Introduction
Congenital heart disease (CHD) is the most common
form of birth defect in humans worldwide, affecting nearly 1% of
all live births, and is the leading cause of infant mortality from
developmental malformations (1).
According to specific anatomic or hemodynamic lesions, CHD is
clinically classified into at least 21 different types, of which
tetralogy of Fallot (TOF), a tetrad of right ventricular outflow
tract obstruction, over-riding of the aortic root, ventricular
septal defect, and right ventricular hypertrophy, is the most
prevalent type of cyanotic CHD, occurring in approximately 3 of
every 10,000 neonates alive, and accounts for roughly 7–10% of all
congenital cardiac abnormalities. Without surgical repair, 25% of
TOF patients with severe obstruction succumb to the disease within
the first year, 40% succumb to the disease by the age of 3, 70% by
the age of 10, and 95% by the age of 40 (1–3).
Various congenital cardiovascular anomalies, such as atrial septal
defect, ventricular septal defect, atrioventricular septal defect,
TOF, patent ductus arteriosus, double outlet right ventricle,
aortic stenosis, and transposition of great arteries, can occur
alone or together, leading to poor quality of life, cardiac
enlargement or hypertrophy, ventricular dysfunction or failure,
delayed fetal brain development, pulmonary hypertension,
Eisenmenger’s syndrome, arrhythmias, and even sudden cardiac death
in the absence of surgical or catheter-based corrections (4–10).
Despite the high prevalence and the important clinical
significance, the etiology responsible for CHD remains to be
identified in an overwhelming majority of patients (11–13).
It is generally understood that normal embryonic
heart development is a complex and dynamic process that requires
the orchestration of cardiac cell commitment, differentiation,
proliferation and migration, and abnormal cardiac morphogenesis
appears to be implicated in both environmental and genetic risk
factors, which disrupt the finely regulated biological
developmental process (11–13). Previous studies demonstrated that
an evolutionarily conserved network of transcription factors that
connect signaling pathways with genes for muscle growth,
patterning, and contractility, including GATA and NK families,
plays a pivotal role in early cardiogenesis (14–17), and a great number of germline
mutations in NKX2-5, GATA4 and GATA6 have been associated with CHD
(18–39). Nevertheless, the genetic
determinants underlying CHD remain largely unclear.
Emerging evidence suggests a novel genetic mosaic
mechanism for CHD. Somatic mutations have been identified in GATA4
and its molecular partners, NKX2-5 and TBX5, as well as the
transcription factor HAND1 and HEY2 in cardiac tissue derived from
hearts with CHD (40–48). GATA6 is another member of the GATA
family, and its expression and function overlap at least partially
with those of GATA4, NKX2-5 and TBX5 during cardiovascular genesis,
which makes it logical to hypothesize that somatic GATA6 mutations
are involved in the pathogenesis of TOF.
In the present study, in order to evaluate the
prevalence and spectrum of somatic GATA6 mutations in patients with
sporadic TOF and explore the mechanism by which mutated GATA6
predisposes to TOF, the entire coding region and splice junctions
of GATA6 was sequenced in patients as comparison to control
individuals, and the functional characteristics of the mutant GATA6
were assessed in comparison with its wild-type counterpart by using
a luciferase reporter assay system.
Materials and methods
Study subjects
A cohort of 52 unrelated patients with TOF, who
underwent cardiac surgery at Shanghai Renji Hospital in China
between January 2009 and December 2011, was recruited. Their age
ranged from 6 months to 7 years, with an average of 1.24 years at
the time of surgery. The patients were evaluated by skillful
cardiologists and the diagnosis was made by echocardiography and
confirmed by direct view during surgical procedure. The patients
with known chromosomal abnormalities or syndromic cardiovascular
defects, such as DiGeorge, Alagille, Noonan, Holt-Oram, Marfan and
Char syndrome, were excluded from the study. The sample size was
adequate to draw the conclusion that the absence of somatic
mutations is significant, according to the report by Draus et
al (42).
A total of 46 unrelated patients (25 males and 21
females) with rheumatic heart disease undergoing cardiac valve
replacement, and 200 unrelated healthy individuals (105 males and
95 females) randomly selected from those undergoing routine
physical examinations, were enrolled as controls. In terms of
individual medical history and echocardiographic record, the
control individuals had no apparent congenital cardiovascular
defects, except for subclinical cardiac aberrations such as
bicuspid aortic valve or patent foramen ovale.
The participants were Chinese Han people. The ethnic
origin of a participant was ascertained by a combination of
self-reported ethnicity and a personal questionnaire regarding
birthplace, language, religion and ancestry. The study protocol was
reviewed and approved by the local Institutional Ethics Committee
and written informed consent was obtained from all participants or
their guardians prior to the study.
Sample collection and storage
The cardiac muscle tissues from the right
ventricular outflow tracts of TOF patients were resected during the
routine cardiac surgery procedures. At the time of resection, the
discarded cardiac tissue sample was collected after cleaning the
blood stain by sterile normal saline and stored at −80°C.
Meanwhile, the patient’s discarded peripheral venous blood with
sodium citrate was collected (the blood samples were mostly used
for activated partial thromboplastin time and prothrombin time
tests before surgery). The discarded cardiac specimens from the
cardiac valves and matched blood samples of the patients undergoing
cardiac valve replacement due to rheumatic valvular disease, and
the peripheral venous blood samples of healthy individuals were
prepared as controls.
