Introduction
Temporomandibular joint disorders (TMD), including
temporomandibular joint osteoarthrosis (TMJ-OA), are a group of
multifactorial joint diseases characterized by progressive joint
degeneration (1,2). Despite intensive research efforts,
many challenges still remain in the treatment of TMJ-OA. The
specific non-vascular property of the condylar cartilage makes
self-repairing and regeneration difficult. Over the last decade,
synovium-derived mesenchymal stem cells (SMSCs) have evoked
interest as a very promising cell source for cartilage regeneration
due to their superiority in proliferation and chondrogenesis
(3–5). Previous studies have shown that
SMSCs may be induced to undergo chondrogenesis when exposed to
certain growth factors in vitro (6,7).
Although there has been great interest in the use of SMSCs for
articular cartilage tissue engineering, the detailed mechanism of
the chondrogenic differentiation of SMSCs remains unclear.
It is well established that the reorganization of
the actin cytoskeleton is one of the earliest cellular responses to
extracellular stimuli (8). During
chondrogenesis, cells undergo changes in cell shape from a
characteristic fibroblast-like structure to a round or polygonal
morphology; these changes accompany the onset of gene expression
levels, such as those of type II collagen and aggrecan (9–11).
Therefore, the actin cytoskeleton appears to act as an integrated
signaling network that plays a crucial role in regulating the
differentiation and function of SMSCs. The transforming growth
factor-β1 (TGF-β1), a member of the TGF-β superfamily, is known to
modulate diverse cellular responses, such as cell growth,
proliferation, differentiation and production of the extracellular
matrix. TGF-β1 is the most extensively examined growth factor for
inducing chondro-genesis and cartilage matrix synthesis (7,12).
Critical steps in the intracellular TGF-β1 signaling pathway are
mediated by different intracellular pathways, including the
canonical Smad and non-Smad pathways (13). The signals are transduced by Smad
and non-Smad pathways to the nucleus, where they regulate the
expression of genes involved in chondrogenesis (14,15). Moreover, TGF-β1 has been shown to
modulate cell growth and morphology in a concerted manner via a
mechanism that controls the actin cytoskeleton in a number of cell
types (16).
The Rho family small GTPases, including RhoA, Rac1
and Cdc42, are the best characterized upstream regulators of the
actin cytoskeleton (17). The
activation of Rho GTPases also controls a number of cellular
processes, such as cell proliferation, cell cycle progression and
gene expression, through a multitude of effector proteins (18,19). The Rho kinases (ROCK)1 and ROCK2
are the most important downstream effectors of RhoA. Certain
studies have shown that RhoA activity negatively affects
chondrogenesis in the ATDC5 chondrogenic cell line and mesenchymal
cells (20,21). On the contrary, the addition of
the ROCK inhibitor, Y27632, to micromass cultures of ATDC5 cells
has been shown to inhibit chondrocyte-specific gene expression
(22). In addition, the RhoA/ROCK
pathway has been implicated in the TGF-β1 matrix responses in
various cells (23,24). However, to the best of our
knowledge, there has been no other report investigating the role of
the RhoA/ROCK signaling pathway in the process of chondrogenesis in
SMSCs.
The aim of the present study was to evaluate the
activation of the RhoA/ROCK pathway in the TGF-β1-induced
chondrogenesis and actin organization of SMSCs, and to further
investigate any interactions between the RhoA/ROCK and Smad
pathways.
Materials and methods
Cell culture and treatment
SMSCs were isolated and expanded as reported by Li
et al (25). Briefly,
synovial membrane tissues were harvested from the temporomandibular
joints of male Sprague-Dawley rats aged 6 weeks in accordance with
the guidelines approved by the Animal Committee of the Zhejiang
University. The excised tissues were vigorously washed in
phosphate-buffered saline (PBS), and then minced into 1
mm3 pieces and plated in a T25 culture flask with a
medium consisting of high-glucose Dulbecco’s modified Eagle’s
medium (DMEM; Gibco-BRL, Gland Island, NY, USA) supplemented with
10% fetal bovine serum (FBS; Gibco-BRL), penicillin (100 U/ml),
streptomycin (100 μg/ml) and 4 mM L-glutamine at 37°C with 5% CO2.
The cells were expanded in a monolayer culture for 3–5 passages. In
order to evaluate SMSC chondrogensis, cells were seeded at a
density of 5×104 cells/cm2 in 6-well plates
and cultured in chondrogenic medium composed of high glucose DMEM
supplemented with 100 nmol/l dexamethasone, 50 mg/l vitamin C, 40
µg/ml proline, 50 µg/ml ascorbate-2-phosphate, 100 µg/ml pyruvate
(Sigma, St. Louis, MO, USA) and 10 ng/ml TGF-β1 (R&D Systems,
Minneapolis, USA), replaced with each medium change. The culture
medium was refreshed once every 2 days until day 7. The cells that
were cultured in chondrogenic medium without TGF-β1 served as the
control. Each experiment was repeated at least 3 times in order to
verify the results.
Pharmacological inhibition study
A pharmacological inhibition study was subsequently
performed to investigate the role of the RhoA/ROCK and Smad
signaling pathways in TGF-β1-stimulated chondrogenesis. Specific
inhibitors of RhoA (C3 transferase, 1 µM), ROCK (Y27632, 5 and 10
µM) (Cytoskeleton, Inc., Denver, CO, USA) and TGF-β type I receptor
(SB431542, 5 and 10 µM) (Tocris Bioscience, Bristol, UK) were added
into the chondrogenic medium. The inhibitor-containing chondrogenic
medium was replenished once every 2 days.
RhoA activation assay
RhoA activation by TGF-β1 was quantified using a
luminescence based G-LISA RhoA activation assay biochemistry kit
(Cytoskeleton). Immediately following chondrogenic stimulation in
the presence of TGF-β1 for different periods of time, the cells
were rinsed with ice-cold PBS, and lysed using the provided cell
lysis buffer. The lysates were then clarified by centrifugation at
10,000 rpm at 4°C for 2 min. After equalizing protein
concentrations in all lysed cell extracts, GTP-bound RhoA levels
were determined according to the manufacturer’s instructions.
