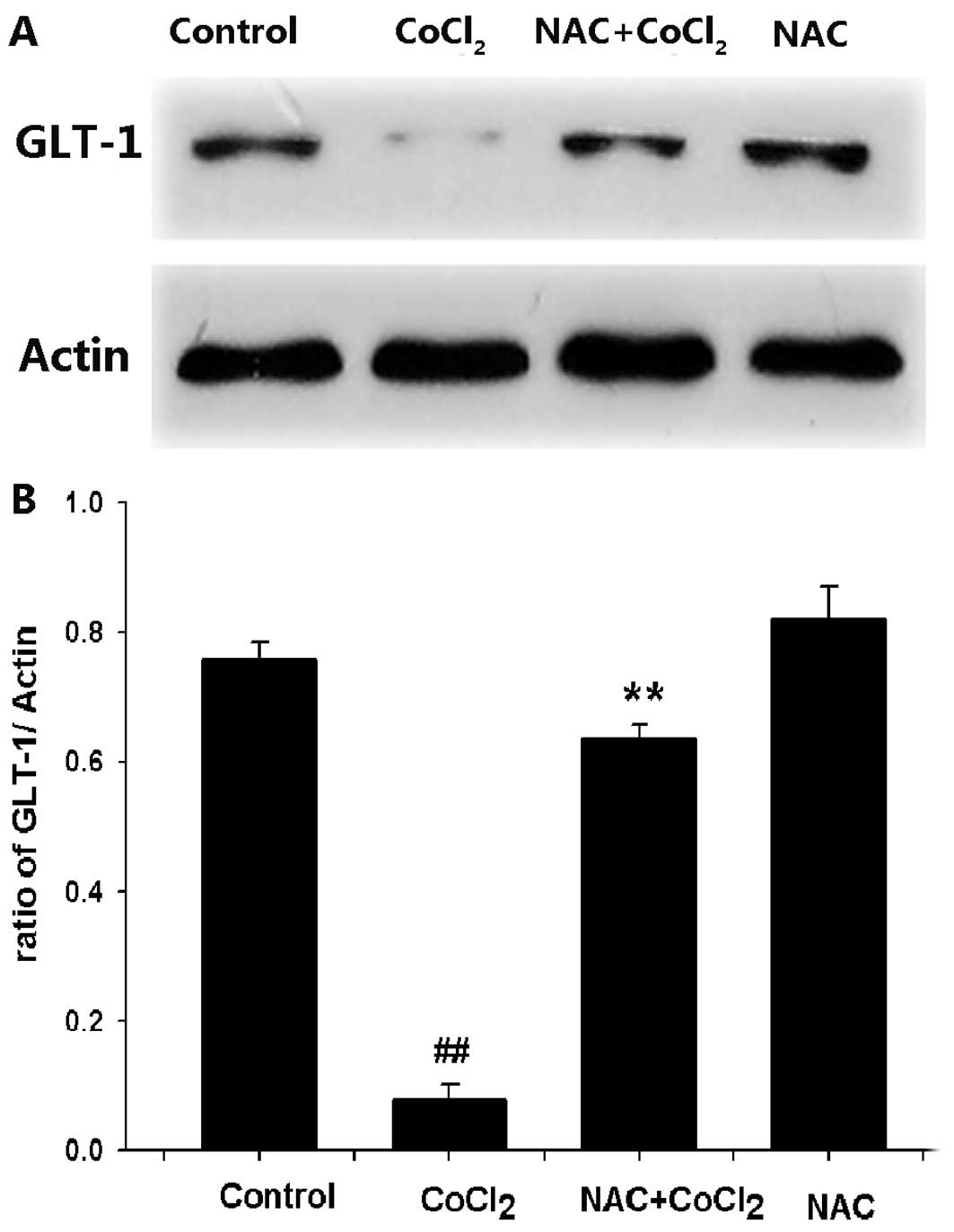
ROS are involved in the
CoCl2-induced downregulation of GLT-1 expression in PC12
cells
Since ROS generation inhibits glutamate uptake
function (22), we examined
whether ROS is involved in the CoCl2-induced
downregulation of GLT-1 protein expression in PC12 cells.
Pretreatment of cells with 500 μM NAC (a ROS scavenger) for 60 min
prior to exposure to 600 μM CoCl2 for 24 h significantly
blocked CoCl2-induced downregulation of GLT-1 expression
(Fig. 6). These data indicate
that the inhibitory effect of CoCl2 on GLT-1 expression
may be associated with oxidative stress.