DNA extraction
The somatic DNA was extracted from freshly frozen
tissues using QIAamp DNA FFPE Tissue kit (Qiagen GmbH, Hilden,
Germany) according to the manufacturer’s instructions. The genomic
DNA was extracted from peripheral blood lymphocytes with Wizard
Genomic DNA purification kit (Promega, Madison, WI, USA).
Genetic studies
The primers for amplification of the coding exons
and flanking splicing sites of the GATA6 gene were designed
as previously described (39).
Polymerase chain reaction (PCR) was performed in an automated
Perkin-Elmer 9700 Thermal Cycler (Applied Biosystems, Foster City,
CA, USA). The reaction mixture for PCR used in the present series
of experiments consisted of 2 μl of genomic DNA (50–100 ng/μl), 2.5
μl of 10× Taq Buffer, 5 μl of 5× Q Solution, 2 μl of dNTP Mixture
(2.5 mM each), 0.5 μl of each primer (20 mM each), 0.25 μl (1.25 U)
of HotStar TaqDNA polymerase (Qiagen), and 12.25 μl of deionized
H2O, with a final volume of 25 μl. PCR was carried out
under the following conditions: pre-denaturation at 95°C for 15
min, followed by 35 cycles of denaturation at 95°C for 1 min,
annealing at 62°C for 30 sec, and extension at 72°C for 1 min, and
final extension at 72°C for 10 min. To avoid potential carry-over
contamination of the PCR mixture, all reactions were performed
under stringent conditions as recommended by Kwok and Higuchi
(49). Amplified products were
analyzed on 1% agarose gels stained with ethidium bromide and
purified with QIAquick Gel Extraction kit (Qiagen). Both strands of
each PCR product were sequenced with a BigDye®
Terminator v3.1 Cycle Sequencing kit under an ABI PRISM 3130XL DNA
Analyzer (were from Applied Biosystems). The sequencing primers
were the same as previously designed for specific region
amplification. The DNA sequences were analyzed with the DNA
Sequencing Analysis Software v5.1 (Applied Biosystems). The
sequence variation was validated by re-sequencing an independent
PCR-generated amplicon from the same subject and met the standard
quality control thresholds with a call rate >99%. Additionally,
an identified GATA6 sequence variation was searched in the single
nucleotide polymorphism (SNP) database of the National Center for
Biotechnology Information to confirm the novelty (http://www.ncbi.nlm.nih.gov/SNP).
Alignment of GATA6 protein sequences
across species
Multiple GATA6 protein sequences across various
species were aligned using the online program of Muscle, version
3.6 (http://www.ncbi.nlm.nih.gov).
Prediction of the causative potential of
a GATA6 sequence variation
The pathogenic potential of a GATA6 sequence
variation was predicted by MutationTaster (an online program at
http://www.mutationtaster.org), which
automatically gave a probability for the variation to be either a
disease-causing mutation or a benign polymorphism. Notably, the
P-value used here is the probability of the correct prediction
rather than the probability of error as used in t-test statistics
(i.e., a value close to 1 indicates a high ‘security’ of the
prediction).
Plasmids and site-directed
mutagenesis
The recombinant expression plasmid pcDNA3-hGATA6 was
kindly provided by Dr Angela Edwards-Ghatnekar, from the Division
of Rheumatology and Immunology, Medical University of South
Carolina, Charleston, SC, USA. The atrial natriuretic factor
(ANF)-luciferase reporter gene, which contains the 2600-bp
5′-flanking region of the ANF gene, i.e., ANF(-2600)-Luc,
was kindly provided by Dr Ichiro Shiojima, from the Department of
Cardiovascular Science and Medicine, Chiba University Graduate
School of Medicine, Chuo-ku, Chiba, Japan. The identified mutation
was introduced into the wild-type GATA6 using a QuickChange
II XL Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA)
with a complementary pair of primers. The mutant was sequenced to
confirm the desired mutation and to exclude any other sequence
variations.
Reporter gene assay
HEK-293 cells were cultured in Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal calf serum. The
ANF(-2600)-Luc reporter vector and an internal control reporter
plasmid pGL4.75 (hRluc/CMV, Promega) were used in transient
transfection assays to evaluate the transcriptional activation
function of the GATA6 mutants. HEK-293 cells were
transfected with 0.4 μg of wild-type or mutant pcDNA3-hGATA6
expression vector, 0.4 μg of ANF(-2600)-Luc reporter construct, and
0.04 μg of pGL4.75 control reporter vector using
Lipofectamine® 2000 Transfection reagent (Invitrogen,
Carlsbad, CA, USA). For co-transfection experiments, 0.2 μg of
wild-type pcDNA3-hGATA6, 0.2 μg of empty pcDNA3 plasmid or 0.2 μg
of mutant pcDNA3-hGATA6, 0.4 μg of ANF(-2600)-Luc, and 0.04 μg of
pGL4.75 were used. Firefly luciferase and Renilla luciferase
activities were measured with the Dual-Glo luciferase assay system
(Promega) 48 h after transfection. The activity of the ANF promoter
was presented as fold activation of Firefly luciferase relative to
Renilla luciferase. Three independent experiments were performed at
minimum for wild-type and mutant GATA6.
