Introduction
An elevated level of oxidized low-density
lipoprotein (ox-LDL) is one of the major risk factors for
atherosclerosis and plays a major role in vascular endothelial
dysfunction. The vascular endothelium acts as a selective barrier,
regulating the exchange of macromolecules between blood and the
underlying tissues (1). The
elevated permeability of the endothelium is the first event in the
cascade of processes leading to atherosclerotic lesion formation,
which leads to lipid infiltration and accumulation within the
arterial wall. Three potential pathways regulate endothelial
permeability: namely the transcellular pathway, by which blood
components pass through the cell; the paracellular pathway, by
which the components cross the endothelial barrier through
intercellular cell-cell junctions; and the leaky junction pathway,
by which components across the endothelium through the gap due to
cells undergoing mitosis or apoptosis (2).
Cathepsins belong to the papain family of cysteine
proteases, which degrade elastin and collagen. Cathepsin L (CATL)
is one of the potent mammalian collagenases and elastases and was
found to be upregulated in the arteries of apolipoprotein E-null
mice fed a Western diet (3).
Inhibition of CATL was able to reduce expression of the adhesion
molecule αVβ3 integrin, disrupting secretion of the proangiogenic
factors fibroblast growth factor (FGF) and vascular endothelial
growth factor (VEGF) (4).
Recently, Mahmoud et al (5) discovered that increased levels of
CATL mRNA and protein induced by peroxisome proliferator-activated
receptor γ (PPARγ) activation, stimulated apoptosis and inhibited
autophagy in human monocyte-derived macrophages. These observations
revealed that CATL contributes to atherosclerosis not simply its
elastinolytic and collagenolytic activities.
In the present study, we investigated the role of
CATL in ox-LDL-induced early atherosclerotic events and the
potential mechanisms. As several studies have indicated that CATL
activation exerts proatherogenic effects, we hypothesized that CATL
activation decreases EC autophagy. Surprisingly, we found that a
CATL inhibitor concentration-dependently decreased EC
autophagy.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), Trypsin,
fluorescein isothiocyanate (FITC)-dextran and bovine serum albumin
(BSA) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
ReverAid™ first-strand cDNA synthesis kit was purchased from
Invitrogen Life Technologies (Carlsbad, CA, USA). CATL fluorogenic
activity kit was purchased from Merck KGaA (Darmstadt, Germany).
CATL inhibitor was purchased from Santa Cruz Biotechnology Inc.
(Santa Cruz, CA, USA). Antibodies for CATL, beclin 1, LC3,
caspase-3, Bcl-2, VE-cadherin, and HRP-labeled and alkaline
phosphatase-labeled goat anti-rabbit IgG were obtained from
Proteintech Biotechnology (Chicago, IL, USA). Ox-LDL was obtained
from Zhongshan University (Guangzhou, China).
Cell culture
Human umbilical vein endothelial cells (ECs) were
purchased from Central South University and cultured in DMEM
supplemented with 10% fetal bovine serum (FBS; Invitrogen), 100
U/ml penicillin, and 100 μg/ml streptomycin. Cell cultures
were maintained in a humidified incubator at 37°C in 5%
CO2 atmosphere. ECs were treated with ox-LDL or with a
combination of pretreatment with a CATL inhibitor.
Cathepsin L activity assay
After treatment, cells were washed twice with
ice-cold phosphate-buffered saline (PBS) and lysed in 40 mmol/l
sodium acetate buffer, pH 5.5, and 0.1% Triton X-100. CATL activity
was determined by the InnoZyme™ Cathepsin L fluorogenic activity
kit. Following the manufacturer’s protocol,
Z-Phe-Arg-7-amido-4-methylcoumarin (AMC) was used as a substrate.
The reaction mixture was incubated at 37°C for 10 min, and AMC
fluorescence intensity was quantified with a fluorescence plate
reader (excitation at 360 nm and emission at 460 nm).
