Introduction
Age-related macular degeneration (AMD) is a
progressive neurodegenerative disease of the central retinal area
(macula lutea) and it represents the most common cause of legal
blindness in industrialized countries (1,2).
Epidemiologic studies from several countries also showed a dramatic
increase in the prevalence and severity of AMD with age. Despite
intensive basic and clinical research, its pathogenesis remains
unclear, likely due to its multifactorial character (3–6).
In addition to a strong age-dependence of the disease, a complex
interaction of metabolic, functional, genetic and environmental
factors appears to create a platform for chronically developing
changes in the ocular structures of the macular region
[choriocapillaries, Bruch’s membrane, retinal pigment epithelium
(RPE), photoreceptors] which may contribute in varying degrees to
the onset of AMD.
Traditionally, two subgroups of AMD can be
distinguished: atrophic (dry form) and exudative (wet form). The
dry form, synonymously known as age-related maculopathy, is
characterized by the presence of small yellowish deposits (drusen)
under the RPE, accompanied with either loss or focal accumulation
of melanin pigment. This form of AMD is typically characterized by
a progressing course leading to degeneration of RPE and
photoreceptors. The exudative form is linked to choroidal
neovascularization directed to the subretinal macular region, with
subsequent bleeding and/or fluid leakage, which may result in a
sudden loss of central vision; it is the most rapidly progressing
form of AMD. Both atrophic and exudative forms are associated with
severe impairment of visual functions (7). The pathophysiology of AMD is complex
and, in addition to genetic predispositions, at least 4 processes
contribute to the disease: lipofuscinogenesis, drusogenesis, local
inflammation and neovascularization (in the case of the wet form)
(3–13). The current pathophysiologic
concept regarding AMD assigns a primary role to the age-related,
cumulative oxidative damage to the RPE due to an imbalance between
generation and elimination of reactive oxygen species (ROS)
(14–16). Specifically, lipofuscin, a
heterogeneous material composed of a mixture of lipids (lipid
peroxides, proteins and different fluorescent compounds derived
mainly from vitamin A), has been hypothesized to be the primary
source of ROS responsible for both cellular and extracellular
matrix alterations in AMD (17–19).
Accumulation of lipofuscin, other lipid peroxides
and potentially toxic substances may dramatically influence the RPE
physiology. In vitro it greatly reduces the phagocytic
capacity, lysosomal enzyme activities and antioxidant potential of
human RPE (20,21). In vivo studies support
findings regarding the in vitro effects of lipofuscin on RPE
metabolism (22).
N-retinylidene-N-retinylethanolamine (A2E), the major
autofluorescent component of lipofuscin, specifically targets
cytochrome c oxidase (23,24), causes caspase activation and RPE
cell apoptosis (18,19). Thus, lipofuscin is thought to be
responsible for oxidative damage to RPE resulting in impaired
metabolism and apoptosis characteristic of late AMD (15).
Mitochondria play a central role in aging and in the
pathogenesis of age-related neurodegenerative diseases (25). Early studies have revealed several
abnormalities in mitochondrial DNA (mtDNA) and subsequent disorders
of respiratory enzyme complexes accompanied by reduced energy
production, generation of excessive ROS and activation of the
apoptosis pathway (26). However,
recent studies suggest that mitochondrial dysfunctions may also
include compositional and structural alterations in mitochondrial
membranes (mtMEM) (27,28). Alterations in membrane lipid
composition, such as decreases in cardiolipin content or changes in
the ω-3/ω-6 ratio, impair the electron transport chain and energy
production (29), ion channel and
Ca2+ homeostasis of the mitochondria and of the cell
(30–33). These alterations can also impair
carnitine-mediated lipid transport, mitochondrial lipid metabolism
(34,35), cholesterol biosynthesis (36–39), activity of the pyruvate
dehydrogenase complex (40,41) and, finally, cause activation of
the apoptosis pathway (42). In
the present study, detailed electron microscopy of human RPE cells
from aged and AMD specimens are described. Based on morphometric
data, we found that progressive deterioration of mtMEM with aging
occurred in association with peroxisome proliferation and
accumulation of lipofuscin in the RPE, and alterations in the RPE
mtMEM and proliferation of peroxisomes were significantly more
severe in AMD compared to normal aging.
Sensory innervation of the eye comes from the first
branch of the trigeminal nerve and it plays an essential role in
the physiology and pathophysiology of both the anterior and
posterior segments of the eye (43,44). Sensory nerves regulate tear
secretion either through the sensory arc of reflex tearing
(45) or directly through
releasing neuropeptides from sensory nerve endings (46). Both mechanisms are involved in the
development of dry eye syndromes (47,48). Although the retina has no sensory
innervations, substance P (SP)- and calcitonin gene-related peptide
(CGRP)-containing amacrine and ganglion cells have been observed in
the retina in various species including humans (49–51). Furthermore, several experimental
studies have suggested that these neuropeptides are involved in
various retinal diseases (52–54). Previously, an increase in SP and
CGRP immunoreactivity was observed in the retina after electric
stimulation of the trigeminal ganglion (55). However, the mechanism by which
these neuropeptides are generated in the retina has yet to be
elucidated. Capsaicin (CAP), a neurotoxic substance for polymodal C
sensory nerve fibers, is widely used in experiments, either for
sensory nerve stimulation or sensory nerve damage, depending on the
dose applied (56). Animals
exposed to CAP exhibit various corneal lesions, including a
reduction in the number of corneal nerve fibers and disintegration
of epithelial cells (57,58). In addition, changes in the
physiology of the ocular surface, such as a reduction in tear fluid
secretion, impairment of corneal epithelial barrier function and a
delay in corneal epithelial wound healing, are apparent in such
animals (59). These CAP-induced
changes thus appear to be similar to those characteristic of
neurotrophic keratouveitis in humans (60). Degeneration of amacrine and
ganglion cells has also been observed after CAP treatment (61). Pigment epithelium-derived factor
(PEDF) has been isolated from the RPE and has also been found in
the vitreous and the cornea (62). PEDF was originally identified as a
neurotrophic factor (63).
Subsequently, it was found to possess potent antiangiogenic
activity (64,65). It has been shown that PEDF is
essential for maintaining the avascularity of the cornea and
vitreous (66,67) and that it influences CAP-induced
neurotrophic keratouveitis. We confirm that retrobulbar
administration of PEDF may attenuate the effect of CAP on tear
secretion, keratouveitis and the retina.
Materials and methods
Sixty-five human eyes (age range 2–87 years) were
selected for these electron microscopic studies. All experiments
were conducted in accordance with the Declaration of Helsinki
(1964) and with the understanding and consent of the human
subjects. The responsible ethics committee approved the
experiments. Three eyes, aged 2, 7 and 27 years, were used only for
qualitative analysis of age-related changes. Thirty-one of the eyes
were affected by early AMD (ages from 42 to 87 years, mean age 70.9
years; 20 female and 11 male) and 31 non-affected eyes were used
for age- and gender-matched controls for both the qualitative and
quantitative morphometric studies. The selection criteria for early
AMD was based on the presence of drusen and/or basement membrane
thickening of the RPE, while in the controls no drusen or basement
membrane thickening of RPE was observed by electron microscopy.
Late forms of AMD (geographic atrophy and/or choroidal
neovascularization) were excluded from these studies. All of the
human eyes had been surgically removed as a result of malignant
tumors or severe ocular trauma, neither of which affected the
posterior pole of the eyeball.
One hundred and forty-four 4-week-old Sprague-Dawley
rats, weighing ~200 g and of both sexes, maintained in standard
laboratory conditions at 22°C temperature, 60% humidity and a 12-h
light/dark cycle, were divided into 6 groups of 24 rats each. Care
and treatment of the animals conformed to the ARVO Statement for
the Use of Animals in Ophthalmic and Vision Research, avoiding
animal suffering at each stage of the experiments. Rats of the
first group (CAP) were given a single retrobulbar injection of CAP
50 mg/kg (Sigma-Aldrich, St. Louis, MO, USA) in a procedure
described elsewhere. Rats in the second (CAP+PEDF 3.2 from Day 0)
and third (CAP+PEDF 6.4 from 0) groups received the same injection
of CAP plus daily retrobulbar injections of 3.2 or 6.4 μg/kg PEDF
starting from Day 0 (BioProducts, Middletown, MD, USA),
respectively. Rats in the fourth (CAP+PEDF 3.2 from Day 14) and
fifth (CAP+PEDF 6.4 from Day 14) group also received the same CAP
plus daily retrobulbar injection of 3.2 or 6.4 μg/kg PEDF starting
at Day 14 after CAP challenge. The sixth (control) group (C)
received the vehicle solution alone. One randomly selected eye of
each rat was used and the untreated fellow eye was used to evaluate
eventual systemic effects. PEDF treatment was repeated daily until
the end of the study.
Electron microscopy
Small pieces of the human retina and choroid were
dissected at the posterior pole <2 min after removal of the
eyeball and fixed at 4°C in 2% buffered glutaraldehyde for 2 h and
postfixed in 2% osmium tetroxide for another 2 h. The postfixation
with osmium tetroxide demonstrates lipid peroxides as effectively
as tetramethylbenzidine but avoids incubation, which might damage
membrane structures (68). The
specimens were dehydrated, embedded in Araldite, sectioned with a
Reichert ultramicrotome (Reichert, Vienna, Austria), contrasted
with lead citrate and uranyl acetate and studied with a Zeiss 109
electron microscope.
Small pieces (1×1 mm) of the rat cornea were
dissected and fixed in buffered 2% glutaraldehyde for 2 h,
postfixed in 2% osmium tetroxide for another 2 h, dehydrated and
embedded in Araldite. Ultrathin sections were cut with an
ultramicrotome (Reichert), contrasted with uranyl acetate and lead
citrate and studied with an electron microscope. After prefixation,
rat tissue samples were oriented and the exposed surface was coated
with gold-carbon vapor and examined with an electron microscope
equipped with a high-resolution scanning device used for
photography (EM Asid JEM-100B; JEOL, Tokyo, Japan).