Statistical analysis
Data are expressed as the means ± SD. Continuous
variables were examined for normality of distribution and the
Student’s unpaired t-test was used for the comparison of numeric
variables between two groups. A comparison of the categorical
variables between two groups was conducted using Pearson’s
χ2 test or Fisher’s exact test when appropriate. A
two-tailed P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Baseline characteristics of the study
population
A cohort of 52 unrelated Han-nationality patients
with sporadic TOF, who underwent cardiac surgery, was enrolled and
clinically evaluated as comparison to 46 unrelated Han-race
patients with rheumatic heart disease undergoing cardiac valve
replacement and 200 ethnically matched, unrelated healthy
individuals used as controls. The subjects had neither positive
family history of CHD nor established environmental risk factors
for CHD, such as maternal illness and drug use in the first
trimester of pregnancy, parental smoking, and chronic exposure to
toxicants and ionizing radiation. The baseline clinical
characteristics of the study population are summarized in Table I.
 | Table IClinical characteristics of the 52
unrelated patients with sporadic tetralogy of Fallot. |
Table I
Clinical characteristics of the 52
unrelated patients with sporadic tetralogy of Fallot.
| Parameter | No. or mean
value | Percentage or
range |
|---|
| Male | 28 | 54 |
| Age at the initial
diagnosis of tetralogy of Fallot (year) | 0.65 | 0–3 |
| Age at the time of
surgery (year) | 1.25 | 0.5–7 |
| Positive family
history of tetralogy of Fallot | 0 | 0 |
| Distribution of
various types of tetralogy of Fallot |
| Isolated tetralogy
of Fallot | 33 | 63 |
| Tetralogy of
Fallot and bicuspid pulmonary valve | 7 | 13 |
| Tetralogy of
Fallot and stenosis of left pulmonary artery | 5 | 10 |
| Tetralogy of
Fallot and right-sided aortic arch | 2 | 4 |
| Tetralogy of
Fallot and atrial septal defect | 1 | 2 |
| Tetralogy of
Fallot and atrioventricular septal defect | 1 | 2 |
| Tetralogy of
Fallot and at least two other anatomical defects | 3 | 6 |
| Incidence of
arrhythmias |
| Atrioventricular
block | 4 | 8 |
| Atrial
fibrillation | 2 | 4 |
| Treatment |
| Surgical
repair | 52 | 100 |
Source of samples
Peripheral venous blood samples were available for
all 298 participants. The malformed myocardial tissue samples were
obtained from 52 unrelated patients with sporadic TOF who underwent
surgical resection of the right ventricular outflow musculature for
relieving the stenosis. Generally, the cardiac muscle fragment
obtained was ~5×5 mm in size. In addition, the cardiac valvular