Luminescence was detected as suggested by the manufacturer using a
microplate spectrophotometer. The results were expressed relative
to the untreated controls.
F-actin staining and fluorescent
intensity
For the immunofluorescence detection of the actin
cytoskeleton, the cells were grown on coverslips in 24-well plates,
and then incubated with or without inhibitors of RhoA/ROCK (1 μM C3
transferase, 10 μM Y27632) and Smads (10 μM SB431542) in the
presence of TGF-β1. The cells were rinsed with PBS and fixed in 4%
paraformaldehyde for 30 min. For F-actin staining, each sample was
stained with fluorescein isothiocyanate (FITC)-phalloidin (Sigma)
for 40 min at room temperature. Representative fluorescence images
were captured using a fluorescence microscope (CX-RFL-2; Olympus,
Tokyo, Japan) at ×400 magnification. The average relative intensity
of F-actin/cell in each group was analyzed using the MetaMorph
software (Universal Imaging Corporation, Downingtown, PA, USA).
Protein isolation and western blot
analysis
Total cell lysates were suspended in
radio-immunoprecipitation assay lysis buffer (Beyotime, Shanghai,
China) containing 1 mM protease inhibitor phenylmethanesulfonyl
fluoride (Beyotime) and incubated on ice for 30 min. The samples
were clarified by centrifugation at 12,000 rpm for 5 min at 4°C and
boiled for 5 min with a sample loading buffer. The protein
concentrations were determined using a commercial bicinchoninic
acid protein assay kit with bovine serum albumin as the standard.
Equivalent protein amounts were fractionated by 10% sodium dodecyl
sulfatepolyacrylamide gels and transferred onto 0.1 μM
polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA).
Blots were blocked with 5% non-fat milk for 1 h, followed by
incubation with specific primary antibodies against phosphorylated
(phospho)-Smad2, phospho-Smad3, Smad2/3 and GAPDH (diluted in
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA)
overnight at 4°C. After washing, the blots were then hybridized
with specific horseradish peroxidase-conjugated secondary
antibodies and visualized using an electrochemiluminescence reagent
(Pierce, Rockford, IL, USA).
RNA extraction and cDNA synthesis
Total RNA was obtained using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The purity and yield of the isolated
RNA were monitored using a NanoDrop ND-2000 Spectrophotometer
(NanoDrop Technologies, Wilmington, DE, USA). The first-strand cDNA
was synthesized from 1 μg total RNA by reverse transcription using
a SYBR PrimeScript™ RT reagent kit (Takara Bio, Inc., Dalian,
China). The samples were diluted in nuclease-free water and stored
at −20°C prior to quantitative real-time polymerase chain reaction
(PCR).
Quantitative real-time PCR analysis
Quantitative real-time RT-PCR was performed using a
SYBR PrimeScript™ RT-PCR kit (Takara) in the ABI PRISM 7500
Real-Time PCR System (Applied Biosystems, Foster City, CA, USA).
The sequence-specific primers used for PCR amplification are listed
in Table I. The thermal cycling
conditions were 1 cycle at 95°C for 30 sec, 40 cycles at 95°C for 5
sec and 60°C for 34 sec. The melting curve analysis for each PCR
reaction was generated to ensure the purity of the amplified
product (data not shown). Each gene was normalized against the
corresponding glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
levels using the ΔΔCT method (normalized CT values were
expressed relative to the untreated controls) (26). The relative gene expression of
each sample was shown.
 | Table I.Primer sequences used during
quantitative real-time RT-PCR analysis. |
Table I.
Primer sequences used during
quantitative real-time RT-PCR analysis.
| Target gene | GenBank accession
no. | Primer sequence
(5′→3′) |
|---|
| ROCK1 | NM_031098.1 | Forward:
AGAGGCTCAAGACATGCTCAATCA |
| Reverse:
CAGTTAGCCGCGCTTTGGTTA |
| ROCK2 | NM_013022.1 | Forward:
GTTCAGTTGGTTCGTCATA |
| Reverse:
ATCATAATTGCTCATCAGGTTA |
| Type I
collagen | NM_053356.1 | Forward:
TGCTGGCCAACCATCCCTCT |
| Reverse:
CGACATCATTGGATCCTTGCAG |
| Type II
collagen | NM_012929.1 | Forward:
AGCGGAGACTACTGGATTGATC |
| Reverse:
CTCTCCAAACCAGATGTGCTTC |
| Aggrecan | NM_022190.1 | Forward:
AGCCATAGCTTCTCCTGAG |
| Reverse:
GGGTATCTGACAGTCTGGTC |
| Sox9 | XM_001081628.2 | Forward:
GAAGAGCAATGGTGACAGAG |
| Reverse:
TGGAATCTCAGCAATCGTTAC |
| GAPDH | NM_017008.3 | Forward:
GAAGGTGAAGGTCGGAGTCG |
| Reverse:
GAAGATGGTGATGGGATTTC |
Statistical analysis
All data were presented as the means ± SD. The
statistical analysis of the data was performed using the SPSS 19.0
software package by one-way ANOVA. Post hoc comparisons were made
using Bonferroni corrections. P<0.05 was considered to indicate
a statistically significant difference.