Light microscopy
Six rats from each group were sacrificed using
carbon dioxide at Days 7, 14, 28 and 42. The eyes were enucleated
and fixed in Karnovsky’s solution for 48 h, dehydrated and embedded
in paraffin, and the 6-μm sections were colored with H&E and
Masson’s trichrome staining for light microscopy. In addition,
myeloid leukocytes labeled with chloroacetate-esterase were counted
in the H&E-stained sections under light microscopy.
Chloroacetate-esterase specifically identifies cells of the
granulocyte lineage, from the early promyelocyte stage to mature
neutrophils. The number of leukocytes was counted in the anterior
chamber, posterior chamber, peripheral retina and peripheral
vitreous at the same microscopic magnification.
Morphometry
In the parafoveal area of the macula, the total
number of mitochondria, number of lipofuscin granules and number of
peroxisomes were counted at a magnification of ×8,000. The number
of well-defined mitochondrial cristae and the area of the
mitochondria and lipofuscin, were measured in 6–8 RPE
cells/specimen at a magnification of ×20,000. Each organelle was
outlined and the area was determined using the NIH Image J program.
Morphometric analysis was performed by two experienced observers,
and the questionable cases were re-evaluated by the senior
researcher.
Tear secretion
Tear fluid secretion was measured without topical
anesthesia by a modified version of the Schirmer’s test. Schirmer
strips cut to dimensions of 20×1 mm were inserted below the lower
eyelid of the test animal for 1 min. The wet length of the strip
was measured to an accuracy of ±0.5 mm. The Schirmer’s test was
performed at Day 1 (before any treatment) and at Days 7, 14, 28 and
42 after CAP injection.
Statistical analysis
Data are expressed as means ± SD and were analyzed
using Statistica 6.0 software (StatSoft, Inc., Tulsa, OK, USA). In
the first experimental model to measure the relation between age
and the evaluated parameters, linear regression analysis was
applied for determining the P-value and Pearson correlation
coefficient, r. We used 2-sided comparisons to detect any
difference between linear regression results of the aged and AMD
groups. In all cases P<0.05 was considered to indicate a
statistically significant result.
In the second experimental model, statistical
analysis was performed with repeated-measures ANOVA using the
Dunnett’s multiple-comparison test for results in the 3 groups.
After Bonferroni adjustment, the significance level was set at
P<0.01.
Results
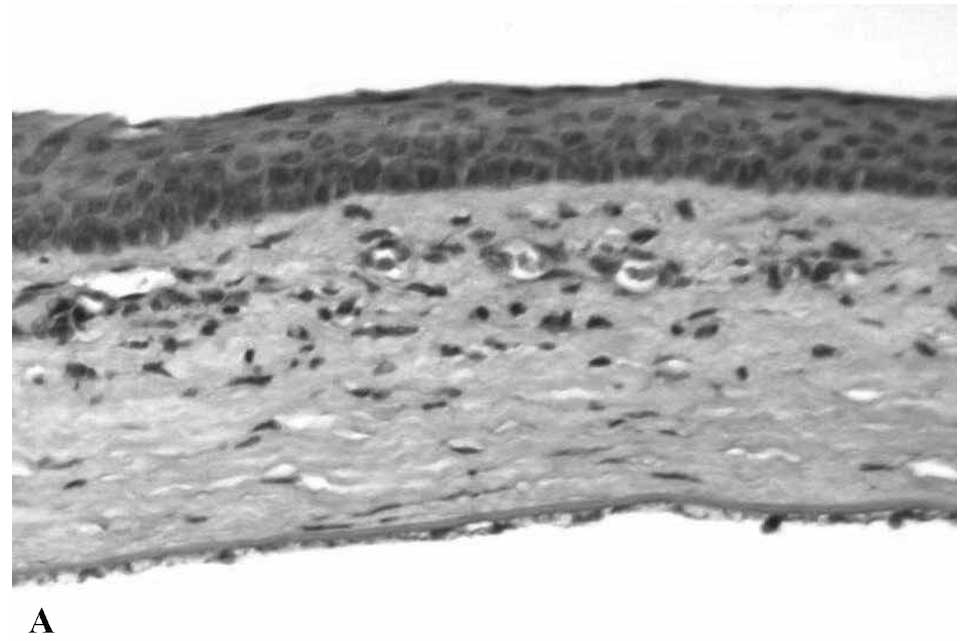
Concerning age-related changes of the RPE and
Bruch’s membrane, electron microscopy showed very marked
differences between young and aged human specimens (Fig. 1A and B). Mitochondria in young RPE
were numerous, mostly bacillus-like shaped and oriented parallel to
the apical-basal axis (Fig. 1A).
They were typically rich in well-preserved cristae. In normal aged
eyes, mitochondria of the RPE clearly decreased in number (Fig. 1B), were variable in size and were
usually oval-shaped without any preferential orientation. In some
instances, there was disorganization of cristae, ranging from focal
to complete loss, in association with decreased electron density of
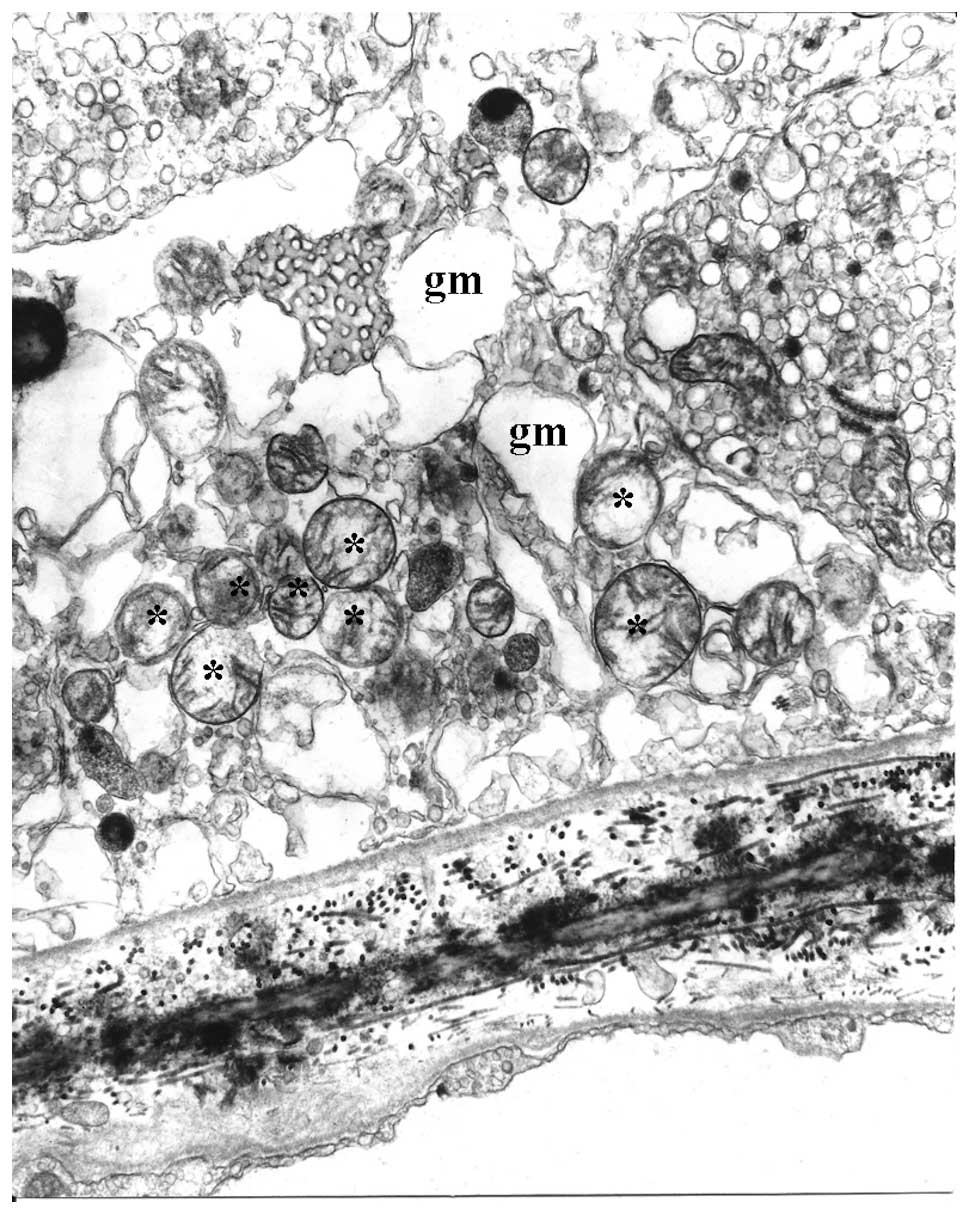
the matrix. Mitochondria of the RPE in AMD eyes (Fig. 2B) appeared to decrease in number
and size when compared to the normal aging eyes (Fig. 2A). They were often round or oval,
with focal or even complete loss of cristae and associated with
more extensive decreases in matrix density (Fig. 3). In some instances, either bleb
formation or interruption of the mitochondrial internal and
external membranes was observed. These may be considered
pre-apoptotic alterations. All of these mitochondrial alterations
were apparently more marked and more extensive in AMD compared to
normal aging. Bruch’s membrane fine structure was clearly visible
in young eyes (Fig. 1A) and
consisted of a core elastic layer, outer and inner collagenous
layers, basement membranes of the RPE and choriocapillaries. During
normal aging, there was an increase in electron density of Bruch’s
membrane (Fig. 1B). Moreover,
both inner and outer collagenous layers contained electron-dense
granular and vesicular material. Bruch’s membrane showed
characteristic differences in AMD compared to normal aged eyes. In
addition to the age-related increase in thickness and electron
density of collagenous layers, AMD specimens usually showed
multiplex, focal thickenings of the inner collagenous layer known
as hard or soft drusen. Apart from drusen, thickenings of the RPE
basement membrane due to basal laminar deposits were also present.
In most cases they were focal, wart-like depositions of filamentous
material in which several electron-dense areas, possibly composed
of lipids, were present. Cytoplasmic processes of the RPE invading
this excessive basement membrane material were frequently
observed.