RNA extraction and analysis
Total cellular RNA was extracted using TRI reagent
following the manufacturer’s instructions. One microgram of the RNA
sample was reversely transcribed to cDNA, and 2 μl cDNA was
added to the PCR amplification system. The primers were as follows:
LC3 sense, 5′-GAGTTACCTCCCGCAGCCGCA-3′ and antisense,
5′-TCCGCCGCTGCTTGAAAGGC-3′; beclin 1 sense,
5′-GGATGGATGTGGAGAAAGGCAAG-3′ and antisense,
5′-TGAGGACACCCAAGCAAGACC-3′; caspase-3 sense,
5′-CAGGGCGCCATCGCCAAGTA-3′ and antisense, 5′-TCA
GCTCTGGCCTCCGGCTG-3′; Bcl-2 sense, 5′-GCATGGAG GGCAGTGACGCA-3′ and
antisense, 5′-TCGCAGGACACC CAGGACCC-3′; glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) sense, 5′-TCACCATCTTCCAGGAGCGAG-3′ and
antisense, 5′-TGTCCCTGTTGAAGTCAGAG-3′. The bands were visualized
under UV light and the relative quantification of mRNA was assessed
from the cycle threshold and normalized to GAPDH mRNA in the same
samples.
Western blotting
ECs were treated with 0, 25, 50 or 75 mg/l ox-LDL
for 24 h or pretreated with 0, 2, 4 or 8 mg/l CATL inhibitors for
24 h and exposed to ox-LDL (50 μg/ml) for an additional 24
h. Protein concentrations were measured using the BSA method as a
standard. Equal amounts of total protein were resolved by SDS-PAGE,
and the blots were probed with antibodies to CATL (1:500), beclin 1
(1:200), LC3 (1:200), caspase-3 (1:100), Bcl-2, VE-cadherin (1:100)
(all were from Protech, USA), and appropriate secondary antibodies
conjugated to alkaline phosphatase. Human β-actin served as the
loading control. Blots were visualized with a chemiluminescence
kit.
Quantification of apoptosis
Apoptotic cells were distinguished using propidium
iodide (PI) staining. After treatment, cells were collected, washed
with ice-cold PBS pH 7.4, centrifuged, and resuspended in 1X
binding buffer. Cells were then incubated with fluorochrome for 15
min at 37°C. Apoptotic rates were determined by flow cytometry (BD
Biosciences, Franklin Lakes, NJ, USA) and analyzed using FlowJo
software.
Permeability assay
Endothelial permeability was analyzed using the
Costar Transwell system using an FITC-labeled dextran tracer. ECs
were cultured to confluency on collagen-coated polycarbonate
membranes. In the final 1 h of the respective treatments,
FITC-labeled dextran (1 mg/ml) was added to the upper chamber.
Fluorescence in the lower compartment was measured after 2 h using
a spectrofluorimeter (LS45; Perkin Elmer Life Sciences) with an
excitation wavelength of 490 nm and emission wavelength of 520
nm.