tissue samples used as controls were obtained from surgical
discards of 46 unrelated patients undergoing cardiac valve
replacement.
GATA6 mutations
Genomic DNA from the malformed cardiac tissues of 52
TOF patients, the cardiac valvular tissues of 46 patients with
rheumatic heart disease, and the peripheral blood lymphocytes of
the 298 participants, were screened for GATA6 mutations. As a
result, 2 heterozygous mutations in GATA6 were identified in
the fresh pathological myocardial tissues of 2 TOF patients,
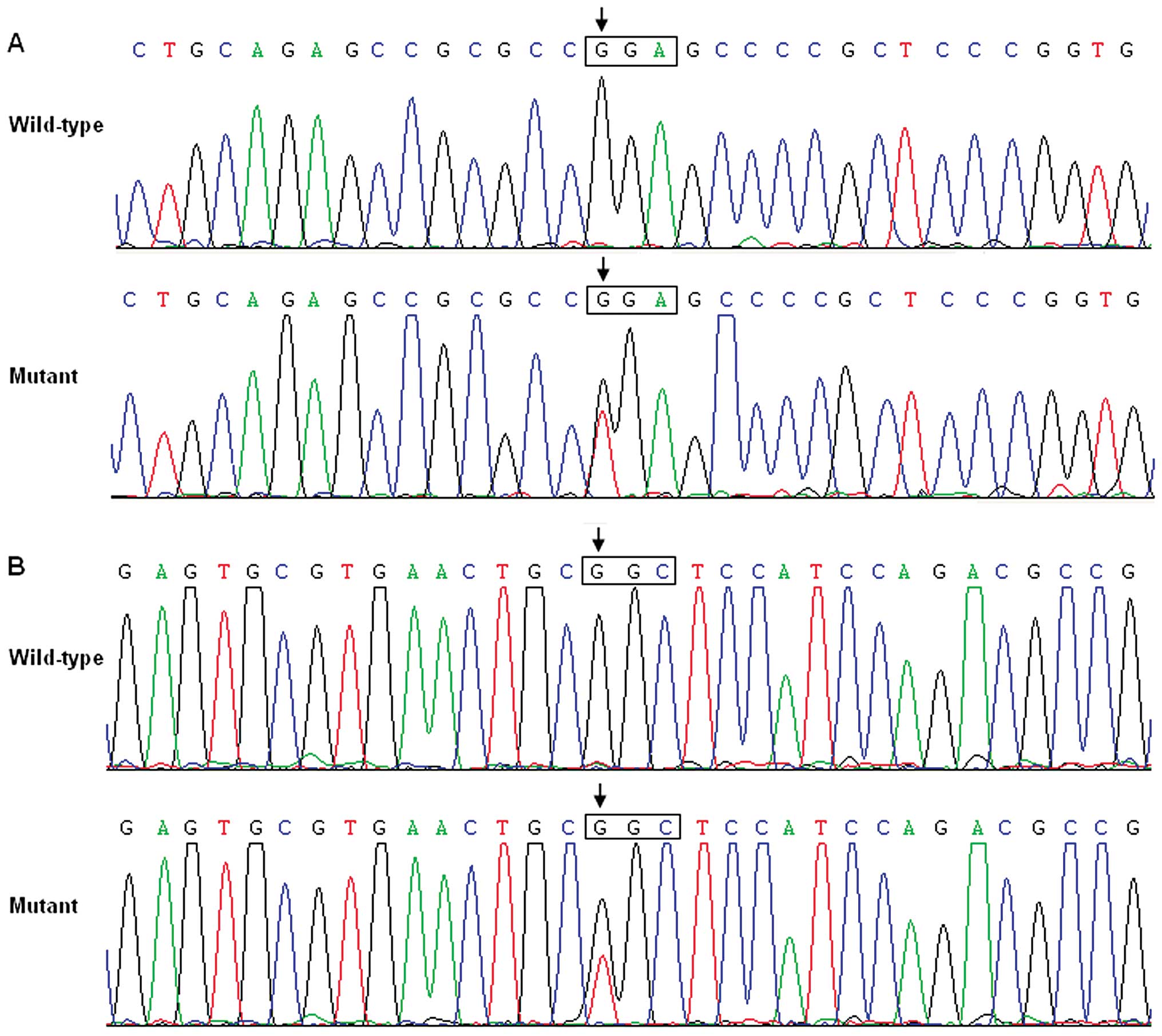
respectively, with a mutational prevalence of ~3.85%. Specifically,
a substitution of thymine for guanine in the first nucleotide of
codon 367 of the GATA6 gene (c.1099G>T), resulting in a
truncated protein with only N-terminal 366 amino acids left
(p.G367X), was identified in the cardiac tissue of a 1-year-old
male TOF patient. A replacement of guanine by thymine at coding
nucleotide 1180 (c.1180G>T), predicting the transition of
glycine to cysteine at amino acid position 394 (p.G394C), was
identified in the cardiac tissue of a 2-year-old female TOF
patient. The sequence chromatograms showing the detected
heterozygous GATA6 mutations as comparison to corresponding
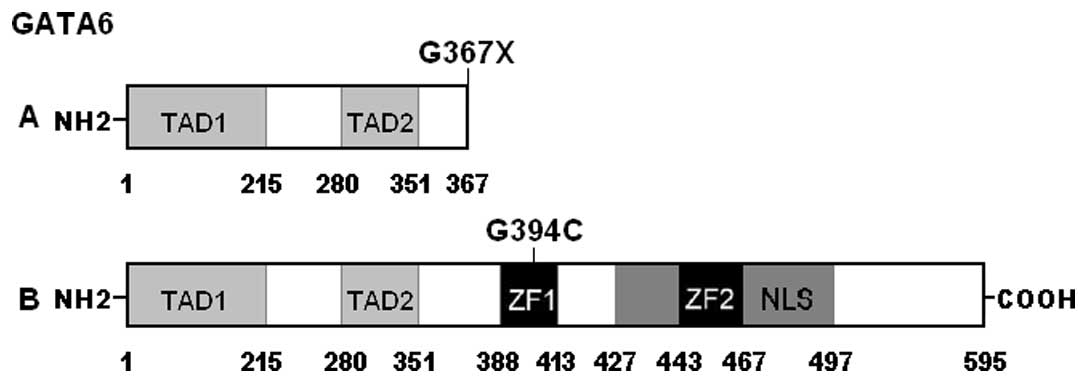
control sequences are depicted in Fig. 1. A schematic diagram of GATA6
showing the structural domains and the locations of the identified
mutations is presented in Fig. 2.
The 2 mutations were neither observed in the cardiac valvular
tissues of 46 patients with rheumatic heart disease nor found in
the peripheral blood samples of the 298 participants. Neither of
the 2 mutations was reported in the SNP database at the National
Center for Biotechnology Information, which was consulted again on
August 20, 2012. Additionally, during a 24-h period of ambulatory
electrocardiographic monitoring, no atrial fibrillation was
observed in these 2 mutation carriers.
Alignment of multiple GATA6 protein
sequences
A cross-species alignment of multiple GATA6 protein
sequences demonstrated that the affected amino acid p.G394 was
completely conserved evolutionarily and the affected amino acid
p.G367 was relatively conserved evolutionarily (Fig. 3). However, the amino acids deleted
by the p.G367× mutation constitute functionally important
structural domains, including 2 zinc fingers and 1 nuclear
localization signal (Fig. 2).
Causative potential of GATA6
mutations
The GATA6 mutations of c.1099G>T and
c.1180G>T were both automatically predicted to be
disease-causing, with P-values of 1.000000 and 0.999997,
respectively. No SNPs in the altered regions were found in the
MutationTaster database.