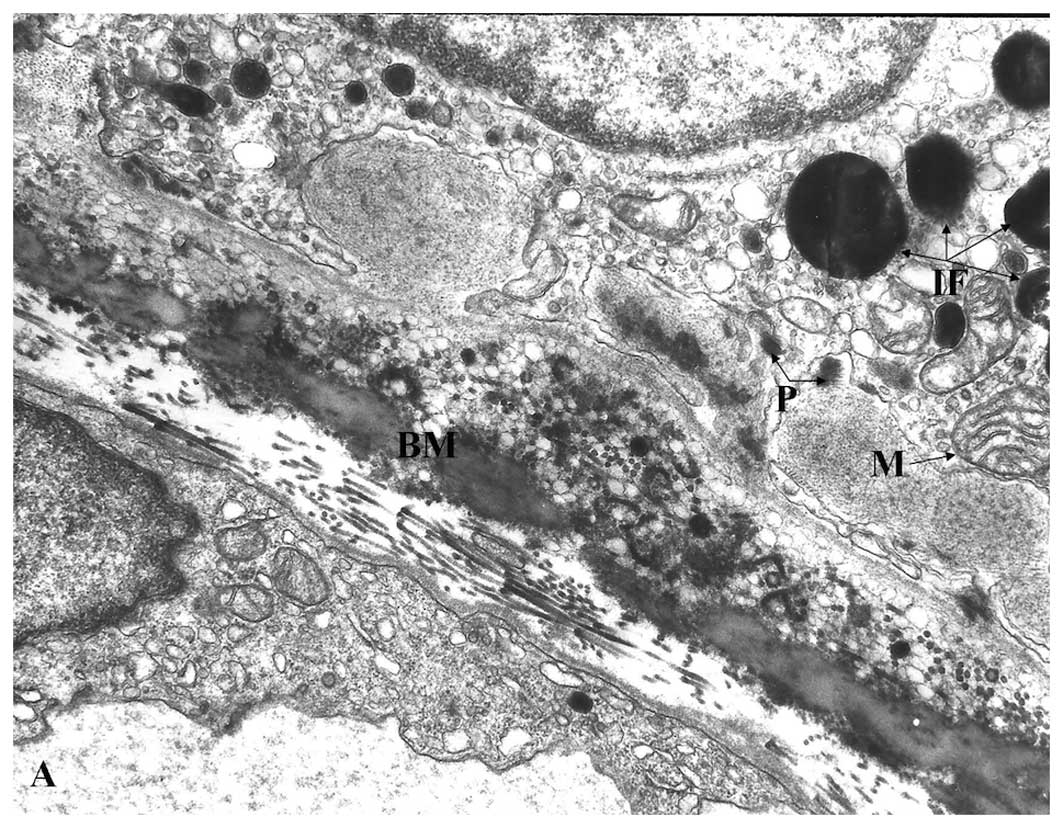
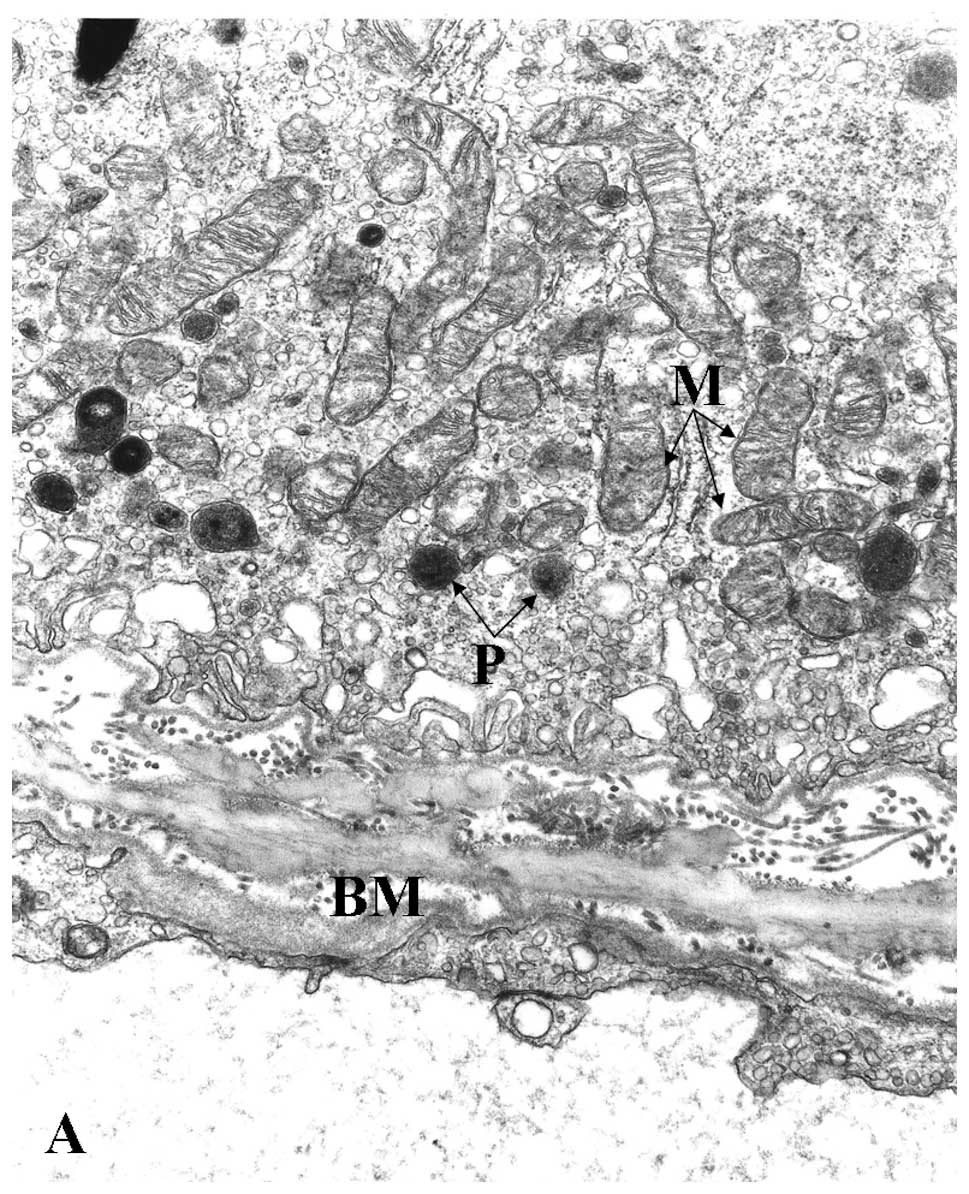 | Figure 1Representative electron micrographs
of the RPE and Bruch’s membrane. (A) Young RPE cells contained
numerous bacillus-like mitochondria (M) with long axes oriented
from the apical to the basal surfaces of the RPE. They were
parallel to one another and closely packed, filling a large portion
of the cytoplasm. The mitochondrial cristae were well preserved and
the matrix was homogeneously electron-dense. The cytoplasm
contained several rough-surface endoplasmic reticulum along with
numerous small vesicles, probably microsomes. Several peroxisomes
(P) appeared as small, round, electron-dense organelles. No
lipofuscin granules were noted. There were numerous basal
infoldings of the plasma membrane. Bruch’s membrane (BM)
ultrastructure was clearly visible and included an elastic core
layer with some interruptions, inner and outer collagenous layers,
basement membrane of the RPE and choriocapillaries. Two-year-old
male; magnification, ×25,000. (B) Aged RPE cells had mitochondria
(M) that showed various degrees of membrane disorganization.
Mostly, there were focal losses of cristae accompanied by decreased
electron density of the matrices. Numerous lipofuscin (IF) and
melano-lipofuscin granules were present in the cytoplasm of the
RPE. Peroxisomes (P) of various density, shape and size were
distributed randomly in the cytoplasm of the RPE cells, even among
the lipofuscin granules. Electron-dense homogeneous or granular
material was present in Bruch’s membrane (BM). Sixty-five-year-old
female; magnification, ×28,000. |
Peroxisomes in the RPE were 0.1–0.3 μm in diameter,
smaller than that in other cells (Fig. 1A and B). Some of these peroxisomes
had electron-dense cores while others had a granular appearance.
They were usually localized in the basal region of the cytoplasm
and occasionally next to the basal and basolateral cell membrane.
In normal aged eyes, peroxisomes were more numerous and more
variable in size, electron-density and distribution than in young
eyes. Furthermore, in young eyes they were dispersed randomly in
the cytoplasm of the RPE, while in aged eyes they formed small
groups containing 4–5 peroxisomes. The distribution of peroxisomes
in AMD eyes was highly variable within each RPE cell. Occasionally,
they were located in the apical cytoplasm among the lipofuscin
granules. More rarely, peroxisomes were in close topographic
correlation with mitochondria and the nucleus of the RPE cells. In
osmium-fixed specimens the peroxisomes varied in electron density.
Lipofuscin granules and residual bodies were exceptionally rare at
an early age, but they clearly increased in number and size with
time (Fig. 1B). In aged adults
they were abundantly distributed in the cytoplasm of the RPE.
Numerous melanolipofuscin granules were also present, formed by
fusion of melanosomes and lipofuscin. These organelles were located
in the apical half of the RPE in normal aged eyes. Lipofuscin
granules and residual bodies in AMD specimens presented a
morphology and distribution similar to control eyes.