Activation of ERK1/2 contributes to the
downregulation of GLT-1 expression induced by CoCl2 in
PC12 cells
Stimulation of ERK1/2MAPK also contributes to the
inhibition of glutamate uptake (23). In order to investigate the effect
of ERK1/2 activation on the downregulation of GLT-1 expression
induced by CoCl2, PC12 cells were pretreated with 10 μM
U0126 (a MEK1/2 inhibitor) for 120 min prior to treatment with 600
μM CoCl2 for 24 h. U0126 significantly reversed the
inhibitory effect of CoCl2 on the expression of GLT-1 in
PC12 cells, suggesting that activation of ERK1/2 contributes to the
downregulation of GLT-1 expression induced by CoCl2 in
PC12 cells (Fig. 2).
Discussion
GLT-1 has been classified as an astroglial
transporter due to its predominant and widespread expression in
astrocytes. In the present study, we found that PC12 cells
expressed GLT-1, suggesting that GLT-1 may be involved in
maintaining a normal level of glutamate in PC12 cells, which is
consistent with a previous study (17). It is well known that GLT-1 plays a
major role in glutamate re-uptake from the synaptic cleft after
neuronal transmission (6,24,25). Lack of GLT-1 has indeed been shown
to promote extracellular glutamate accumulation, excitotoxicity
and, ultimately, cell death (26,27). GLT-1 has been estimated to
represent up to 1% of total brain protein (6). The expression of GLT-1 is reduced in
several animal models of neurodegenerative diseases, including
traumatic brain injury (28) and
hypoxic/ischemic insults (16,29). The levels of the GLT-1 and/or
GLAST protein are also lower in the brain tissue from the patients
with Alzheimer’s disease (AD) (30) and Huntington’s disease (31). The results of the present study
showed that CoCl2, a well-known hypoxia mimetic agent,
attenuates expression of GLT-1 in a time-dependent manner. Our
findings are comparable with a study showing that transient global
ischemia reduces GLT-1 expression (16). Similarly, it was reported that
GLT-1 protein levels are reduced in the brain in various models of
central hypoxia/ischemia (16,29,32). Under hypoxic conditions (2.5 and
1% O2 exposure for 24 h), glutamate uptake and GLT-1
protein levels are significantly decreased in astrocytes (33). These studies all support our
results. By contrast, Kobayashi and Millhorn (17) indicated that exposure of PC12
cells to hypoxia (1% O2) for 6 to 24 h increases GLT-1
protein levels. Therefore, it is likely that the effects of
hypoxia/ischemia on the expression of GLT-1 may be affected by many
factors, including tissue or cell types, the level of hypoxia,
manner of hypoxia induction and also the period of
hypoxia/ischemia.
To clarify the mechanisms underlying the inhibitory
effect of chemical hypoxia on GLT-1 expression, we tested the
possible involvement of ROS. Several previous studies have shown
that oxidative stress is implicated in glutamate clearance
impairment and reduction of GLT-1 expression (18,21,34). Our recent studies have
demonstrated the promotive effects of CoCl2 on ROS
production (19,20). In this study, we found that NAC, a
ROS scavenger, can significantly block the inhibition of GLT-1
expression induced by CoCl2, revealing that ROS partly
contribute to the inhibitory effect of chemical hypoxia on the
expression of GLT-1 in PC12 cells. We provide novel evidence for
the role of ROS in CoCl2-induced neuronal injury.
Additionally, there is currently a lot of data demonstrating that
oxidative stress may trigger and modulate the MAPK signaling
pathways (21,35,36). We recently demonstrated that ROS
can activate the MAPK pathway (20), linking to the possibility of an
altered ERK1/2 activation that ultimately affects the expression of
GLT-1. To confirm this possibility, we observed the effects of
pretreatment of PC12 cells with UO126 (an inhibitor of MEK1/2) on
the inhibition of GLT-1 expression by CoCl2. Our results
showed that U0126 clearly suppressed the CoCl2-induced
decrease in the expression of GLT-1, suggesting that the ERK1/2
pathway is involved in the inhibitory effect of CoCl2 on
GLT-1 expression. This is also a novel finding showing that the
ROS-activated ERK1/2 pathway plays a role in the inhibition of
GLT-1 expression by CoCl2. Our findings are supported by
previous studies (18,35). Lu et al (18) reported that PD98059, a specific
ERK1/2 inhibitor, significantly reverses the reduction of
trafficking of GLT-1 from cytoplasma to plasma membrane.