Results
TGF-β1-induces RhoA/ROCK activation in
SMSCs
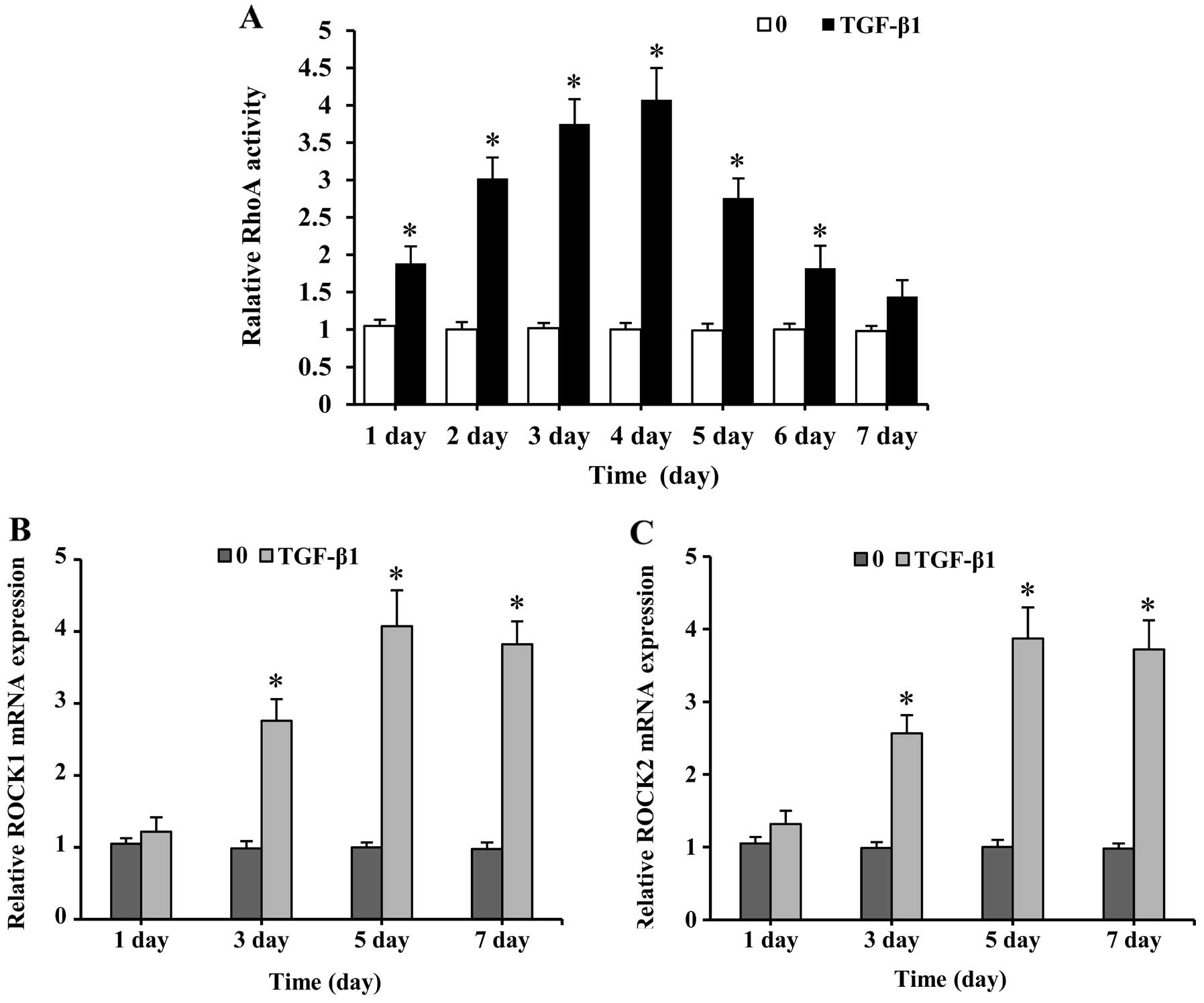
TGF-β1 addition caused a rapid increase in RhoA
activation after 1 day of stimulation, reaching its peak on day 4,
but gradually decreased as the time proceeded (Fig. 1A). RhoA activation leads to the
activation of Rho kinase family members. Thus, these downstream
effector kinases were assessed by quantitative real-time PCR
analysis with emphasis on determining ROCK1 and ROCK2 mRNA
expression. The treatment of SMSCs with TGF-β1 resulted in an
upregulation of the ROCK1 and ROCK2 genes (Fig. 1B and C). The expression of these
genes was low on day 1, but increased as chondrogenesis proceeded,
peaked after 5 days, and was sustained at relatively high levels
until day 7. These results demonstrate that TGF-β1 induces the
activation of the RhoA/ROCK pathway in SMSCs.