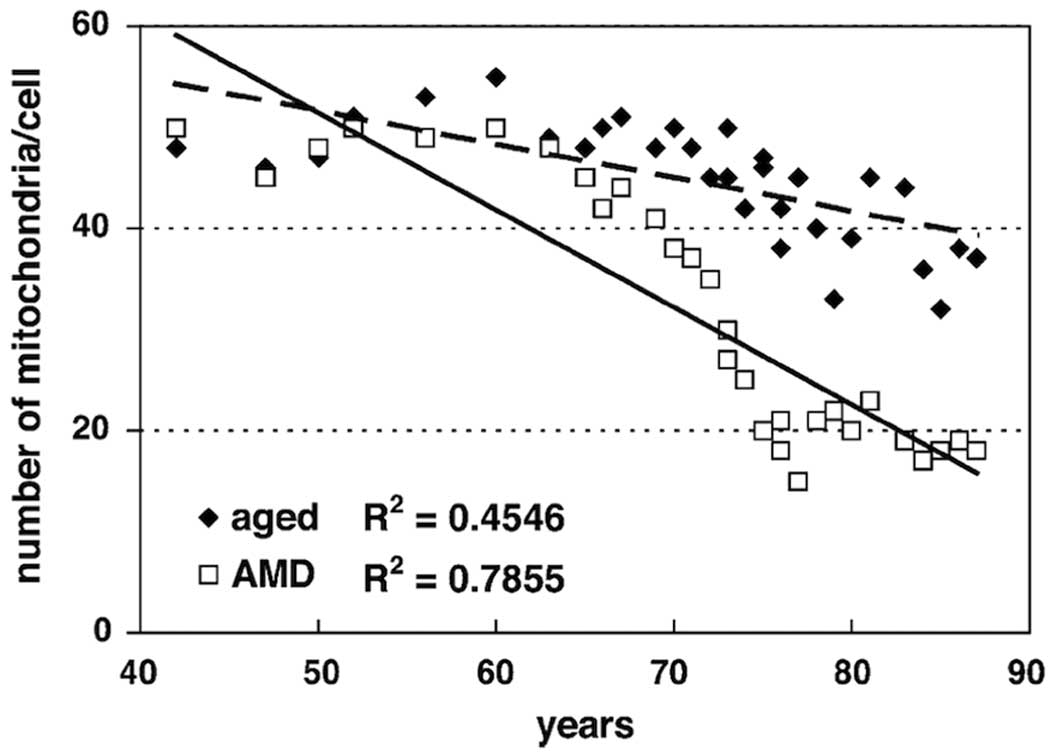
Morphometric analysis of the electron micrographs
quantified the mitochondrial and peroxisomal alterations. The total
number of mitochondria decreased significantly in both aged
(R2=0.455; P<0.001) and AMD (R2=0.778;
P<0.001) groups (Fig. 4). The
decrease in the AMD group was more severe and the difference was
statistically significant (P=0.038). The number of mitochondria at
age 75 in the AMD group was equivalent to that at age 85 in the
control group. These findings suggest that with age the number of
mitochondria decreased more rapidly in AMD compared to normal aged
patients.
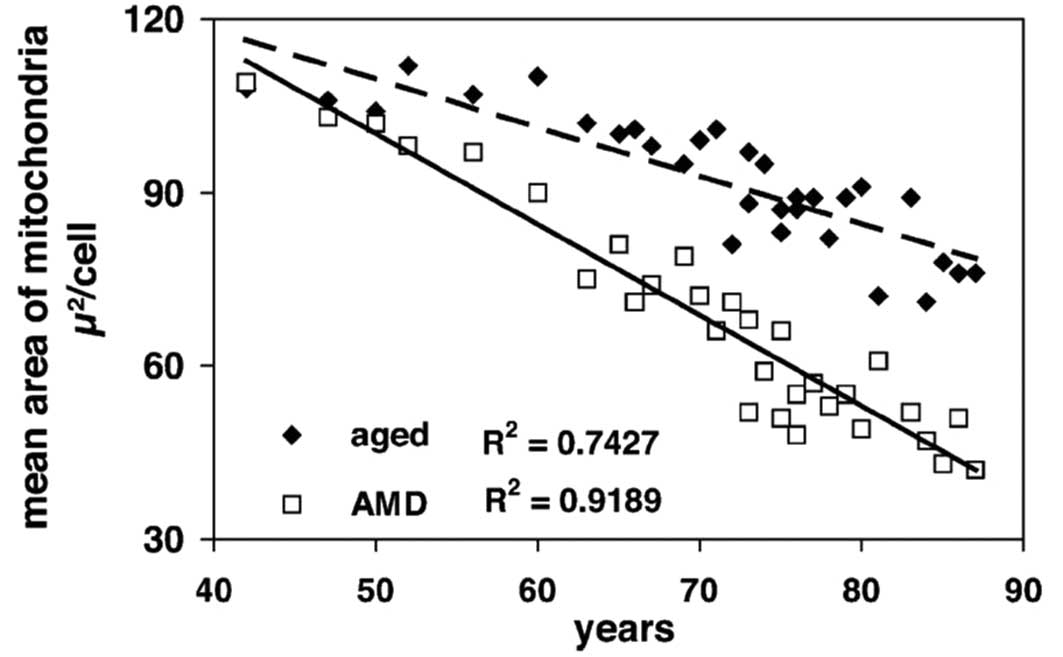
The area of the mitochondria also decreased
significantly with age in both control (R2=0.743;
P<0.001) and AMD (R2=0.919; P<0.001) groups
(Fig. 5). The decrease in the AMD
group was again more severe and the difference between the control
and AMD groups was statistically significant (P=0.019). The area of
mitochondria at 85 years in the control group was similar to that
of 70 years in the AMD patients; the decrease in area of
mitochondria developing 15 years earlier in the AMD patients.
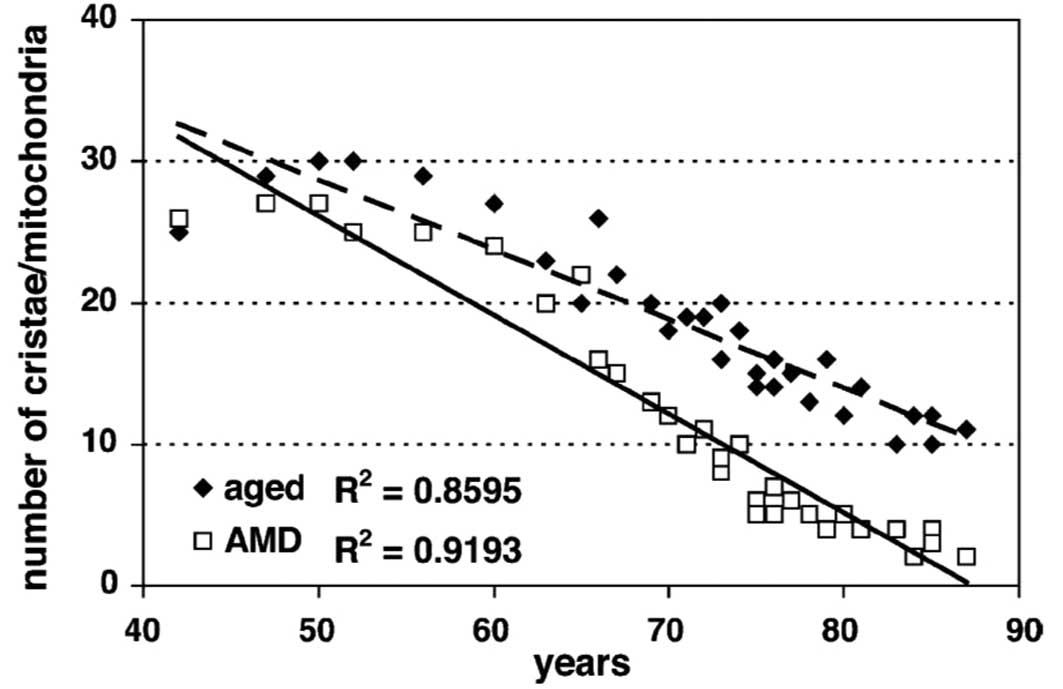
The number of well-defined mitochondrial cristae was
also counted and it showed a significant decrease in both the
control (R2=0.861; P<0.001) and AMD
(R2=0.918; P<0.001) groups (Fig. 6). However, the difference between
controls and AMD was not significant (P=0.28). Comparison of data
from the control group at 85 years showed again that the same loss
of well-defined mitochondrial cristae developed 15 years earlier in
AMD. The area of reduced matrix density corresponded to the loss of
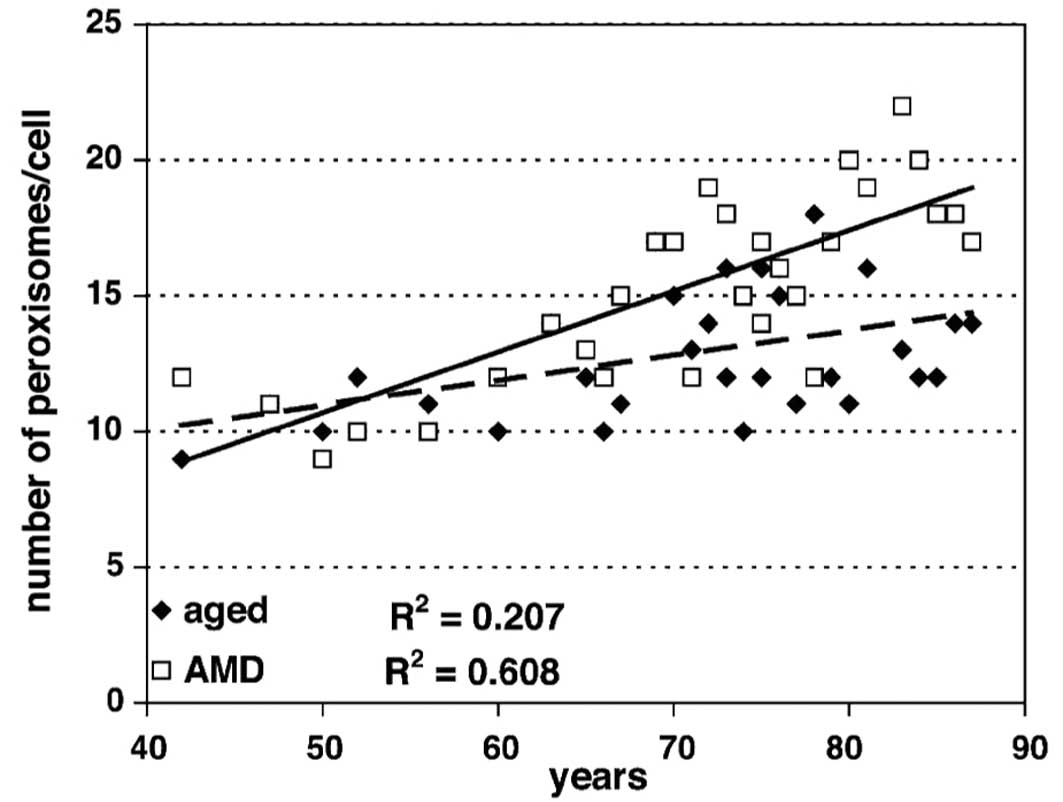
cristae (data not shown). Morphometric analysis showed a
significant increase in the number of peroxisomes in both the
control (R2=0.207; P<0.001) and AMD
(R2=0.608; P<0.001) groups (Fig. 7). Moreover, the difference between
the controls and AMD was statistically significant (P=0.044).