Although research on the regulatory mechanisms for
GLT-1 expression has intensified, scarce data are available
regarding the regulatory effect of gasotransmitter on the
expression of GLT-1. H2S, recently recognized as the
third gasotransmitter alongside nitric oxide (NO) and carbon
monoxide (CO) (39), has
attracted extensive attention due to its multiple physiological and
pathophysiological roles in various body systems (18–20,37–41). Kimura and Kimura (38) demonstrated the nueroprotective
effect of H2S against oxidative stress-induced injury in
primary rat cortical neurons. H2S.also.protects.astro.
also protects astrocytes from H2O2-induced
neural injury (18). We recently
found that H2S protects PC12 cells against
CoCl2-induced damage by enhancing heat shock protein 90
(HSP90) (19), inhibiting the
ROS-activated ERK1/2 and p38MAPK signaling pathways (20) and scavenging ROS (19,20). In the present study, we provide
evidence for the first time that NaHS (a donor of H2S)
pretreatment prevents the CoCl2-induced downregulation
of GLT-1 expression in PC12 cells. Our results are in line with a
recent study that H2S protects astrocytes against
oxidative stress-induced neural damage by increasing glutamate
uptake (18). Based on our recent
results (19,20,39–41) and other studies (18,21,35,36,38,42), there are several possible
mechanisms responsible for the regulatory effect of H2S
on the expression of GLT-1: i) its antioxidation, by which
H2S can protect PC12 cells from CoCl2-induced
suppression of GLT-1 expression; ii) its inhibitory effect on the
ERK1/2 pathway (20); and iii)
H2S functions as an ATP-sensitive potassium
(KATP) channel opener (43). Hu et al (42) indicated that iptakalim, a
KATP channel opener, can reverse the inhibition of
glutamate uptake induced by N-methyl-4-4-phenylpyridinium (MPP+)
[used to stimulate Parkinson’s disease (PD)-like conditions],
revealing a role of the KATP channel opener in the
functional regulation of glutamate transporter. Further research is
required to confirm these findings.
We further explored the role of GLT-1 in the
neuroprotection of H2S against chemical hypoxia-induced
injury. We found that pretreatment with DHK, a selective inhibitor
of GLT-1, significantly reversed the protective effect of
H2S against CoCl2-induced injuries, evidenced
by a decrease in cell viability and an increase in apoptotic PC12
cells as well as MMP loss, suggesting that upregulation of GLT-1
expression may play an important role in the neuroprotective
effects of H2S.
In summary, in the present study, we have
demonstrated for the first time that: i) both ROS and the ERK1/2
pathway contribute to the downregulation of GLT-1 expression
induced by CoCl2; ii) H2S, a novel gaseous
neuromodulator, reverses CoCl2-induced downregulation of
GLT-1 expression; and iii) upregulation of GLT-1 expression may
play a crucial role in the neuroprotective effects of
H2S against chemical hypoxia- induced neuronal injury in
PC12 cells. The findings of the present study may provide a
potential neuroprotective therapeutic approach for treatment of
hypoxia/ischemia-related neuronal injury. In addition, based on the
notable findings that both levels of endogenous H2S and
GLT-1 are reduced in neurodegenerative diseases, such as AD and PD,
we speculate that endogenous H2S may be an important
modulator of GLT-1. These findings remain to be confirmed in future
studies.
Acknowledgements
The present study was supported by the
Guangdong Science and Technology Planning project (nos.
2010B080701105, 2009B080701014 and 2007B080701030).
References
|
1.
|
GE NilssonPL LutzRelease of inhibitory
neurotransmitters in response to anoxia in turtle brainAm J
Physiol261R32R3719911677540
|
|
2.
|
DW RichterPM LalleyO
PierreficheIntracellular signal pathways controlling respiratory
neuronsRespir
Physiol110113123199710.1016/S0034-5687(97)00077-79407605
|
|
3.
|
D NichollsD AttwellThe release and uptake
of excitatory amino acidsTrends Pharmacol
Sci11462468199010.1016/0165-6147(90)90129-V1980041
|
|
4.
|
SM RothmanJW OlneyGlutamate and the
pathophysiology of hypoxic - ischemic brain damageAnn
Neurol19105111198610.1002/ana.4101902022421636
|
|
5.