TGF-β1 causes RhoA/ROCK-dependent
cytoskeletal responses in SMSCs
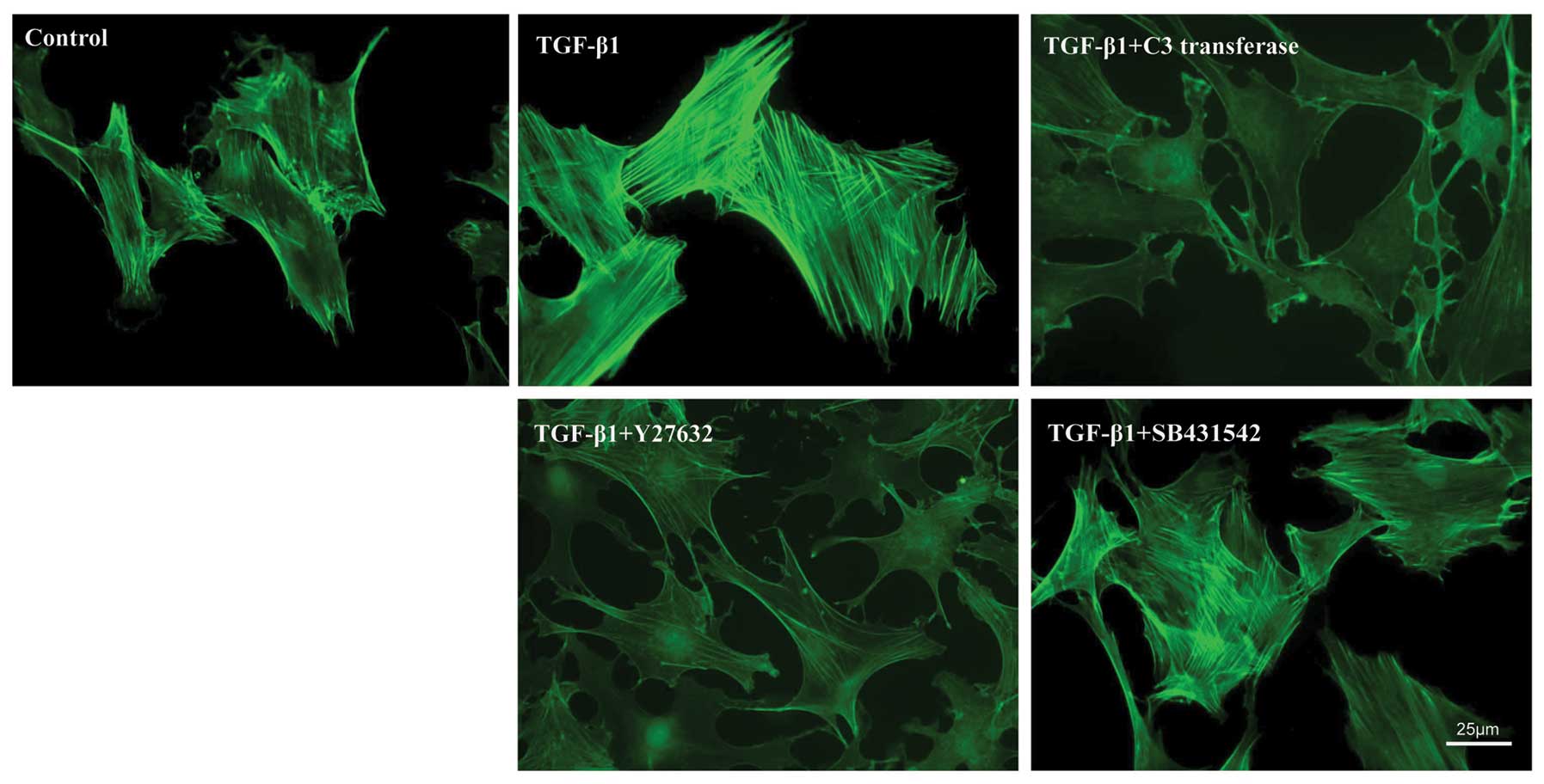
To identify the actin cytoskeletal characteristics
of SMSCs, cells were treated with or without pharmacological
inhibitors (see above) in addition to TGF-β1 and visualized by
fluorescence microscopy using FITC-phalloidin staining. After 7
days of in vitro expansion, the untreated control cells
exhibited a relatively weak cytoplasmic staining of F-actin fibers,
which were arranged randomly (Fig.
2). Compared with the control cells, TGF-β1-stimulated cells
showed a dramatically increased cytoplasmic staining of F-actin
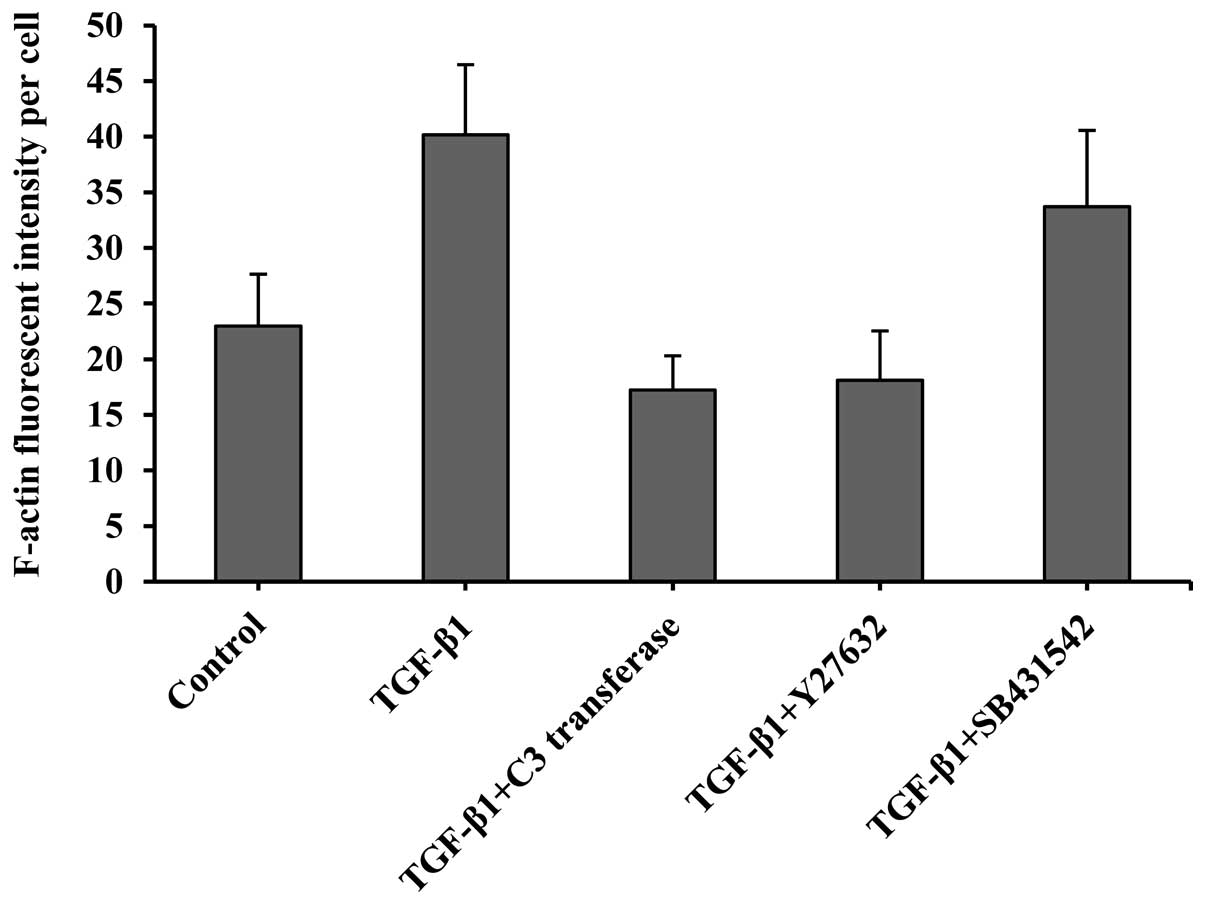
fibers, primarily arranged in parallel. The value expressed as an
arbitrary unit also showed a dramatic increase of F-actin staining
over the controls, indicating more filamentous actin formation
(Fig. 3).