Comparison of data from the normal aged group at 85 years showed
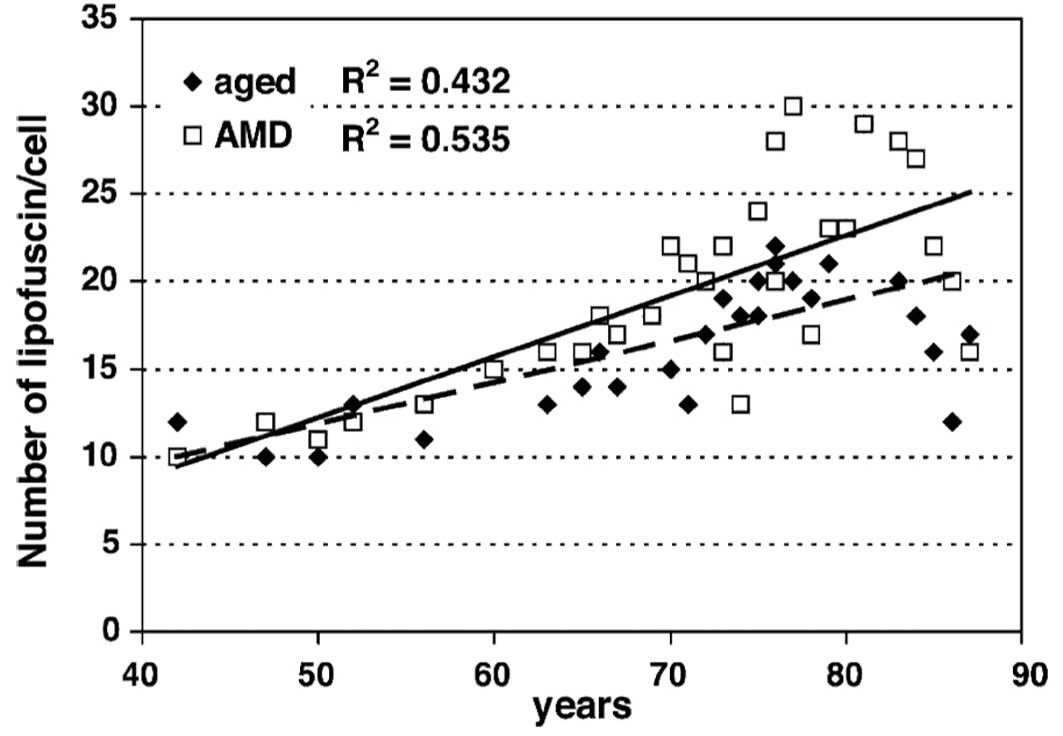
the same levels ~10 years earlier in the AMD group. There was a
significant increase in lipofuscin granules in both the control
(R2=0.432; P<0.001) and AMD (R2=0.535;
P<0.001) groups (Fig. 8).
However, the difference between the two groups was not
statistically significant (P=0.61). Comparison of data from the
normal aged group at 85 years showed that the same increase in the
number of lipofuscin granules developed ~15 years earlier in the
AMD group. Our study did not reveal any substantial differences
between females and males in both the aged and AMD groups.
In the second experimental model, we evaluated the
tear fluid secretion by Schirmer’s test in animals treated by
retrobulbar injection of CAP and PEDF. CAP treatment resulted in a
statistically significant decrease in tear secretion at Days 1
(2.50±0.55), 7 (3.17±0.41), 14 (3.50±0.55), 28 (3.50±0.55) and 42
(3.67±0.52) compared with the control values (6.83±1.33),
(6.50±1.38), (6.67±0.52), (6.67±0.52) and (7.17±0.98),
respectively. Treatment with 3.2 or 6.4 μg/kg PEDF from Day 0
attenuated the effect of CAP at Days 28 and 42 (P<0.001) only,
and a significantly decreased tear secretion was measured in group
CAP+PEDF 3.2 at Days 1 (2.67±0.52), 7 (3.33±0.82), 14 (4.17±0.75),
28 (4.83±0.41) and 42 (5.33±0.52) and in group CAP+PEDF 6.4 at Days
1 (2.50±0.55), 7 (3.83±0.41), 14 (4.33±0.52) and 28 (5.33±0.52)
compared with the controls. PEDF treatment initiated at Day 14 also
attenuated the effect of CAP pretreatment on tear secretion, but
these effects were not significant at any time point.
A single retrobulbar injection of CAP into young
rats caused a clinically marked inflammation of the anterior
segment progressing from slight punctate vacuolization in the
epithelium at the second or third days to diffuse edematous
opacities and neovascularization in the stroma, at the third or
fourth week, persisting at least for 6 weeks. These alterations in
the cornea showed continuous progression by the end of follow-up.
In contrast, by administration of PEDF, both corneal opacities and
scar formation were prevented and the cornea became completely
transparent at the end of the treatment. The most prominent
histopathologic feature of the affected corneas was a marked
disorganization of the epithelium, followed by marked
polymorphonuclear leukocyte influx as well as by edema and
disorganization of the corneal stroma with degeneration and loss of
the central epithelium. These alterations were accompanied by
fibrinous and cellular exudation into the anterior chamber
(Fig. 9A). Intercellular edema in
the corneal epithelium was particularly evident, even in the basal
and intermediate layers of the epithelium of CAP-treated animals.
There was extensive corneal neovascularization occurring
approximately on Day 14. Myeloid cell infiltration was also evident
within the angle, the anterior chamber and the iris. Treatment with
PEDF prevented and/or significantly attenuated corneal epithelial
and stromal damage in a dose-dependent manner (Fig. 9B). These data are provided in
detail in Table I.
 | Table INumber of myeloid cells in the eyes
at different time points of the study. |
Table I
Number of myeloid cells in the eyes
at different time points of the study.
| Group | Localization | Day 7 | Day 14 | Day 28 | Day 42 |
|---|
| Control | AC | 0.17±0.40 | 0.33±0.51 | 0.33±0.51 | 0.5±0.55 |
| PC | 0.16±0.41 | 0.33±0.51 | 0.33±0.51 | 0.5±0.55 |
| Vitreous | 0.16±0.41 | 0.33±0.51 | 0.33±0.51 | 0.5±0.55 |
| Retina | 0.16±0.41 | 0.33±0.51 | 0.33±0.51 | 0.5±0.55 |
| CAP-treated | AC | 10.5±2.16a | 12.5±2.95a | 17.83±3.92a | 22.16±2.14a |
| PC | 9.17±0.75a | 12.5±1.38a | 17.83±1.72a | 21.5±2.07a |
| Vitreous | 2.66±0.52a | 4.67±1.21a | 7.67±0.82a | 9.33±1.75a |
| Retina | 5.83±1.47a | 9.83±2.14a | 15±2.61a | 19±2.83a |
| CAP+3.2 μg PEDF
from Day 0 | AC | 7±0.89a,b | 9±0.89a,b | 5.83±1.16a,b | 5.16±0.75a,b |
| PC | 6.5±.05a,b | 7.67±0.82a,b | 5.5±0.83a,b | 4.5±0.55a,b |
| Vitreous | 2.5±0.55a | 2.83±0.41a,b | 3.5±1.22a,b | 1.83±0.41b |
| Retina | 3±0.63a,b | 3.67±1.03a,b | 4.33±0.82a,b | 2.67±1.03b |
| CAP+6.4 μg PEDF
from Day 0 | AC | 3.83±0.41a,b | 4.33±0.52a,b | 4.16±1.47a,b | 2.33±1.51b |
| PC | 3.83±0.41a,b | 3.83±0.75a,b | 3.5±0.83a,b | 2±1.26b |
| Vitreous | 1.66±0.82a | 2.66±0.52a,b | 2±1.26b | 0.33±0.52b |
| Retina | 3.67±0.52a,b | 2.67±0.51a,b | 2.5±1.04b | 1±0.89b |
| CAP+3.2 μg PEDF
from Day 14 | AC | 10.5±0.89a | 12.8±0.89a | 11.4±0.89a,b | 10.0±0.89a,b |
| PC | 9.8±1.03a | 12.6±0.98a | 12±0.89a,b | 10.3±0.83a,b |
| Vitreous | 3.1±0.75a | 5.3±0.83a | 5±0.83a,b | 4.8±0.89a,b |
| Retina | 6.8±0.89a | 10.6±0.89a | 9.4±0.98a,b | 8.5±0.98a,b |
| CAP+6.4 μg PEDF
from Day 14 | AC | 11±0.98a | 13.1±0.89a | 11.6±0.89a,b | 9.7±0.89a,b |
| PC | 9.9±0.89a | 12.4±1.14a | 11.8±0.89a,b | 9.3±0.82a,b |
| Vitreous | 4±0.83a | 6.0±0.82a | 5.1±0.75a,b | 4.3±0.83a,b |
| Retina | 7.2±0.89a | 11.2±0.89a | 9.7±0.98a,b | 7.6±0.89a,b |
Discussion
Our electron microscopic study demostrated that
mitochondria of the RPE undergo significant morphological changes
with age as a result of marked decreases in the number and area of
mitochondria, significantly more severe in ADM compared to
age-matched controls. These changes include a partial to complete
loss of cristae and decrease in the density of the mitochondrial
matrix in both normal aging and AMD groups. To our knowledge, this
is the first study to demonstrate alterations in mtMEM related to
age and AMD in human RPE. It also revealed significant differences
between normal aging and AMD. Ultrastructural and morphometric
studies showed similar alterations in mtMEM in certain age-related
human diseases, such as Alzheimer’s disease (69), in the skeletal muscle of patients
affected by type 2 diabetes or obesity (70) and in heart failure with chronic
obstructive pulmonary disease (71), as well as in schizophrenia
(72). Our findings suggest that
RPE in aging and AMD should be included in that list. These data
provide morphological support to the concept that mtMEM and
subsequent mitochondrial dysfunctions may play a crucial role in
the development of retinal degeneration, in particular of AMD
(16,73). Our data suggest that loss of
mitochondrial structure is one of the differences between normal
aging and AMD. Photoreceptors and RPE have an intimate morphologic
and functional partnership in order to maintain adequate metabolic
support for survival, excitability and turnover of photoreceptor
cells. Mitochondrial β-oxidation, which is present in the RPE
(74), is considered to be the
major pathway that metabolizes fatty acids as a primary source of
energy production (75,76). Since the RPE is involved in the
light-induced retinoid cycle (77,78), one of the possible consequences of
mitochondrial dysfunction in AMD may manifest as a disorder of
light-induced retinoid recycle. Mitochondrial alterations were also
accompanied by proliferation of peroxisomes. The morphometrical
study showed that an age-related increase in peroxisome number in
AMD specimens was significantly greater than the number in
age-matched controls. Peroxisomes are membrane-bound organelles
that play an essential role in lipid metabolism. They shorten the
very-long-chain fatty acids, and the resulting long-chain fatty
acids preferentially move back to the endoplasmic reticulum where
they are used for membrane lipid synthesis (79), or they are then handed over to
mitochondria for completion of oxidation (80). Intracellular elevation of
naturally occurring fatty acids and eicosanoids activates
peroxisome-proliferator-activated receptors (PPARs) resulting in
peroxisome proliferation and activation of lipid metabolism
(81). Activation of PPAR-α
upregulates genes of lipid catabolism, while activation of PPAR-γ
upregulates genes of lipogenesis (82,83). In a previous study (84), PPAR-γ ligands inhibited vascular
endothelial growth factor-induced choroidal angiogenesis in
vitro and choroidal neovascularization in vivo. This
suggests the potential involvement of PPAR-γ in AMD, at least in
the development of the late form of this disease. The increased
number of peroxisomes in aging and AMD may be a morphologic
manifestation for activation of an alternative pathway for lipid
degradation. This hypothesis is supported by observations that
induction of PPAR-α was associated with a strong stimulation of the
enzymes involved in peroxisomal β-oxidation (85,86). However, only PPRA-γ has been
detected in human RPE (84).