|
R SattlerZ XiongWY LuDistinct roles of
synaptic and extrasynaptic NMDA receptors in excitotoxicityJ
Neurosci202233200010627577
|
|
6.
|
KP LehreNC DanboltThe number of glutamate
transporter subtype molecules at glutamatergic synapses: chemical
and stereological quantification in young adult rat brainJ
Neurosci18875187571998
|
|
7.
|
K TanakaExpression cloning of a rat
glutamate transporterNeurosci
Res16149153199310.1016/0168-0102(93)90082-28387171
|
|
8.
|
D AttwellB BarbourM SzatkowskiNonvesicular
release of
neurotransmitterNeuron11401407199310.1016/0896-6273(93)90145-H
|
|
9.
|
Y KanaiCP SmithMA HedigerA new family of
neurotransmitter transporters: the high-affinity glutamate
transportersFASEB J71450145919937903261
|
|
10.
|
CM AndersonRA SwansonAstrocyte glutamate
transport: review of properties, regulation, and physiological
functionsGlia32114200010.1002/1098-1136(200010)32:1%3C1::AID-GLIA10%3E3.0.CO;2-W10975906
|
|
11.
|
G GegelashviliA SchousboeHigh affinity
glutamate transporters: regulation of expression and activityMol
Pharmacol5261519979224806
|
|
12.
|
JW PhillisJ RenMH O’ReganTransporter
reversal as a mechanism of glutamate release from the ischemic rat
cerebral cortex: studies with DL-threo-beta-benzyloxyaspartateBrain
Res868105112200010.1016/S0006-8993(00)02303-9
|
|
13.
|
VLR RaoA DoganJG ToddAntidense knockdown
of the glial glutamate transporter GLT-1, but not the neuronal
glutamate transporter EAAC1, exacerbates transient focal cerebral
ischemia-induced neuronal damage in rat brainJ
Neurosci21187618832001
|
|
14.
|
G ZhangYS RaolFC HsuAR
Brooks-KayalLong-term alterations in glutamate receptor and
transporter expression following early-life seizures are associated
with increased seizure susceptibilityJ
Neurochem8891101200410.1046/j.1471-4159.2003.02124.x
|
|
15.
|
K MimuraT TomimatsuK MinatoCeftriaxone
preconditioning confers neuroprotection in neonatal rats through
glutamate transporter 1 upregulationReprod
Sci1811931201201110.1177/193371911141071021693777
|
|
16.
|
VL Raghavendra RaoAM RaoA DoganGlial
glutamate transporter GLT-1 downregulation precedes delayed
neuronal death in gerbil hippocampus following transient global
cerebral ischemiaNeurochem Int365315372000
|
|
17.
|
S KobayashiDE MillhornHypoxia regulates
glutamate metabolism and membrane transport in rat PC12 cellsJ
Neurochem7619351948200110.1046/j.1471-4159.2001.00214.x11259512
|
|
18.
|
M LuLF HuG HuJS BianHydrogen sulfide
protects astrocytes against H202-induced neural injury via
enhancing glutamate uptakeFree Radic Biol
Med417051713200810.1016/j.freeradbiomed.2008.09.01418848879
|
|
19.
|
JL MengWY MeiYF DongHeat shock protein 90
mediates cytoprotection by H2S against chemical
hypoxia-induced injury in PC12 cellsClin Exp Pharmacol
Physiol384249201121083699
|
|
20.
|
A LanX LiaoL MoHydrogen sulfide protects
against chemical hypoxia-induced injury by inhibiting ROS-activated
ERK1/2 and p38MAPK signaling pathways in PC12 cellsPLoS
One6e25921201110.1371/journal.pone.002592121998720
|
|
21.
|
M MatosE AugustoCR OiveiraP
AgostinhoAmyloid-beta peptide decreases glutamate uptake in
cultured astrocytes: involvement of oxidative stress and
mitogen-activated protein kinase
cascadesNeuroscience156898910200810.1016/j.neuroscience.2008.08.022
|
|
22.
|
XL SunXN ZengF ZhouK(ATP) channel openers
facilitate glutamate uptake by GluTs in rat primary cultured
astrocytesNeuropsychopharmacology3313361342200810.1038/sj.npp.130150117609675
|
|
23.
|
M FigielT MaucherJ RozyczkaRegulation of
glial glutamate transporter expression by growth factorsExp
Neurol183124135200310.1016/S0014-4886(03)00134-112957496
|
|
24.