Statistical analysis
All experiments were repeated independently at least
3 times. Values were expressed as means ± SE. Student’s unpaired
t-test was used to establish significance between groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
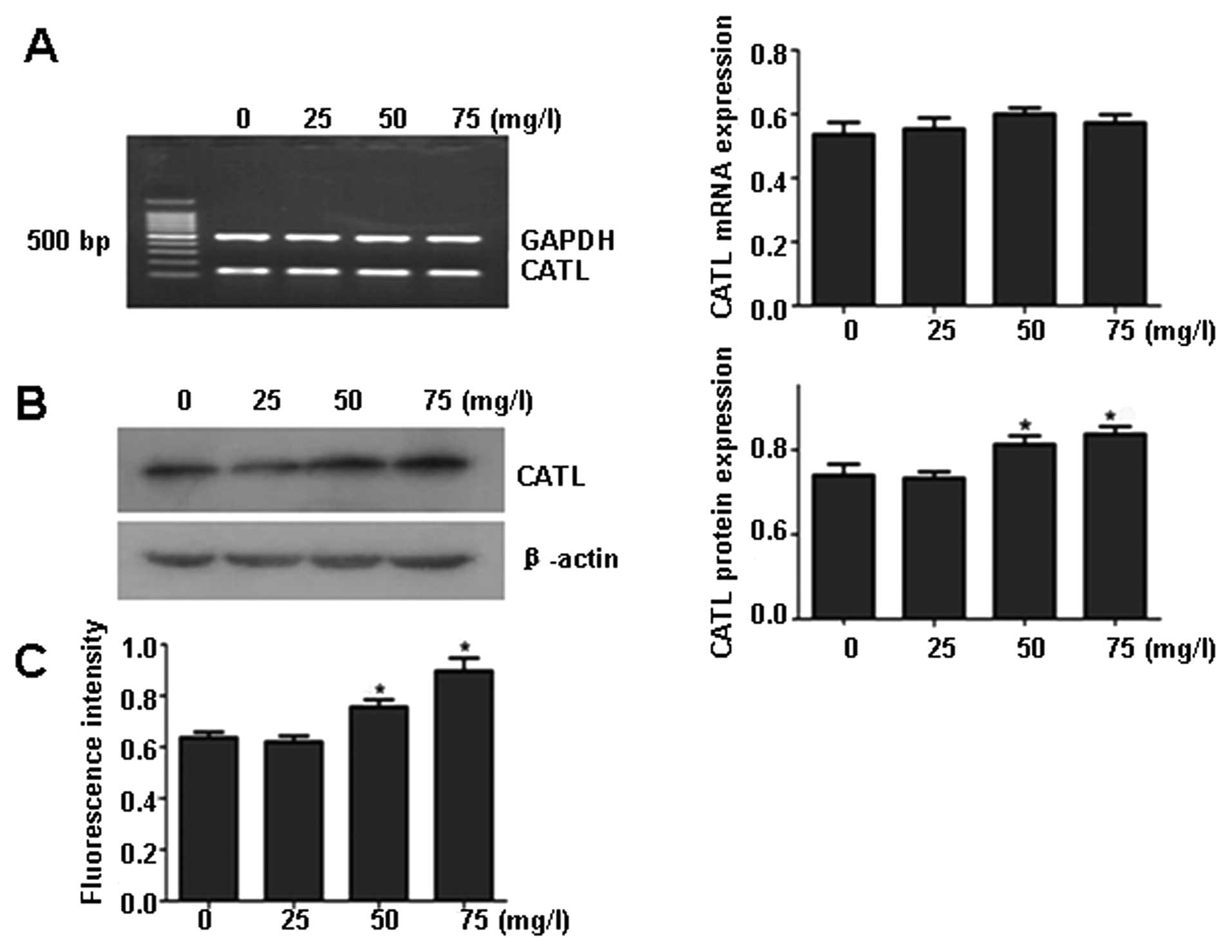
ox-LDL upregulates CATL protein
expression and activation
After treatment of ECs with 0, 25, 50 or 75
μg/ml ox-LDL for 24 h, CATL protein levels (Fig. 1B) and activity (Fig. 1C) were increased in response to
ox-LDL in a concentration-dependent manner. However, there was no
significant change in the mRNA levels of CATL (Fig. 1A).