Accumulation of partially metabolized lipids such as lipofuscin and
lipid peroxides in RPE may be another possible consequence of
mitochondrial dysfunction (20,87). Lipofuscin may be responsible for
oxidative damage to the RPE that results in impaired metabolism and
apoptosis characteristic of late AMD (15). Studies suggest that lipofuscin is
a manifestation of the balance between production and elimination
of partially metabolized substances, mostly lipid
peroxide-containing materials (88,89). Thus, the age-related accumulation
of intracellular lipofuscin is an indicator for impairment of lipid
degradation processes (90). A
previous autofluorescent study (91) showed a higher lipofuscin content
in AMD compared to age-matched controls. Lipofuscin and other
metabolized lipids containing ROS may certainly compromise RPE
functions. However, the exact role of lipofuscin in AMD remains to
be elucidated. Alterations in mtMEM influence the carnitine system
located in the mitochondrial outer and inner membrane resulting in
impaired lipid metabolism (35).
mtMEM play an essential role in cholesterol metabolism in the liver
and in several extrahepatic cells including macrophages (38), which have several features in
common with RPE. A peripheral type benzodiazepine receptor, a
channel-forming mitochondrial protein, is involved in the
regulation of cholesterol transport from the outer to the inner
mtMEM. This is the rate-determining step in steroid biosynthesis.
The lipid composition of mtMEM might be involved directly in ion
channel regulation (31). A
decrease in mtMEM potential occurs in a variety of aging cell types
from several mammalian species (32,33). When mitochondria are subjected to
oxidative stress and relatively high Ca2+, they may
undergo a permeability transition in which the inner membrane
becomes freely permeable to low-molecular-weight substances
resulting in impairment of all mitochondrial functions (30). Mitochondrial ion channels are
critically involved in apoptotic changes in mitochondria (42). In addition to the well-documented
changes in mtDNA, our data suggest that alterations in mtMEM also
play a crucial role in the development of mitochondrial
dysfunctions in AMD and, possibly, in other age-related diseases
(92). This has an immediate
clinical relevance as shown by several in vitro and in
vivo studies. In fact, mtMEM could be a target for treatment of
mitochondrial dysfunctions. In vitro modification of mtMEM
composition by ω-3 fatty acids and addition of carnitine or
acetyl-L-carnitine resulted in a subsequent improvement in
mitochondrial lipid metabolism (93). An in vivo study showed that
dietary ω-3 fatty acids directly increase membrane ω-3/ω-6 ratio,
restore mtMEM cardiolipin content and membrane potential, as well
as subsequently restore alterated mitochondrial Ca2+
flux and Ca2+-dependent processes such as activity of
pyruvate dehydrogenase complex (94).
Furthermore, research demonstrated that a
retrobulbar injection of CAP into young rats resulted in decreased
tear secretion and neurotrophic keratouveitis characterized by
epithelial alteration, stromal edema and scar formation accompanied
by neovascularization of the cornea (60). In contrast to this study, we
observed leukocyte infiltration in the posterior chamber,
peripheral retina and vitreous body. These CAP-induced alterations
were attenuated in a dose-dependent manner by retrobulbar injection
of PEDF. CAP exerts its effects through binding to transient
receptor potential vanilloid type 1 (TRPV1), which is a
Ca2+-permeable ion channel. CAP may act on TRPV1
receptors of non-neuronal cells or sensory nerves of the cornea,
conjunctiva, lacrimal glands, ciliary body and choroids. The
activation of TRPV1 in the sensory nerve endings induces the
release of the proinflammatory neuropeptides SP and CGRP, resulting
in neurogenic inflammation, that may have been a leading
contributing factor to the CAP-induced keratouveitis in our model.
Non-neuronal cells include epithelial cells (keratinocytes,
urothelium, gastric epithelial cells, enterocytes and pneumocytes),
vascular endothelium and cells of the immune system as well as
smooth muscle cells, fibroblasts and hepatocytes (95). PEDF accelerated the recovery of
tear secretion and prevented neurotrophic keratouveitis and
peripheral vitreoretinal inflammation by neurotrophic and
antiangiogenic mechanisms. In vitro studies have shown that
neuronal growth factors exert their effects through modulating
TRPV1 expression and activity of sensory nerve cells (96). Recently, topical treatment with
nerve growth factor (NGF) was also shown to restore corneal
integrity in humans with corneal neurotrophic ulcers or keratitis
(97). Moreover, there is
accumulating evidence that in normal conditions there may be a
balance between the release of PEDF and vascular endothelial growth
factor (VEGF) (98). A decrease
in the levels of PEDF or an increase in VEGF may be responsible for
neovascularization in the exudative form of age-related macular
degeneration (AMD), diabetic retinopathy and ischemia-induced
retinal neovascularization (99).
These findings suggest a certain antagonism between PEDF and VEGF
(98). Contoversial studies have
shown direct neurotrophic effects of VEGF similar to nerve growth
factors suggesting a synergy between them in providing
neuroprotection (100–102). Further studies are certainly
needed to reveal the molecular mechanism of the association between
PEDF and VEGF in both neuroprotection and angiogenesis.
In conclusion, our study on restoration of
mitochondrial function are certainly very promising as they open up
a new therapeutic approach to AMD. However, the correlation between
mitochondrial dysfunction and Bruch’s membrane alterations remains
to be elucidated through ongoing studies. Clinical studies
confirmed that a combination of acetyl-L-carnitine, ω-3 fatty acids
and Coenzyme Q10, after an initial improvement, stabilized several
visual functions in early AMD (103). However, further studies are also
necessary to reveal how the clinically known risk factors of AMD
are different between normal aging and AMD and why alterations of
the RPE common to both normal aging and AMD groups occur at a
younger age in AMD. This is also the first experimental study to
suggest a neuroprotective and antiangiogenic effect of PEDF in
CAP-induced keratouveitis. Although the involvement of TRPV1 is
presumed, the molecular mechanisms of PEDF remain to be elucidated.
PEDF is certainly related to neuroprotection and angiogenesis, but
its role in the pathophysiology of the ocular compartment remains
somewhat unclear.
Acknowledgements
The authors are very grateful to Ida Bozso for the
technical assistance in preparing the microscopy specimens and to
Sharon Hobby for the English language editing.
References
|
1
|
Friedman DS, O’Colmain BJ, Munoz B, Tomany
SC, McCarty C, de Jong PT, Nemesure B, Mitchell P and Kempen J; Eye
Diseases Prevalence Research Group. Prevalence of age-related
macular degeneration in the United States. Arch Ophthalmol.
122:564–572. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klaver CC, Wolfs RC, Vingerling JR, Ofman
A and De Jong PT: Age specific prevalence and causes of blindness
and visual impairment in an older population: the Rotterdam Study.
Arch Ophthalmol. 116:653–658. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fine SL, Berger JW, Maguire MG and Ho AC:
Age-related macular degeneration. N Engl J Med. 342:483–492. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McConnell V and Silvestri G: Age-related
macular degeneration. Ulster Med J. 74:82–92. 2005.PubMed/NCBI
|
|
5
|
Nowak JZ: Role of lipofuscin in
pathogenesis of age-related macular degeneration (AMD). Mag Okul.
2:103–114. 2005.(In Polish).
|
|
6
|
Nowak JZ: Drusen, basal deposits,
inflammation and age-related macular degeneration (AMD). Mag Okul.
2:174–186. 2005.(In Polish).
|
|
7
|
Bird AC, Bressler NM, Bressler SB,
Chisholm IH, Coscas G, Davis MD, de Jong PT, Klaver CC, Klein BE,
Klein R, et al: An international classification and grading system
for age-related maculopathy and age-related macular degeneration.
The International ARM Epidemiological Study Group. Surv Ophthalmol.
39:367–374. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anderson DH, Mullins RF, Hageman GS and
Johnson LV: A role for local inflammation in the formation of
drusen in the aging eye. Am J Ophthalmol. 134:411–431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Campochiaro PA: Ocular neovascularization
and excessive vascular permeability. Expert Opin Biol Ther.