Transcriptional activity of the GATA6
mutants
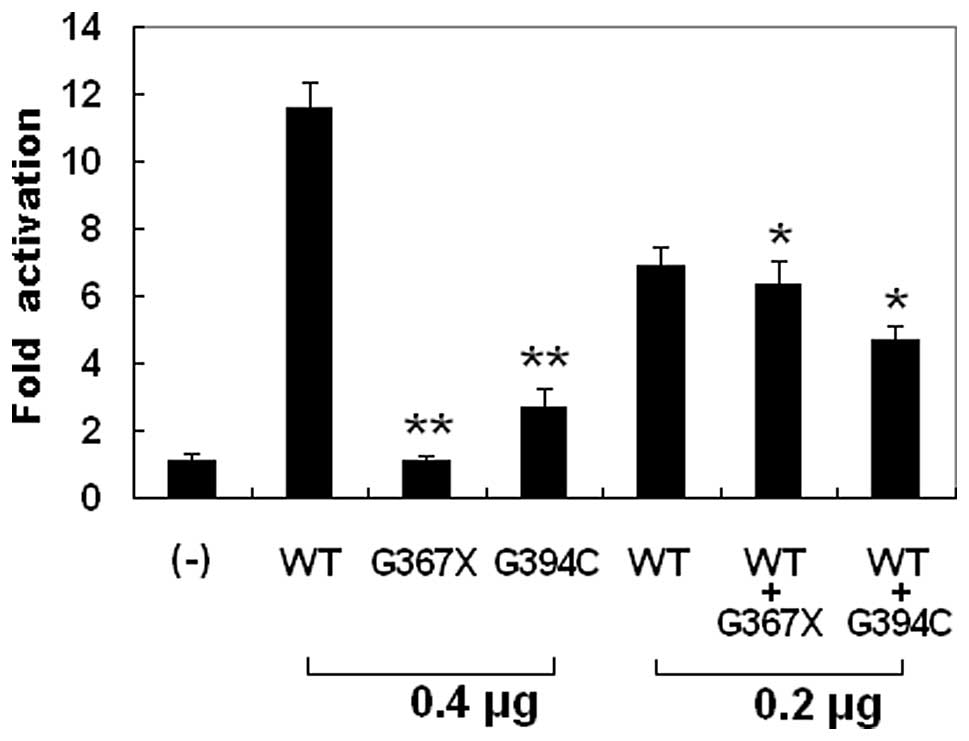
The wild-type GATA6, the G367X-mutant, and the
G394C-mutant GATA6 activated the ANF promoter by ~12-, 1- and
3-fold, respectively. When wild-type GATA6 was co-expressed with
the same amount of G367X-mutant or G394C-mutant GATA6, the induced
activation of the ANF promoter was ~6- or 5-fold, respectively.
These results indicate that both GATA6 mutants are associated with
significantly reduced activation activity compared with their
wild-type counterpart (Fig.
4).
Discussion
Somatic cell is defined as any cell, other than a
germ cell, which is involved in the formation of the body,
differentiating into the various tissues, organs. Mutations derived
from somatic cells can occur in people, but are not transmitted to
offspring (50). Diseases
associated or causing somatic mutations can be identified by
exploring the genetic substance from diseased tissue cells as
comparison to that from healthy tissue cells. Somatic mutations can
not be detected by genetic analysis of lymphocytic DNA alone, and
mosaicism may reduce the likelihood of detection in the affected
tissue. Therefore, mutations in cardiac tissues may be absent or
sporadic in blood lymphocytes of the same person (40). In the present study, 2 novel
heterozygous mutations of GATA6 (p.G367× and p.G394C) were
identified in the malformed heart tissues of 2 out of 52 patients
with TOF. The mutant alleles were absent in the cardiac tissues of
46 patients with rheumatic heart disease and in the peripheral
blood samples of the 298 participants, including 200 ethnically
matched healthy individuals. A cross-species alignment of multiple
GATA6 protein sequences showed that the altered amino acids were
highly conserved evolutionarily. The 2 variants were both predicted
to be pathogenic, and the functional experiments substantiated that
the mutant GATA6 proteins were associated with significantly
decreased or absent transcriptional activity. Therefore, somatic
GATA6 mutations may contribute to the pathogenesis of TOF in the 2
mutation-harboring patients.
The GATA6 gene, mapped to human chromosome
18q11.1-q11.2 by fluorescence in situ hybridization (a
powerful cytogenetic technique used to detect and localize the
specific DNA sequences on chromosomes), encodes a zinc
finger-containing protein with 595 amino acids (51). By alignment of GATA6 with GATA4,
the structural domains of GATA6 comprise 2 transcriptional
activation domains (TAD1, amino acids 1–215; TAD2, amino acids
280–351), 2 adjacent zinc fingers (ZF1, amino acids 388–413; ZF2,
amino acids 443–467), and 1 nuclear localization signal (NLS, amino
acids 427–497). The two TADs are both essential for the
transcriptional activity of GATA6. The C-terminal ZF1 is required
for DNA sequence recognition and binding to the consensus motif,
while the N-terminal ZF2 is responsible for sequence specificity
and stability of protein-DNA binding. The NLS is associated with
the sub-cellular trafficking and distribution of GATA6 (39). The GATA6 mutation p.G367×
eliminates the functionally pivotal domains of ZF1, ZF2 and NLS as
well as the carboxyl terminus, and may thus be expected to nullify
the function of GATA6. Another GATA6 mutation p.G394C identified in
this study is located at ZF1, hence it is likely to induce loss of
transcriptional activity of GATA6 by interfering with the specific
binding of GATA6 to target gene promoters.
GATA6 is an upstream transcriptional regulator of
multiple genes expressed during embryogenesis and cardiac
morphogenesis, including the genes that encode atrial natriuretic
factor (ANF), brain natriuretic peptide, α-myosin heavy chain, β
myosin heavy chain, and gap junction protein connexin-40 (52). Therefore, the functional
characteristics of the GATA6 mutations may be delineated by
assessing the transcriptional activity of the ANF promoter
in cultured cells. In this study, the functional effect of the
novel GATA6 mutations identified in TOF patients was characterized
by transcriptional activation assay, and the results showed a
significantly decreased transcriptional activity on a downstream
gene. These data suggest that dysfunctional GATA6 caused by
loss-of-function mutations is potentially an alternative pathogenic
mechanism implicated in TOF.