The cells that were treated with RhoA/ROCK
inhibitors (1 µM C3 transferase or 10 µM Y27632) displayed a
significant decrease in fluorescence intensity in comparison to
TGF-β1, indicating a reduced number of actin fibers. By contrast,
treatment with TβRI inhibitors (10 µM SB431542) did not lead to
further alterations in actin fiber formation induced by TGF-β1,
showing no apparent change in the F-actin intensity of the SB431542
group. This finding demonstrates that RhoA/ROCK activation plays a
specific role in the control of TGF-β1-induced actin cytoskeletal
reorganization.
Involvement of RhoA signaling in
TGF-β1-induced chondro-genesis in SMSCs
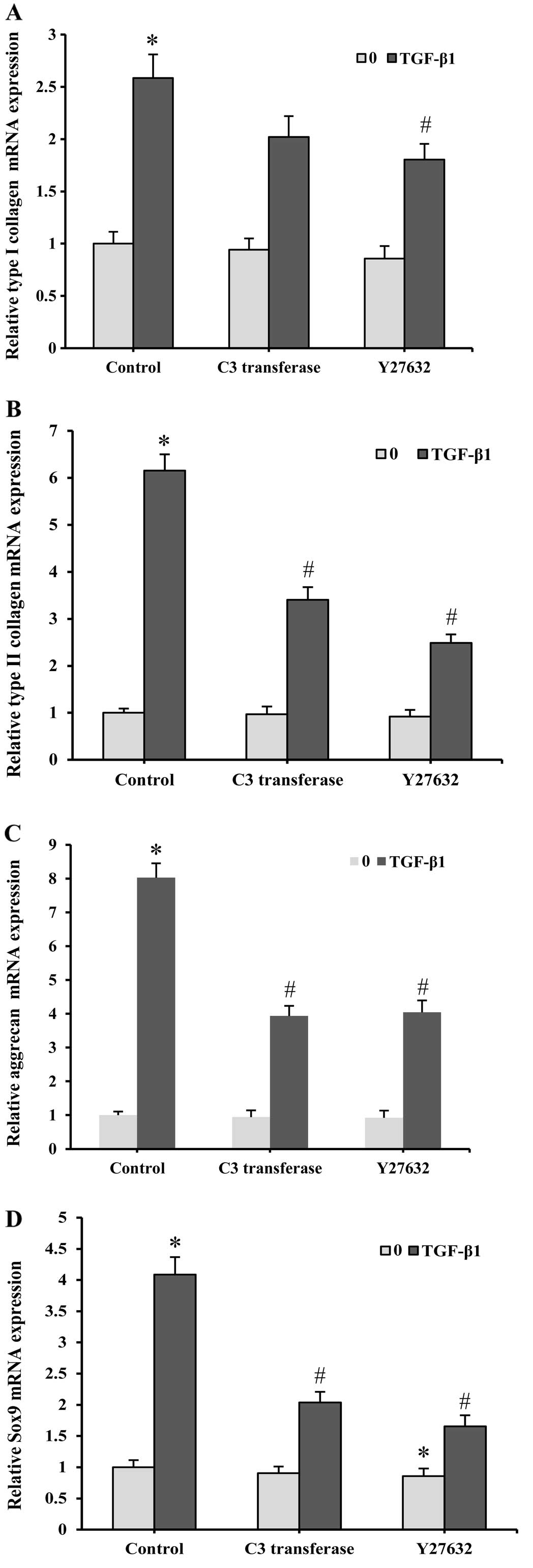
By quantitative real-time PCR analysis, the
chondrogenic potential of SMSCs was confirmed by assessing the
chondrocyte-specific gene expressions of type I collagen, type II
collagen and aggrecan. The mRNA levels of type I collagen, type II
collagen and aggrecan significantly increased 7 days after the
TGF-β1 treatment compared with the untreated controls (Fig. 4A–C). Further treatment with the
ROCK inhibitors resulted in a significantly reduced transcription
of the cartilage-specific genes. Specifically, TGF-β1-induced type
I collagen, type II collagen and aggrecan mRNA levels decreased by
30, 44 and 51%, respectively, after treatment with 1 µM C3
transferase. When treated with 10 µM Y27632, the TGF-β1-induced
gene expressions levels of type I collagen, type II collagen and
aggrecan were reduced by 37, 59 and 49%, respectively.
Sox9 has been shown to be a major transcription
regulator in the progression of chondrogenesis. Correlating the
above observations in type I collagen, type II collagen and
aggrecan expression levels, Sox9 mRNA levels were significantly
upregulated in the presence of TGF-β1 (Fig. 4D). The induction of the Sox9 mRNA
expression was also repressed by C3 transferase and Y27632. The
gene expression levels of type I collagen, type II collagen,
aggrecan and Sox9 were also affected by C3 transferase and Y27632
in the absence of TGF-β1. However, the influence of these
inhibitors on gene expression was relatively minor compared with
the samples cultured with TGF-β1. These results suggest the
involvement of RhoA/ROCK signaling in the regulation of
TGF-β1-induced chondrocyte-specific gene expression in SMSCs.