4:1395–1402. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kijlstra A, La Heij EC and Hendrikse F:
Immunological factors in the pathogenesis and treatment of
age-related macular degeneration. Ocular Immunol Inflam. 13:3–11.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klein R, Peto T, Bird A and Vannewkirk MR:
The epidemiology of age-related macular degeneration. Am J
Ophthalmol. 137:486–495. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sparrow JR and Boulton M: RPE lipofuscin
and its role in retinal pathobiology. Exp Eye Res. 80:595–606.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wiktorowska-Owczarek A and Nowak JZ:
Oxidative damage in age-related macular degeneration (AMD) and
antioxidant protection as a therapeutic strategy. Pol J Environ
Stud. 15:69–72. 2006.
|
|
14
|
Beatty S, Koh H, Phil M, Henson D and
Boulton M: The role of oxidative stress in the pathogenesis of
age-related macular degeneration. Surv Ophthalmol. 45:115–134.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dunaief JL, Dentchev T, Ying GS and Milam
AH: The role of apoptosis in age-related macular degeneration. Arch
Ophthalmol. 120:1435–1442. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Winkler BS, Boulton ME, Gottsch JD and
Sternberg P: Oxidative damage and age-related macular degeneration.
Mol Vis. 5:321999.PubMed/NCBI
|
|
17
|
Hollyfield JG, Salomon RG and Crabb JW:
Proteomic approaches to understanding age-related macular
degeneration. Adv Exp Med Biol. 533:83–89. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin GF, Hurst JS and Godley BF: Rod outer
segments mediate mitochondrial DNA damage and apoptosis in human
retinal pigment epithelium. Curr Eye Res. 23:11–19. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wolf G: Lipofuscin and macular
degeneration. Nutr Rev. 61:342–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shamsi FA and Boulton M: Inhibition of the
RPE lysosomal and antioxidant activity by the age pigment
lipofuscin. Invest Ophthalmol Vis Sci. 42:3041–3046.
2001.PubMed/NCBI
|
|
21
|
Sundelin S, Wihlmark U, Nilsson SE and
Brunk UT: Lipofuscin accumulation in cultured retinal pigment
epithelial cells reduces their phagocytic capacity. Curr Eye Res.
17:851–857. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rakoczy PE, Zhang D, Robertson T, Barnett
NL, Papadimitriou J, Constable IJ and Lai CM: Progressive
age-related changes similar to age-related macular degeneration in
a transgenic mouse model. Am J Pathol. 161:1515–1524. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shaban H, Borras C, Vina J and Richter C:
Phosphatidylglycerol potently protects human retinal pigment
epithelial cells against apoptosis induced by A2E, a compound
suspected to cause age-related macula degeneration. Exp Eye Res.
75:99–108. 2002. View Article : Google Scholar
|
|
24
|
Suter M, Remé C, Grimm C, Wenzel A,
Jaattela M, Esser P, Kociok N, Leist M and Richter C: Age-related
macular degeneration. The lipofusion component
N-retinyl-N-retinylidene ethanolamine detaches proapoptotic
proteins from mitochondria and induces apoptosis in mammalian
retinal pigment epithelial cells. J Biol Chem. 275:39625–39630.
2000. View Article : Google Scholar
|
|
25
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar
|
|
26
|
Schon EA and Manfredi G: Neuronal
degeneration and mitochondrial dysfunction. J Clin Invest.
111:303–312. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
James AM and Murphy MP: How mitochondrial
damage affects cell function. J Biomed Sci. 9:475–487. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Toescu EC, Myronova N and Verkhratsky A:
Age-related structural and functional changes of brain
mitochondria. Cell Calcium. 28:329–338. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paradies G, Ruggiero FM, Petrosillo G and
Quagliariello E: Age-dependent decline in the cytochrome c
oxidase activity in rat heart mitochondria: role of cardiolipin.
FEBS Lett. 406:136–138. 1997.PubMed/NCBI
|
|
30
|
Crompton M: Mitochondria and aging: a role
for the permeability transition. Aging Cell. 3:3–6. 2004.
View Article : Google Scholar
|
|
31
|
Gabbita SP, Subramaniam R, Allouch F,
Carney JM and Butterfield DA: Effects of mitochondrial respiratory
stimulation on membrane lipids and proteins: an electron
paramagnetic resonance investigation. Biochim Biophys Acta.
1372:163–173. 1998. View Article : Google Scholar
|
|
32
|
Rottenberg H and Wu S: Mitochondrial
dysfunction in lymphocytes from old mice: enhanced activation of
the permeability transition. Biochem Biophys Res Commun. 240:68–74.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sugrue MM and Tatton WG: Mitochondrial
membrane potential in aging cells. Biol Signals Recept. 10:176–188.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Battelli D, Bellei M, Arrigoni-Martelli E,
Muscatello U and Bobyleva V: Interaction of carnitine with
mitochondrial cardiolipin. Biochim Biophys Acta. 1117:33–36. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zammit VA, Corstorphine CG, Kolodziej MP
and Fraser F: Lipid molecular order in liver mitochondrial outer
membranes, and sensitivity of carnitine palmitoyltransferase I to
malonyl-CoA. Lipids. 33:371–376. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Beaumont K, Skowronski R, Vaughn DA and
Fanestil DD: Interactions of lipids with peripheral-type
benzodiazepine receptors. Biochem Pharmacol. 37:1009–1014. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Campbell AM, Capuano A and Chan SH: A
cholesterol-binding and transporting protein from rat liver
mitochondria. Biochim Biophys Acta. 1567:123–132. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hansson M, Ellis E, Hunt MC, Schmitz G and
Babiker A: Marked induction of sterol 27-hydroxylase activity and
mRNA levels during differentiation of human cultured monocytes into
macrophages. Biochim Biophys Acta. 1593:283–289. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Papadopoulos V: Peripheral benzodiazepine
receptor: structure and function in health and disease. Ann Pharm
Fr. 61:30–50. 2003.PubMed/NCBI
|
|
40
|
Denton RM, Randle PJ, Bridges BJ, Cooper
RH, Kerbey AL, Pask HT, Severson DL, Stansbie D and Whitehouse S:
Regulation of mammalian pyruvate dehydrogenase. Mol Cell Biochem.
9:27–53. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Robison WG Jr and Kuwabara T:
Microperoxisomes in the retinal pigment epithelium. Invest
Ophthalmol. 14:866–872. 1975.PubMed/NCBI
|
|
42
|
Szabo II, Adams C and Gulbins E: Ion
channels and membrane rafts in apoptosis. Pflugers Arch.
448:304–312. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Beuerman RW and Stern ME: Neurogenic
inflammation: a first line of defense for the ocular surface. Ocul
Surf. 3(Suppl 4): S203–S206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Troger J, Kieselbach G, Teuchner B,
Kralinger M, Nguyen QA, Haas G, Yayan J, Gottinger W and Schmid E:
Peptidergic nerves in the eye, their source and potential
pathophysiological relevance. Brain Res Rev. 53:39–62.
2007.PubMed/NCBI
|
|
45
|
Stern ME, Gao J, Siemasko KF, Beuerman RW
and Pflugfelder SC: The role of the lacrimal functional unit in the
pathophysiology of dry eye. Exp Eye Res. 78:409–416. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kovacs I, Ludany A, Koszegi T, Fehér J,
Kovacs B, Szolcsanyi J and Pintér E: Substance P released from
sensory nerve endings influences tear secretion and goblet cell
function in the rat. Neuropeptides. 39:395–402. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Feher J: Contribution of neurogenic
inflammation to irritable eye syndrome. Adv Exp Med Biol.
506:1047–1050. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Baudouin C: A new approach for better
comprehension of diseases of the ocular surface. J Fr Ophtalmol.
30:239–246. 2007.(In French).
|
|
49
|
Brecha NC, Sternini C, Anderson K and
Krause JE: Expression and cellular localization of substance
P/neurokinin A and neurokinin B mRNAs in the rat retina. Vis
Neurosci. 3:527–535. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Caruso DM, Owczarzak MT and Pourcho RG:
Colocalization of substance P and GABA in retinal ganglion cells: a
computer-assisted visualization. Vis Neurosci. 5:389–394. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bagnoli P, Dal Monte M and Casini G:
Expression of neuropeptides and their receptors in the developing
retina of mammals. Histol Histopathol. 18:1219–1242.
2003.PubMed/NCBI
|
|
52
|
May A, Shepheard SL, Knorr M, Effert R,
Wessing A, Hargreaves RJ, Goadsby PJ and Diener HC: Retinal plasma
extravasation in animals but not in humans: implications for the
pathophysiology of migraine. Brain. 121:1231–1237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gaspar MN, Ribeiro CA, Cunha-Vaz JG and
Macedo TR: Effects of neuropeptides on the sumatriptan-disturbed
circulation in the optic nerve head of rabbits. Pharmacology.
70:152–159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nucci C, Gasperi V, Tartaglione R, Cerulli
A, Terrinoni A, Bari M, De Simone C, Agrò AF, Morrone LA,
Corasaniti MT, Bagetta G and Maccarrone M: Involvement of the
endocannabinoid system in retinal damage after high intraocular
pressure-induced ischemia in rats. Invest Ophthalmol Vis Sci.
48:2997–3004. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bronzetti E, Artico M, Kovacs I, Felici
LM, Magliulo G, Vignone D, D’Ambrosio A, Forte F, Di Liddo R and
Feher J: Expression of neurotransmitters and neurotrophins in
neurogenic inflammation of the rat retina. Eur J Histochem.
51:251–260. 2007.PubMed/NCBI
|
|
56
|
Szolcsanyi J: Forty years in capsaicin
research for sensory pharmacology and physiology. Neuropeptide.
38:377–384. 2004.PubMed/NCBI
|
|
57
|
Fujita S, Shimizu T, Izumi K, Fukuda T,
Sameshima M and Ohba N: Capsaicin-induced neuroparalytic
keratitis-like corneal changes in the mouse. Exp Eye Res.
38:165–175. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ogilvy CS, Silverberg KR and Borges LF:
Sprouting of corneal sensory fibers in rats treated at birth with
capsaicin. Invest Ophthalmol Vis Sci. 32:112–121. 1991.PubMed/NCBI
|
|
59
|
Gallar J, Pozo MA, Rebollo I and Belmonte
C: Effects of capsaicin on corneal wound healing. Invest Ophthalmol
Vis Sci. 31:1968–1974. 1990.PubMed/NCBI
|
|
60
|
Waldrep JC and Crosson CE: Induction of
keratouveitis by capsaicin. Curr Eye Res. 7:1173–1182. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ritter S and Dinh TT: Capsaicin-induced
neuronal degeneration in the brain and retina of preweanling rats.