It has previously been reported that germline
mutations of the GATA6 gene are responsible for CHD,
including TOF. Kodo et al scanned GATA6 in 21
unrelated patients with persistent truncus arteriosus, and
identified 2 heterozygous mutations of p.E486GfsX10 and p.N466H in
2 patients, respectively, with a prevalence of 9.52%. Functional
analysis demonstrated both GATA6 mutations led to defects in
nuclear localization and transcription activity (36). Lin et al (37) performed sequence analysis of
GATA6 in 270 unrelated patients with CHD, and detected a
novel heterozygous mutation of p.S184N in 3 unrelated patients,
including 1 with TOF and 2 with atrial septal defect, with a
prevalence of 1.11%. Biochemical assays unveiled that the mutation
had significantly decreased transcriptional activity. Maitra et
al (38) genotyped
GATA6 in 310 unrelated patients with CHD and 2 heterozygous
mutations of p.A178V and p.L198V were identified in 2 patients,
respectively, with a prevalence of 0.65%. The p.L198V carrier was
affected with TOF whereas the p.A178V carrier was affected with
atrioventricular septal defect, hypoplastic left ventricle, and
ventricular septal defect. Functional evaluation revealed the
p.A178V mutation had increased transcriptional activity while the
p.L198V mutation had no effect on the transcriptional activity.
Zheng et al (39) made
mutational analysis of GATA6 in 130 unrelated patients with
ventricular septal defect, and found a loss-of-function mutation
p.G220S in a patient, with a mutation prevalence of 0.77%. To date,
6 germline GATA6 mutations have been identified in 8 of 731 index
patients with CHD, with a total mutation prevalence of 1.09%
(36–39). However, in the current study, no
germline GATA6 mutations were discovered except that 2 somatic
GATA6 mutations were found, which highlights a genetic mosaic basis
for TOF in a subset of cases.
Association of functionally compromised GATA6 with
enhanced vulnerability to CHD has been revealed in animals.
Homozygous GATA6 knockout mice die shortly after embryonic
implantation due to defects in visceral endoderm function and
extra-embryonic development (53,54). Although the mice heterozygous for
either GATA4 or GATA6 deletion are viable without
overt cardiovascular defects, the mice that are compound
heterozygous for both GATA4 and GATA6 targeted
disruptions die by E13.5 with 100% penetrance, exhibiting a
phenotypic spectrum of cardiovascular defects, including
ventricular septal defect, persistent truncus arteriosus,
myocardial hypoplasia, reduced myocardial proliferation, and
impaired differentiation of vascular smooth muscle cells (53–55). Similarly, compound null of a
GATA5 and a GATA6 allele also gives rise to double
outlet right ventricle and ventricular septal defect in mice
(56). These results from
experimental animals demonstrate an exquisite sensitivity of the
developing cardiovascular system to the levels of GATA4, GATA5 and
GATA6, and indicate that these GATA factors act synergistically to
regulate downstream target genes.
Atrial fibrillation has been documented in some CHD
patients harboring the germline mutations of GATA4, GATA5 and GATA6
(57–64), which suggests that atrial
fibrillation may share a common genetic origin with CHD. However,
in the current investigation, no atrial fibrillation was documented
in the 2 somatic GATA6 mutation carriers, which may be explained by
insufficient electrocardiographic monitoring duration of only 24 h
for paroxysmal AF, different genetic background, distinct
mutational source, delayed or incomplete penetrance, epigenetic
modifiers, or environmental factors (61).
In conclusion, this is the first report on the
association of somatic GATA6 loss-of-function mutation with
increased susceptibility to TOF, which provides novel insight into
the molecular pathogenesis of CHD, and suggests the potential
implications for the early diagnosis and personalized treatment of
CHD.
Acknowledgements
The authors thank the participants for their
devotion to the study. This study was supported in part by grants
from the National Natural Science Foundation of China (81270161,
81070153 and 30570768), the Personnel Development Foundation of
Shanghai, China (2010019), the Natural Science Foundation of
Shanghai, China (10ZR1428000), and the Key Program of Basic
Research of Shanghai, China (10JC1414002).
References
|
1
|
Roger VL, Go AS, Lloyd-Jones DM, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS,
Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela
BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM,
Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino
ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N,
Turan TN, Virani SS, Wong ND, Woo D and Turner MB; American Heart
Association Statistics Committee and Stroke Statistics
Subcommittee. Heart disease and stroke statistics - 2012 update: a
report from the American Heart Association. Circulation.
125:e2–e220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van der Linde D, Konings EE, Slager MA,
Witsenburg M, Helbing WA, Takkenberg JJ and Roos-Hesselink JW:
Birth prevalence of congenital heart disease worldwide: a
systematic review and meta-analysis. J Am Coll Cardiol.
58:2241–2247. 2011.PubMed/NCBI
|
|
3
|
Starr JP: Tetralogy of Fallot: yesterday
and today. World J Surg. 34:658–668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Müller J, Hess J and Hager A: Exercise
performance and quality of life is more impaired in Eisenmenger
syndrome than in complex cyanotic congenital heart disease with
pulmonary stenosis. Int J Cardiol. 150:177–181. 2011.PubMed/NCBI
|
|
5
|
Teixeira FM, Coelho RM, Proença C, Silva
AM, Vieira D, Vaz C, Moura C, Viana V, Areias JC and Areias ME:
Quality of life experienced by adolescents and young adults with
congenital heart disease. Pediatr Cardiol. 32:1132–1138. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Müller J, Hess J and Hager A: Minor
symptoms of depression in patients with congenital heart disease
have a larger impact on quality of life than limited exercise
capacity. Int J Cardiol. 154:265–269. 2012.PubMed/NCBI
|
|
7
|
Shedeed SA and Elfaytouri E: Brain
maturity and brain injury in newborns with cyanotic congenital
heart disease. Pediatr Cardiol. 32:47–54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perry JC: Sudden cardiac death and
malignant arrhythmias: the scope of the problem in adult congenital
heart patients. Pediatr Cardiol. 33:484–490. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Silka MJ and Bar-Cohen Y: A contemporary
assessment of the risk for sudden cardiac death in patients with
congenital heart disease. Pediatr Cardiol. 33:452–460. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cheng HH, Almodovar MC, Laussen PC, Wypij
D, Polito A, Brown DW, Emani SM, Pigula FA, Allan CK and Costello
JM: Outcomes and risk factors for mortality in premature neonates
with critical congenital heart disease. Pediatr Cardiol.