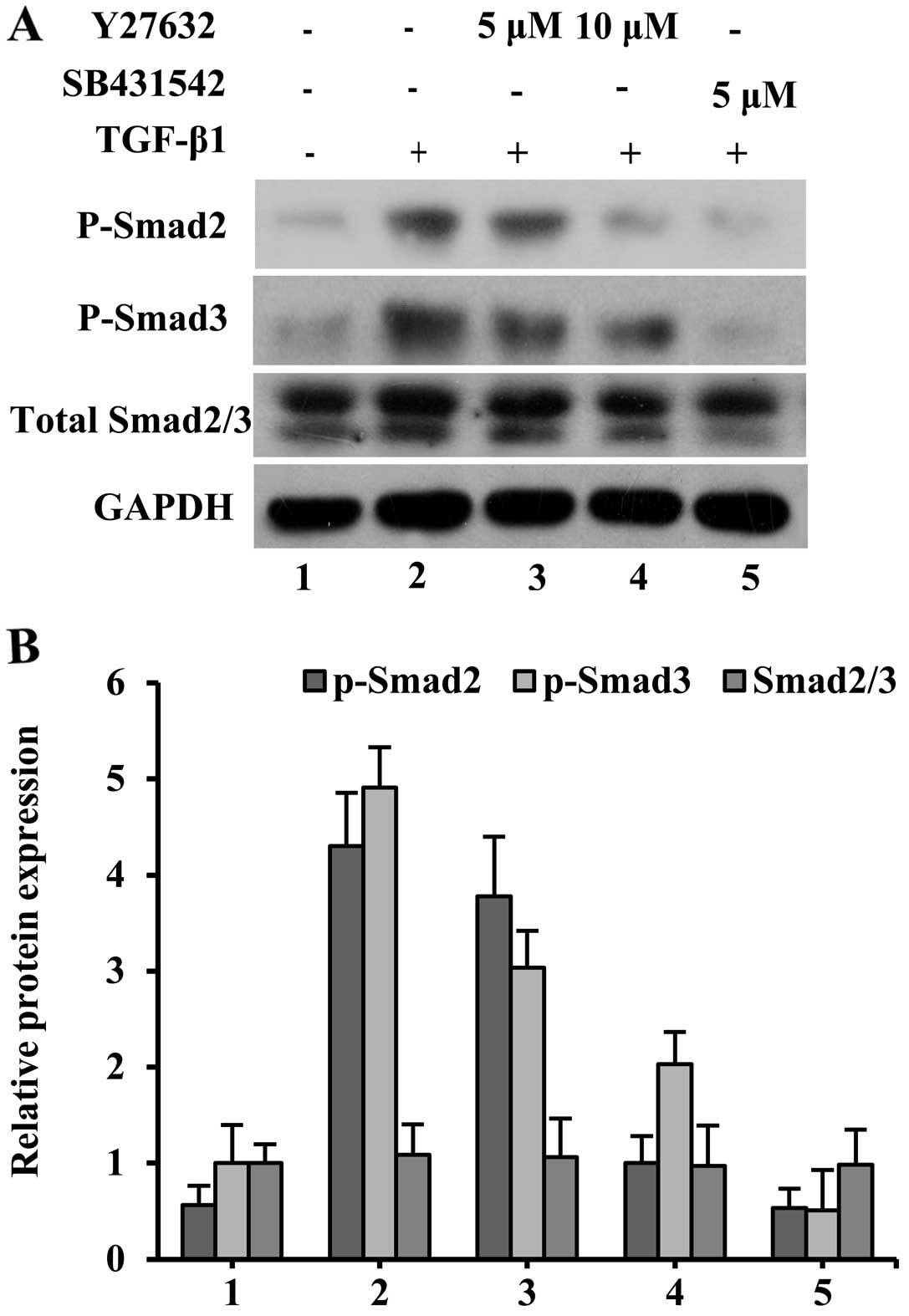
Effect of ROCK inhibitor on
phosphorylation activity of Smads induced by TGF-β1
To obtain insight into the possible interaction
between RhoA/ROCK and TGF-β1 signaling cascades, ROCK activation
was specifically blocked with Y27632 at the final concentrations of
5 and 10 µM. Relatively high levels of phospho-Smad2 and
phospho-Smad3 were maintained after 7 days of TGF-β1 treatment
(Fig. 5). After adding 5 µM
SB431542 to the TGF-β1 group, the Smad2 and Smad3 phosphorylation
levels dramatically decreased. When Y27632 was used at 5 µM, the
TGF-β1-induced phosphorylation of Smad2 and Smad3 in SMSCs was
slightly reduced. In addition, treatment with 10 µM Y27632 together
with TGF-β1 resulted in a marked reduction of Smad2 and Smad3
phosphorylation compared with the TGF-β1 group, which indicated
effective inhibition by this pharmacological inhibitor.
Discussion
SMSCs have previously been shown to have remarkable
potential to undergo chondrogenic differentiation and to contribute
toward tissue engineering and cartilage repair (3,7).
In the present study, the chondrogenic induction of SMSCs by TGF-β1
was initially performed in vitro in a monolayer culture. Our
study demonstrates the regulation of the RhoA/ROCK pathway and its
interaction with the Smad signaling pathway during chondrogenic
differentiation in response to TGF-β1.
TGF-β1 is a well-documented potent chondrogenic
factor, which may induce extracellular matrix synthesis associated
with cartilage regeneration (7,12).
The present results indicated that TGF-β1 promoted chondrogenesis,
as evidenced by the increased gene expression levels of type I
collagen, type II collagen, aggrecan and Sox9. Concomitant to these
enhanced expression levels, TGF-β1-stimulated cells exhibited
strong cytoplasmic actin fiber formation. Image analysis of
FITC-phalloidin stained SMSCs showed that TGF-β1 treatment caused a
dramatic increase in fluorescent intensity compared with the
untreated controls. Therefore, TGF-β1 induces a detailed phenotypic
modification of SMSCs. However, the intracellular signaling
pathways that mediate the functions and differentiation status of
SMSCs by altering the actin cytoskeleton warrant further
investigation.