|
KP LehreLM LevyOP OttersenDifferential
expression of two glial glutamate transporters in the rat brain:
quantitative and immunocytochemical observationsJ
Neurosci5183518531995
|
|
25.
|
P KuglerA SchmittGlutamate transporter
EAAC1 is expressed in neurons and glial cells in the rat nervous
systemGlia27129142199910.1002/(SICI)1098-1136(199908)27:2%3C129::AID-GLIA3%3E3.0.CO;2-Y10417812
|
|
26.
|
K TanakaK WataseT ManabeEpilepsy and
exacerbation of brain injury in mice lacking the glutamate
transporter
GLT-1Science27616991702199710.1126/science.276.5319.16999180080
|
|
27.
|
CK VorwerkR NaskarF SchuettaufDepression
of retinal glutamate transporter function leads to elevated
intravitreal glutamate levels and ganglion cell deathInvest
Ophthalmol Vis Sci41361536212000
|
|
28.
|
VL RaoMK BaşkayaA DoğanTraumatic brain
injury downregulates glial glutamate transporter (GLT-1 and GLAST)
proteins in rat brainJ Neurochem702020202719989572288
|
|
29.
|
R TorpD LekieffreLM LevyReduced
postischemic expression of a glial glutamate transporter, GLT1, in
the rat hippocampusExp Brain
Res1035158199510.1007/BF002419647615037
|
|
30.
|
S LiM MalloryM AlfordS TanakaE
MasliahGlutamate transporter alterations in Alzheimer disease are
possibly associated with abnormal APP expressionJ Neuropathol Exp
Neurol56901911199710.1097/00005072-199708000-000089258260
|
|
31.
|
SA LiptonPA RosenbergExcitatory amino
acids as a final common pathway for neurologic disordersN Engl J
Med330613622199410.1056/NEJM1994030333009077905600
|
|
32.
|
LJ MartinAM BrambrinkC
LehmannHypoxia-ischemia causes abnormalities in glutamate
transporters and death of astroglia and neurons in newborn
striatumAnn Neurol42335348199710.1002/ana.4104203109307255
|
|
33.
|
M DallasHE BoycottL AtkinsonHypoxia
suppresses glutamate transport in astrocytesJ
Neurosci2739463955200710.1523/JNEUROSCI.5030-06.200717428968
|
|
34.
|
B BreraA SerranoML de Ceballosbeta-amyloid
peptides are cytotoxic to astrocytes in culture: a role for
oxidative stressNeurobiol
Dis7395405200010.1006/nbdi.2000.031310964610
|
|
35.
|
JA McCubreyMM LahairRA FranklinReactive
oxygen species-induced activation of the MAP kinase signaling
pathwaysAntioxid Redox
Signal817751789200610.1089/ars.2006.8.177516987031
|
|
36.
|
X ZhuHG LeeAK RainaThe role of
mitogen-activated protein kinase pathways in Alzheimer’s
diseaseNeurosignals112702812002
|
|
37.
|
R WangThe gasotransmitter role of hydrogen
sulfideAntioxid Redox
Signal5493501200310.1089/15230860376829524913678538
|
|
38.
|
Y KimuraH KimuraHydrogen sulfide protects
neurons from oxidative stressFASEB J1811651167200415155563
|
|
39.
|
Z YangC YangL XiaoNovel insights into the
role of HSP90 in cytoprotection of H2S against chemical
hypoxia-induced injury in H9c2 cardiac myocytesInt J Mol
Med28397403201121519787
|
|
40.
|
SL ChenCT YangZL YangHydrogen sulphide
protects H9c2 cells against chemical hypoxia-induced injuryClin Exp
Pharmacol
Physiol37316321201010.1111/j.1440-1681.2009.05289.x19769612
|
|
41.
|
CT YangZL YangMF ZhangHydrogen sulfide
protects against chemical hypoxia-induced cytotoxicity and
inflammation in HaCaT cells through inhibition of ROS/NFB/COX-2
pathway PLOS One6e21971201110.1371/journal.pone.002197121779360
|
|
42.
|
LF HuS WangXR ShiATP-sensitive potassium
channel opener iptakalim protected against the cytotoxicity of MPP+
on SH-SY5Y cells by decreasing extracellular glutamate levelJ
Neurochem9415701579200516000145
|
|
43.
|
D JohansenK YtrehusGF BaxterExogenous
hydrogen sulfide (H2S) protects against regional
myocardial ischemia-reperfusion injury - evidence for a role of
KATP channelsBasic Res Cardiol10153602006
|