32:1139–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bruneau BG: The developmental genetics of
congenital heart disease. Nature. 451:943–948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cecchetto A, Rampazzo A, Angelini A,
Bianco LD, Padalino M, Stellin G and Daliento L: From molecular
mechanisms of cardiac development to genetic substrate of
congenital heart diseases. Future Cardiol. 6:373–393. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Benson DW: Genetic origins of pediatric
heart disease. Pediatr Cardiol. 31:422–429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Olson EN: Gene regulatory networks in the
evolution and development of the heart. Science. 313:1922–1927.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu DL, Chen FK, Liu YQ, Sheng YH, Yang R,
Kong XQ, Cao KJ, Gu HT and Qian LM: GATA-4 promotes the
differentiation of P19 cells into cardiac myocytes. Int J Mol Med.
26:365–372. 2010.PubMed/NCBI
|
|
16
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bartlett H, Veenstra GJ and Weeks DL:
Examining the cardiac NK-2 genes in early heart development.
Pediatr Cardiol. 31:335–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schott JJ, Benson DW, Basson CT, Pease W,
Silberbach GM, Moak JP, Maron BJ, Seidman CE and Seidman JG:
Congenital heart disease caused by mutations in the transcription
factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Xin YF, Liu XY, Liu ZM, Wang XZ
and Yang YQ: A novel NKX2-5 mutation in familial ventricular septal
defect. Int J Mol Med. 27:369–375. 2011.PubMed/NCBI
|
|
20
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC and Srivastava D:
GATA4 mutations cause human congenital heart defects and reveal an
interaction with TBX5. Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okubo A, Miyoshi O, Baba K, Takagi M,
Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N
and Matsumoto N: A novel GATA4 mutation completely segregated with
atrial septal defect in a large Japanese family. J Med Genet.
41:e972004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sarkozy A, Conti E, Neri C, D’Agostino R,
Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A and
Dallapiccola B: Spectrum of atrial septal defects associated with
mutations of NKX2.5 and GATA4 transcription factors. J Med Genet.
42:e162005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hirayama-Yamada K, Kamisago M, Akimoto K,
Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A,
Nakazawa M and Matsuoka R: Phenotypes with GATA4 or NKX2.5
mutations in familial atrial septal defect. Am J Med Genet A.
135:47–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nemer G, Fadlalah F, Usta J, Nemer M,
Dbaibo G, Obeid M and Bitar F: A novel mutation in the GATA4 gene
in patients with tetralogy of Fallot. Hum Mutat. 27:293–294. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tomita-Mitchell A, Maslen CL, Morris CD,
Garg V and Goldmuntz E: GATA4 sequence variants in patients with
congenital heart disease. J Med Genet. 44:779–783. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rajagopal SK, Ma Q, Obler D, Shen J,
Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V,
Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB and Pu
WT: Spectrum of heart disease associated with murine and human
GATA4 mutation. J Mol Cell Cardiol. 43:677–685. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang W, Li X, Shen A, Jiao W, Guan X and
Li Z: GATA4 mutations in 486 Chinese patients with congenital heart
disease. Eur J Med Genet. 51:527–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hamanoue H, Rahayuningsih SE, Hirahara Y,
Itoh J, Yokoyama U, Mizuguchi T, Saitsu H, Miyake N, Hirahara F and
Matsumoto N: Genetic screening of 104 patients with congenitally
malformed hearts revealed a fresh mutation of GATA4 in those with
atrial septal defects. Cardiol Young. 19:482–485. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen MW, Pang YS, Guo Y, Pan JH, Liu BL,
Shen J and Liu TW: GATA4 mutations in Chinese patients with
congenital cardiac septal defects. Pediatr Cardiol. 31:85–89. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Y, Mao J, Sun Y, Zhang Q, Cheng HB,
Yan WH, Choy KW and Li H: A novel mutation of GATA4 in a familial
atrial septal defect. Clin Chim Acta. 411:1741–1745. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Butler TL, Esposito G, Blue GM, Cole AD,
Costa MW, Waddell LB, Walizada G, Sholler GF, Kirk EP, Feneley M,
Harvey RP and Winlaw DS: GATA4 mutations in 357 unrelated patients
with congenital heart malformation. Genet Test Mol Biomarkers.
14:797–802. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu XY, Wang J, Zheng JH, Bai K, Liu ZM,
Wang XZ, Liu X, Fang WY and Yang YQ: Involvement of a novel GATA4
mutation in atrial septal defects. Int J Mol Med. 28:17–23.
2011.PubMed/NCBI
|
|
33
|
Wang J, Fang M, Liu XY, Xin YF, Liu ZM,
Chen XZ, Wang XZ, Fang WY, Liu XS and Yang YQ: A novel GATA4
mutation responsible for congenital ventricular septal defects. Int
J Mol Med. 28:557–564. 2011.PubMed/NCBI
|
|
34
|
Yang YQ, Li L, Wang J, Liu XY, Chen XZ,
Zhang W, Wang XZ, Jiang JQ, Liu XS and Fang WY: A novel GATA4
loss-of-function mutation associated with congenital ventricular
septal defect. Pediatr Cardiol. 33:539–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang YQ, Wang J, Liu XY, Chen XZ, Zhang W,
Wang XZ, Liu XS and Fang WY: Novel GATA4 mutations in patients with
congenital ventricular septal defects. Med Sci Monit.