Changes in the actin cytoskeleton may regulate
cellular responses to TGF-β1 by changes in receptor expression,
focal adhesion signaling and nuclear signaling, which may alter
cell differentiation and function (16). Over the last decade, RhoA
signaling through the ROCK1/2 kinases has been implicated in
regulating the shape-dependent control of mesenchymal cell
differentiation via intrinsic mechanisms (20,21,27). In this study, the TGF-β1
stimulation of SMSCs was found to upregulate RhoA activity rapidly
in the early stage of chondrogenesis, but gradually decreased after
4 days. The upregulation of the gene expression of its downstream
effectors, namely, ROCK1 and ROCK2, was also activated and
sustained at a relatively higher level. To the best of our
knowledge, this is the first study to show that RhoA activation,
which may result in actin cytoskeleton reorganization, is also
initiated by extrinsic growth factors and plays a significant role
in the chondrogenic differentiation of SMSCs.
Notably, the results from our study demonstrate that
type I collagen, type II collagen and aggrecan gene expression
levels are significantly reduced by the inhibition of RhoA and ROCK
activity with C3 transferase and Y27632, respectively. These
findings suggest that the TGF-β1-induced RhoA/ROCK activation plays
a positive role in the chondrogenesis of SMSCs. When the RhoA/ROCK
inhibitors were added, the TGF-β1-induced cytoskeletal
reorganization was interrupted, and chondrocyte-specific genes were
downregulated. This suggests that chondrogenic differentiation
requires an organized actin network that may be achieved by
biochemically inducing RhoA activation. Additionally, the
increasing cytoplasmic stress fiber formation is associated with
the downstream target transcription of the chondrocyte-specific
gene.
Sox9 is a key transcription factor in chondrogenic
differentiation, and its expression directly activates the
transcription of collagen II and aggrecan genes (28). Thus, whether or not the upstream
transcription factor, Sox9, was affected by the inhibition of
RhoA/ROCK signaling was further assessed. In the current study,
treatment with C3 transferase or Y27632 was found to suppress the
influence of TGF-β1 on transcription factor Sox9 mRNA levels. This
result is in agreement with a previous observation by Haudenschild
et al (29), demonstrating
that ROCK directly phosphorylated Sox9 at Ser181 and resulted in
the enhancement of Sox9 activity in chondrocytes. Woods and Beier
(22) demonstrated that ROCK
inhibition reduced the activity of the Sox9-responsive reporter
gene during chondro-genesis, but it was not in cellular context
(20). However, the specific
transcription factors connecting RhoA/ROCK to the Sox9
transcription have not been identified. The differences in these
results may be due to the differences among cell types,
chondrogenic culture conditions, and the presence of specific
growth factors and cytokines. Therefore, there is a compelling need
to examine the roles of RhoA and its effectors in vivo and
to identifiy the upstream and downstream components.
Since both RhoA/ROCK and Smad pathways are known to
mediate TGF-β1 signaling during chondrogenesis, we investigated
their potential interaction in chondrogenesis. A number of studies
have attempted to link Smads with RhoA/ROCK activity. In human
breast cancer cells, RhoA activation by TGF-β has been demonstrated
to be independent of Smads (30),
whereas various studies have conversely shown a potential
regulation of Smad signaling by RhoA/ROCK (23,31). In this regard, we observed a
decrease of TGF-β-induced Smad2/3 phosphorylation with an
increasing concentration of Y2763, suggesting that Y2763 inhibits
the activation of the TGF-β/Smad signaling pathway. The regulatory
effect of the RhoA/ROCK pathway on Smad activity was consistent
with the chondrocyte-specific genes. Therefore, this study provides
evidence that RhoA/ROCK mediates the chondrogenesis of SMSCs by
interacting with the Smad2/3 pathway. However, in addition to
further investigation of alternative cytoskeletal organizations,
such as microtubular and intermediate filaments, TGF-β1 possibly
initiates other signaling cascades involved in chondrogenesis.
Therefore, further research is required to fully elucidate the
interaction between RhoA/ROCK and other signaling pathways in
TGF-β1 stimulation.
In conclusion, to the best of our knowledge, we
report for the first time that the RhoA/ROCK activation initiated
by TGF-β1 has great potential to regulate cytoskeletal
organization, as well as control the chondrogenic gene expression
of SMSCs by interacting with the TGF-β/Smad signaling pathway.
These findings provide novel insights into the regulatory
mechanisms that define the chondrogenetic differentiation of SMSCs,
making the RhoA/ROCK pathway an interesting therapeutic target for
the successful use of SMSCs as a cell source for cartilage tissue
engineering.
Acknowledgements
The present study was supported by
grants from the National Natural Science Foundation of China (nos.
81170979 and 30901687), the Natural Science Foundation of Zhejiang
Province (nos. Y2090262 and Y2080370), and the Young Scientist
Project from the Health Bureau of Zhejiang (no. 2009QN018).
References
|
1.
|
SJ ScrivaniDA KeithLB
KabanTemporomandibular disordersN Engl J
Med35926932705200810.1056/NEJMra080247219092154
|
|
2.
|
E TanakaMS DetamoreLG MercuriDegenerative
disorders of the temporomandibular joint: etiology, diagnosis, and
treatmentJ Dent
Res87296307200810.1177/15440591080870040618362309
|
|
3.
|
J FanRR VarshneyL RenD CaiDA
WangSynovium-derived mesenchymal stem cells: a new cell source for
musculoskeletal regenerationTissue Eng Part B
Rev157586200919196118
|
|
4.
|
Y SakaguchiI SekiyaK YagishitaT
MunetaComparison of human stem cells derived from various
mesenchymal tissues: superiority of synovium as a cell
sourceArthritis Rheum5225212529200516052568
|
|
5.
|
H YoshimuraT MunetaA NimuraA YokoyamaH
KogaI SekiyaComparison of rat mesenchymal stem cells derived from
bone marrow, synovium, periosteum, adipose tissue, and muscleCell
Tissue Res327449462200710.1007/s00441-006-0308-z17053900
|
|
6.
|
SR SampatGD O’ConnellJV FongE
Alegre-AguaronGA AteshianCT HungGrowth factor priming of
synovium-derived stem cells for cartilage tissue engineeringTissue
Eng Part A1722592265201110.1089/ten.tea.2011.015521542714
|
|
7.