J Comp Neurol. 296:447–461. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tombran-Tink J, Shivaram SM, Chader GJ,
Johnson LV and Bok D: Expression, secretion, and age-related
downregulation of pigment epithelium-derived factor, a serpin with
neurotrophic activity. J Neurosci. 15:4992–5003. 1995.PubMed/NCBI
|
|
63
|
Becerra SP, Palmer I, Kumar A, Steele F,
Shiloach J, Notario V and Chader GJ: Overexpression of fetal human
pigment epithelium-derived factor in Escherichia coli: a
functionally active neurotrophic factor. J Biol Chem.
268:23148–23156. 1993.PubMed/NCBI
|
|
64
|
Dawson DW, Volpert OV, Gillis P, Crawford
SE, Xu H, Benedict W and Bouck NP: Pigment epithelium-derived
factor: a potent inhibitor of angiogenesis. Science. 285:245–258.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chung C, Doll JA, Stellmach VM, Gonzales
J, Surapureddi S, Cornwell M, Reddy JK and Crawford SE: Pigment
epithelium-derived factor is an angiogenesis and lipid regulator
that activates peroxisome proliferator-activated receptor alpha.
Adv Exp Med Biol. 617:591–597. 2008. View Article : Google Scholar
|
|
66
|
Bouck N: PEDF: anti-angiogenic guardian of
ocular function. Trends Mol Med. 8:330–334. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Spranger J, Osterhoff M, Reimann M, Mohlig
M, Ristow M, Francis MK, Cristofalo V, Hammes HP, Shith G, Boulton
M and Pfeiffer AF: Loss of the antiangiogenic pigment
epithelium-derived factor in patients with angiogenic eye disease.
Diabetes. 50:2641–2645. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zingsheim HP and Plattner H: Electron
microscopic methods in membrane biology. Methods in Membrane
Biology. Korn ED: Plenum Press; New York, NY: 1. –146. 1976,
View Article : Google Scholar
|
|
69
|
Hirai K, Aliev G, Nunomura A, Fujioka H,
Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M,
Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB,
Perry G and Smith MA: Mitochondrial abnormalities in Alzheimer’s
disease. J Neurosci. 21:3017–3023. 2001.
|
|
70
|
Kelley DE, He J, Menshikova EV and Ritov
VB: Dysfunction of mitochondria in human skeletal muscle in type 2
diabetes. Diabetes. 51:2944–2950. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Gosker HR, Wouters EFM, van der Vusse GJ
and Schols AMWJ: Skeletal muscle dysfunction in chronic obstructive
pulmonary disease and chronic heart failure: underlying mechanisms
and therapy perspectives. Am J Clin Nutr. 71:1033–1047.
2000.PubMed/NCBI
|
|
72
|
Ben-Shachar D: Mitochondrial dysfunction
in schizophrenia: a possible linkage to dopamine. J Neurochem.
83:1241–1251. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fox DA, Poblenz AT, He L, Harris JB and
Medrano CJ: Pharmacological strategies to block rod photoreceptor
apoptosis caused by calcium overload: a mechanistic target-site
approach to neuroprotection. Eur J Ophthalmol. 13(Suppl 3):
S44–S56. 2003.PubMed/NCBI
|
|
74
|
Tyni T, Johnson M, Eaton S, Pourfarzam M,
Andrews R and Turnbull DM: Mitochondrial fatty acid beta-oxidation
in the retinal pigment epithelium. Pediatr Res. 52:595–600.
2002.PubMed/NCBI
|
|
75
|
Andrews RM, Griffiths PG, Johnson MA and
Turnbull DM: Histochemical localisation of mitochondrial enzyme
activity in human optic nerve and retina. Br J Ophthalmol.
83:231–235. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hiltunen JK and Qin Y:
Beta-oxidation-strategies for the metabolism of a wide variety of
acyl-CoA esters. Biochem Biophys Acta. 1484:117–128.
2000.PubMed/NCBI
|
|
77
|
Kuksa V, Imanishi Y, Batten M, Palczewski
K and Moise AR: Retinoid cycle in the vertebrate retina:
experimental approaches and mechanisms of isomerization. Vision
Res. 43:2959–2981. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sparrow JR, Fishkin N, Zhou J, Cai B, Jang
YP, Krane S, Itagaki Y and Nakanishi K: A2E, a byproduct of the
visual cycle. Vision Res. 43:2983–2990. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sprecher H: Metabolism of highly
unsaturated n-3 and n-6 fatty acids. Biochim Biophys Acta.
1486:219–231. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hettema EH and Tabak HF: Transport of
fatty acids and metabolites across the peroxisomal membrane.
Biochim Biophys Acta. 1486:18–27. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kliewer SA, Sundseth SS, Jones SA, Brown
PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Wilson TM, Lenhard JM
and Lehmann JM: Fatty acids and eicosanoids regulate gene
expression through direct interactions with peroxisome
proliferator-activated receptors alpha and gamma. Proc Natl Acad
Sci USA. 94:4318–4323. 1997. View Article : Google Scholar
|
|
82
|
Kersten S, Desvergne B and Wahli W: Roles
of PPARs in health and disease. Nature. 405:421–424. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Nagy L, Tontonoz P, Alvarez JGA, Chen H
and Evans RM: Oxidized LDL regulates macrophage gene expression
through ligand activation of PPARgamma. Cell. 93:229–240. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Murata T, He S, Hangai M, Ishibashi T, Xi
XP, Kim S, Hsueh WA, Ryan SJ, Law RE and Hinton DR: Peroxisome
proliferator-activated receptor-gamma ligands inhibit choroidal
neovascularization. Invest Ophthalmol Vis Sci. 41:2309–2317.
2000.PubMed/NCBI
|
|
85
|
Forman BM, Chen J and Evans RM:
Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids
are ligands for peroxisome proliferator-activated receptors alpha
and delta. Proc Natl Acad Sci USA. 94:4312–4317. 1997. View Article : Google Scholar
|
|
86
|
Keller H, Dreyer C, Medin J, Mahfoudi A,
Ozato K and Wahli W: Fatty acids and retinoids control lipid
metabolism through activation of peroxisome proliferator-activated
receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci USA.
90:2160–2164. 1993. View Article : Google Scholar
|
|
87
|
Brunk UT and Terman A: The
mitochondrial-lysosomal axis theory of aging: accumulation of
damaged mitochondria as a result of imperfect autophagocytosis. Eur
J Biochem. 269:1996–2002. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Katz ML, Rice LM and Gao CL: Reversible
accumulation of lipofuscin-like inclusions in the retinal pigment
epithelium. Invest Ophthalmol Vis Sci. 40:175–181. 1999.PubMed/NCBI
|
|
89
|
Von Ruckmann A, Schmidt KG, Fitzke FW,
Bird AC and Jacobi KW: Dynamics of accumulation and degradation of
lipofuscin in retinal pigment epithelium in senile macular
degeneration. Klin Monbl Augenheilkd. 213:32–37. 1998.(In
German).
|
|
90
|
Kennedy CJ, Rakoczy PE and Constable IJ:
Lipofuscin of the retinal pigment epithelium: a review. Eye (Lond).
9:763–771. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Marmorstein AD, Marmorstein LY, Sakaguchi
H and Hollyfield JG: Spectral profiling of autofluorescence
associated with lipofuscin, Bruch’s membrane, and sub-RPE deposits
in normal and AMD eyes. Invest Ophthalmol Vis Sci. 43:2435–2441.
2002.PubMed/NCBI
|
|
92
|
Feher J, Kovacs I, Artico M, Cavallotti C,
Papale A and Balacco Gabrieli C: Mitochondrial aterations of
retinal pigment epithelium in age-related macular degeneration.
Neurobiol Aging. 27:983–993. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Furuno T, Kanno T, Arita K, Asami M,
Utsumi T, Doi Y, Inoue M and Utsumi K: Roles of long chain fatty
acids and carnitine in mitochondrial membrane permeability
transition. Biochem Pharmacol. 62:1037–1046. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Pepe S: Mitochondrial function in
ischaemia and reperfusion of the ageing heart. Clin Exp Pharmacol
Physiol. 27:745–750. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gunthorpe MJ and Szallasi A: Peripheral
TRPV1 receptors as targets for drug development: new molecules and
mechanisms. Curr Pharm Des. 14:32–41. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Lázár J, Szabó T, Marincsák R, Kovács L,
Blumberg PM and Bíró T: Sensitization of recombinant vanilloid
receptor-1 by various neurotrophic factors. Life Sci. 75:153–163.
2004.PubMed/NCBI
|
|
97
|
Aloe L, Tirassa P and Lambiase A: The
topical application of nerve growth factor as a pharmacological
tool for human corneal and skin ulcers. Pharmacol Res. 57:253–258.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Feher J, Kovacs I, Pacella E, Keresz S,
Spagnardi N and Balacco Gabrieli C: Pigment epithelium-derived
factor (PEDF) attenuated capsaicin-induced neurotrophic
keratouveitis. Invest Ophthalmol Vis Sci. 50:5173–5180. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Gao G, Li Y, Zhang D, Gee S, Crosson C and
Ma J: Unbalanced expression of VEGF and PEDF in ischemia-induced
retinal neovascularization. FEBS Lett. 489:270–276. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Jin KL, Mao XO and Greenberg DA: Vascular
endothelial growth factor: direct neuroprotective effect in in
vitro ischemia. Proc Natl Acad Sci USA. 97:10242–10247. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zachary I: Neuroprotective role of
vascular endothelial growth factor: signalling mechanisms,
biological function, and therapeutic potential. Neurosignals.
14:207–221. 2005. View Article : Google Scholar
|
|
102
|
Khaibullina AA, Rosenstein JM and Krum JM:
Vascular endothelial growth factor promotes neurite maturation in
primary CNS neuronal cultures. Brain Res Dev Brain Res. 148:59–68.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Feher J, Papale A, Mannino G, Gualdi G and
Balacco Gabrieli C: Mitotropic compounds for the treatment of
age-related macular degeneration. The metabolic approach and a
pilot study. Ophthalmologica. 217:351–357. 2003. View Article : Google Scholar : PubMed/NCBI
|