18:CR344–CR350. 2012.PubMed/NCBI
|
|
36
|
Kodo K, Nishizawa T, Furutani M, Arai S,
Yamamura E, Joo K, Takahashi T, Matsuoka RS and Yamagishi H: GATA6
mutations cause human cardiac outflow tract defects by disrupting
semaphorin-plexin signaling. Proc Natl Acad Sci USA.
106:13933–13938. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lin X, Huo Z, Liu X, Zhang Y, Li L, Zhao
H, Yan B, Liu Y, Yang YS and Chen YH: A novel GATA6 mutation in
patients with tetralogy of Fallot or atrial septal defect. J Hum
Genet. 55:662–667. 2010. View Article : Google Scholar
|
|
38
|
Maitra M, Koenig SN, Srivastava DS and
Garg V: Identification of GATA6 sequence variants in patients with
congenital heart defects. Pediatr Res. 68:281–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ
and Liu XY: A novel GATA6 mutation associated with congenital
ventricular septal defect. Int J Mol Med. 29:1065–1071.
2012.PubMed/NCBI
|
|
40
|
Reamon-Buettner SM and Borlak J: Somatic
NKX2-5 mutations as a novel mechanism of disease in complex
congenital heart disease. J Med Genet. 41:684–690. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reamon-Buettner SM, Hecker H,
Spanel-Borowski K, Craatz S, Kuenzel ES and Borlak J: Novel NKX2-5
mutations in diseased heart tissues of patients with cardiac
malformations. Am J Pathol. 164:2117–2125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Draus JM Jr, Hauck MA, Goetsch M, Austin
EH III, Tomita-Mitchell AS and Mitchell ME: Investigation of
somatic NKX2-5 mutations in congenital heart disease. J Med Genet.
46:115–122. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Reamon-Buettner SM and Borlak J: GATA4
zinc finger mutations as a molecular rationale for septation
defects of the human heart. J Med Genet. 42:e322005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Reamon-Buettner SM, Cho SH and Borlak J:
Mutations in the 39-untranslated region of GATA4 as molecular
hotspots for congenital heart disease (CHD). BMC Med Genet.
8:382007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Salazar M, Consoli F, Villegas V, Caicedo
V, Maddaloni V, Daniele P, Caianiello G, Pachón S, Nuñez F,
Limongelli G, Pacileo G, Marino B, Bernal JE, De Luca AS and
Dallapiccola B: Search of somatic GATA4 and NKX2.5 gene mutations
in sporadic septal heart defects. Eur J Med Genet. 54:306–309.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Reamon-Buettner SM and Borlak J: TBX5
mutations in non-Holt-Oram syndrome (HOS) malformed hearts. Hum
Mutat. 24:1042004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang J, Lu Y, Chen H, Yin M, Yu TS and Fu
Q: Investigation of somatic NKX2-5, GATA4 and HAND1 mutations in
patients with tetralogy of Fallot. Pathology. 43:322–326. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Reamon-Buettner SM and Borlak J: HEY2
mutations in malformed hearts. Hum Mutat. 27:1182006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kwok S and Higuchi R: Avoiding false
positives with PCR. Nature. 339:237–238. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Feero WG, Guttmacher AE and Collins FS:
Genomic medicine - an updated primer. N Engl J Med. 362:2001–2011.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Suzuki E, Evans T, Lowry J, Truong L, Bell
DW, Testa JR and Walsh K: The human GATA-6 gene: structure,
chromosomal location, and regulation of expression by
tissue-specific and mitogen-responsive signals. Genomics.
38:283–290. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rémond MC, Iaffaldano G, O’Quinn MP,
Mezentseva NV, Garcia V, Harris BS, Gourdie RG, Eisenberg CA and
Eisenberg LM: GATA6 reporter gene reveals myocardial phenotypic
heterogeneity that is related to variations in gap junction
coupling. Am J Physiol Heart Circ Physiol. 301:H1952–H1964.
2011.PubMed/NCBI
|
|
53
|
Zhao R, Watt AJ, Battle MA, Li J, Bondow
BJ and Duncan SA: Loss of both GATA4 and GATA6 blocks cardiac
myocyte differentiation and results in acardia in mice. Dev Biol.
317:614–619. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Morrisey EE, Tang Z, Sigrist K, Lu MM,
Jiang F, Ip HS and Parmacek MS: GATA6 regulates HNF4 and is
required for differentiation of visceral endoderm in the mouse
embryo. Genes Dev. 12:3579–3590. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xin M, Davis CA, Molkentin JD, Lien CL,
Duncan SA, Richardson JA and Olson EN: A threshold of GATA4 and
GATA6 expression is required for cardiovascular development. Proc
Natl Acad Sci USA. 103:11189–11194. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Laforest B and Nemer M: GATA5 interacts
with GATA4 and GATA6 in outflow tract development. Dev Biol.
358:368–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
58
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang YQ, Wang MY, Zhang XL, Tan HW, Shi
HF, Jiang WF, Wang XH, Fang WY and Liu X: GATA4 loss-of-function
mutations in familial atrial fibrillation. Clin Chim Acta.
412:1825–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Posch MG, Boldt LH, Polotzki M, Richter S,
Rolf S, Perrot A, Dietz R, Ozcelik C and Haverkamp W: Mutations in
the cardiac transcription factor GATA4 in patients with lone atrial
fibrillation. Eur J Med Genet. 53:201–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|