|
M PeiF HeG
Vunjak-NovakovicSynovium-derived stem cell-based
chondrogenesisDifferentiation7610441056200810.1111/j.1432-0436.2008.00299.x18637024
|
|
8.
|
EA PapakonstantiC StournarasCell responses
regulated by early reorganization of actin cytoskeletonFEBS
Lett58221202127200810.1016/j.febslet.2008.02.06418325339
|
|
9.
|
F BeierRF LoeserBiology and pathology of
Rho GTPase, PI-3 kinase-Akt, and MAP kinase signaling pathways in
chondrocytesJ Cell
Biochem110573580201010.1002/jcb.2260420512918
|
|
10.
|
A WoodsG WangF BeierRegulation of
chondrocyte differentiation by the actin cytoskeleton and adhesive
interactionsJ Cell Physiol21318200710.1002/jcp.2111017492773
|
|
11.
|
EJ BlainInvolvement of the cytoskeletal
elements in articular cartilage homeostasis and pathologyInt J Exp
Pathol90115200910.1111/j.1365-2613.2008.00625.x19200246
|
|
12.
|
S YamaneAH ReddiInduction of
chondrogenesis and superficial zone protein accumulation in
synovial side population cells by BMP-7 and TGF-beta1J Orthop
Res26485492200810.1002/jor.2052117972329
|
|
13.
|
Y ShiJ MassagueMechanisms of TGF-beta
signaling from cell membrane to the
nucleusCell113685700200310.1016/S0092-8674(03)00432-X12809600
|
|
14.
|
LA McMahonPJ PrendergastVA CampbellA
comparison of the involvement of p38, ERK1/2 and PI3K in growth
factor-induced chondrogenic differentiation of mesenchymal stem
cellsBiochem Biophys Res
Commun368990995200810.1016/j.bbrc.2008.01.16018267113
|
|
15.
|
R TuliS TuliS NandiX HuangPA MannerWJ
HozackTransforming growth factor-beta-mediated chondrogenesis of
human mesenchymal progenitor cells involves N-cadherin and
mitogen-activated protein kinase and Wnt signaling cross-talkJ Biol
Chem2784122741236200310.1074/jbc.M305312200
|
|
16.
|
A MoustakasCH HeldinDynamic control of
TGF-beta signaling and its links to the cytoskeletonFEBS
Lett58220512065200810.1016/j.febslet.2008.03.02718375206
|
|
17.
|
S Etienne-MannevilleA HallRho GTPases in
cell biologyNature420629635200210.1038/nature01148
|
|
18.
|
A MammotoS HuangK MooreP OhDE IngberRole
of RhoA, mDia, and ROCK in cell shape-dependent control of the
Skp2-p27kip1 pathway and the G1/S transitionJ Biol
Chem2792632326330200410.1074/jbc.M40272520015096506
|
|
19.
|
G WangF BeierRac1/Cdc42 and RhoA GTPases
antagonistically regulate chondrocyte proliferation, hypertrophy,
and apoptosisJ Bone Miner
Res2010221031200510.1359/JBMR.05011315883643
|
|
20.
|
A WoodsG WangF BeierRhoA/ROCK signaling
regulates Sox9 expression and actin organization during
chondrogenesisJ Biol
Chem2801162611634200510.1074/jbc.M40915820015665004
|
|
21.
|
R McBeathDM PironeCM NelsonK BhadrirajuCS
ChenCell shape, cytoskeletal tension, and RhoA regulate stem cell
lineage commitmentDev
Cell6483495200410.1016/S1534-5807(04)00075-915068789
|
|
22.
|
A WoodsF BeierRhoA/ROCK signaling
regulates chondrogenesis in a context-dependent mannerJ Biol
Chem2811313413140200610.1074/jbc.M50943320016565087
|
|
23.
|
SC HubchakCE RunyanJI KreisbergHW
SchnaperCytoskeletal rearrangement and signal transduction in
TGF-beta1-stimulated mesangial cell collagen accumulationJ Am Soc
Nephrol1419691980200310.1097/01.ASN.0000076079.02452.9212874450
|
|
24.
|
S WangX WuTM LincolnJE
Murphy-UllrichExpression of constitutively active cGMP-dependent
protein kinase prevents glucose stimulation of thrombospondin 1
expression and TGF-beta
activityDiabetes5221442150200310.2337/diabetes.52.8.214412882934
|
|
25.
|
J LiX LongJ KeQG MengW FangIdentification
and characterization of synovial mesenchymal stem cells in
temporomandibular jointZhonghua Kou Qiang Yi Xue Za
Zhi403623642005(In Chinese).
|
|
26.
|
KJ LivakTD SchmittgenAnalysis of relative
gene expression data using real-time quantitative PCR and the
2(-Delta Delta C(T))
methodMethods25402408200110.1006/meth.2001.126211846609
|
|
27.
|
G WangA WoodsS SabariL PagnottaLA StantonF
BeierRhoA/ROCK signaling suppresses hypertrophic chondrocyte
differentiationJ Biol
Chem2791320513214200410.1074/jbc.M31142720014726536
|
|
28.
|
W BiJM DengZ ZhangRR BehringerB de
CrombruggheSox9 is required for cartilage formationNat
Genet228589199910.1038/8792
|
|
29.
|
DR HaudenschildJ ChenN PangMK LotzDD
D’LimaRho kinase-dependent activation of SOX9 in
chondrocytesArthritis
Rheum62191200201010.1002/art.2505120039424
|
|
30.
|
AK KamarajuAB RobertsRole of Rho/ROCK and
p38 MAP kinase pathways in transforming growth factor-beta-mediated
Smad-dependent growth inhibition of human breast carcinoma cells in
vivoJ Biol Chem28010241036200510.1074/jbc.M40396020015520018
|
|
31.
|
S ChenM CrawfordRM DayVR BrionesJE
LeaderPA JoseRhoA modulates Smad signaling during transforming
growth factor-beta-induced smooth muscle differentiationJ Biol
Chem28117651770200610.1074/jbc.M50777120016317010
|