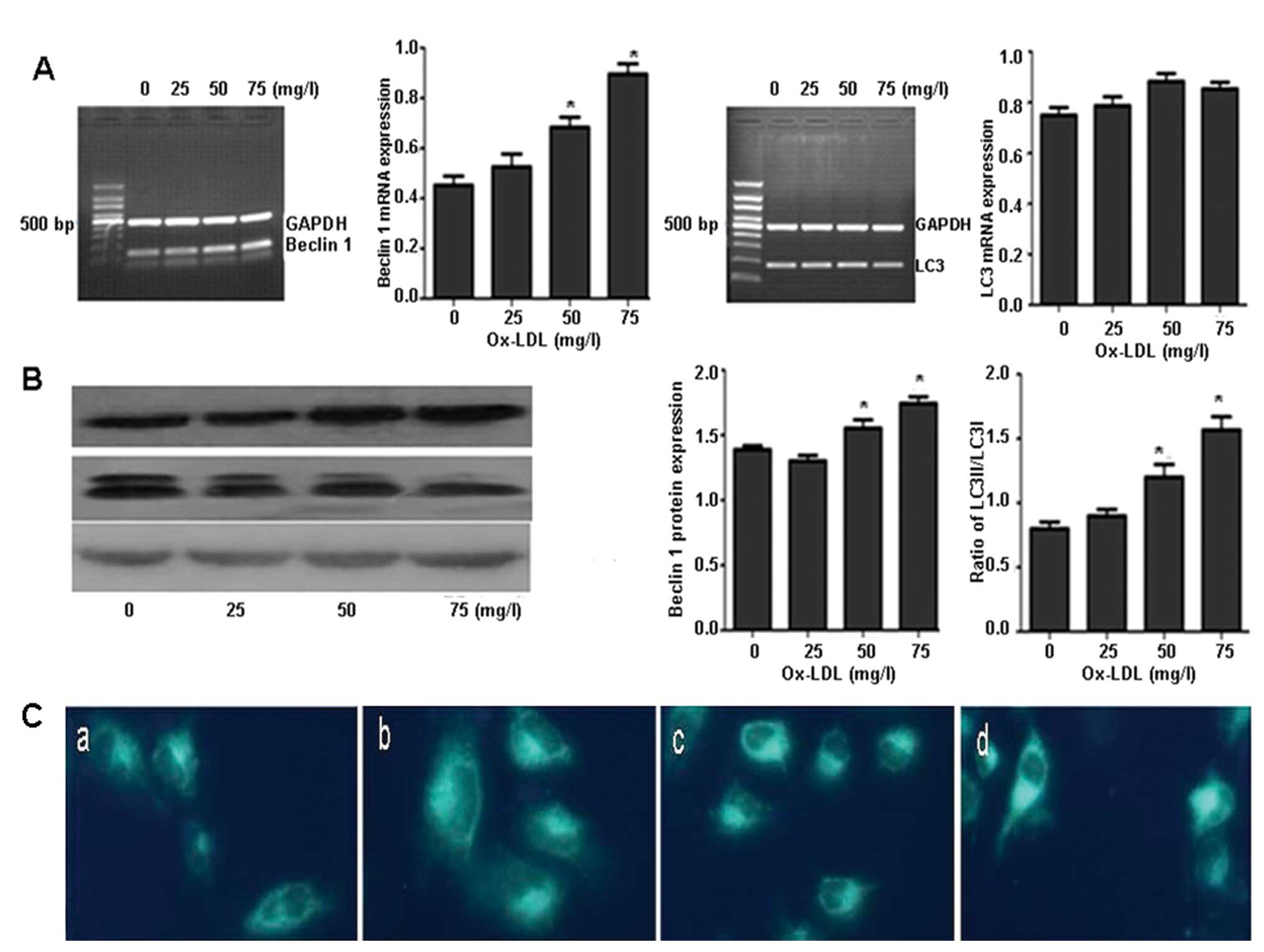
CATL inhibitor decreases ox-LDL-induced
autophagy
After treatment with various concentrations of
ox-LDL for 24 h, the mRNA and protein expression of beclin 1 was
increased in a concentration-dependent manner (Fig. 2A and B). The ratio of LC3II/LC3I
was also increased (Fig. 2B).
Furthermore, monodansylcadaverine (MDC) staining revealed the
accumulation of large MDC-stained vesicles (Fig. 2C).
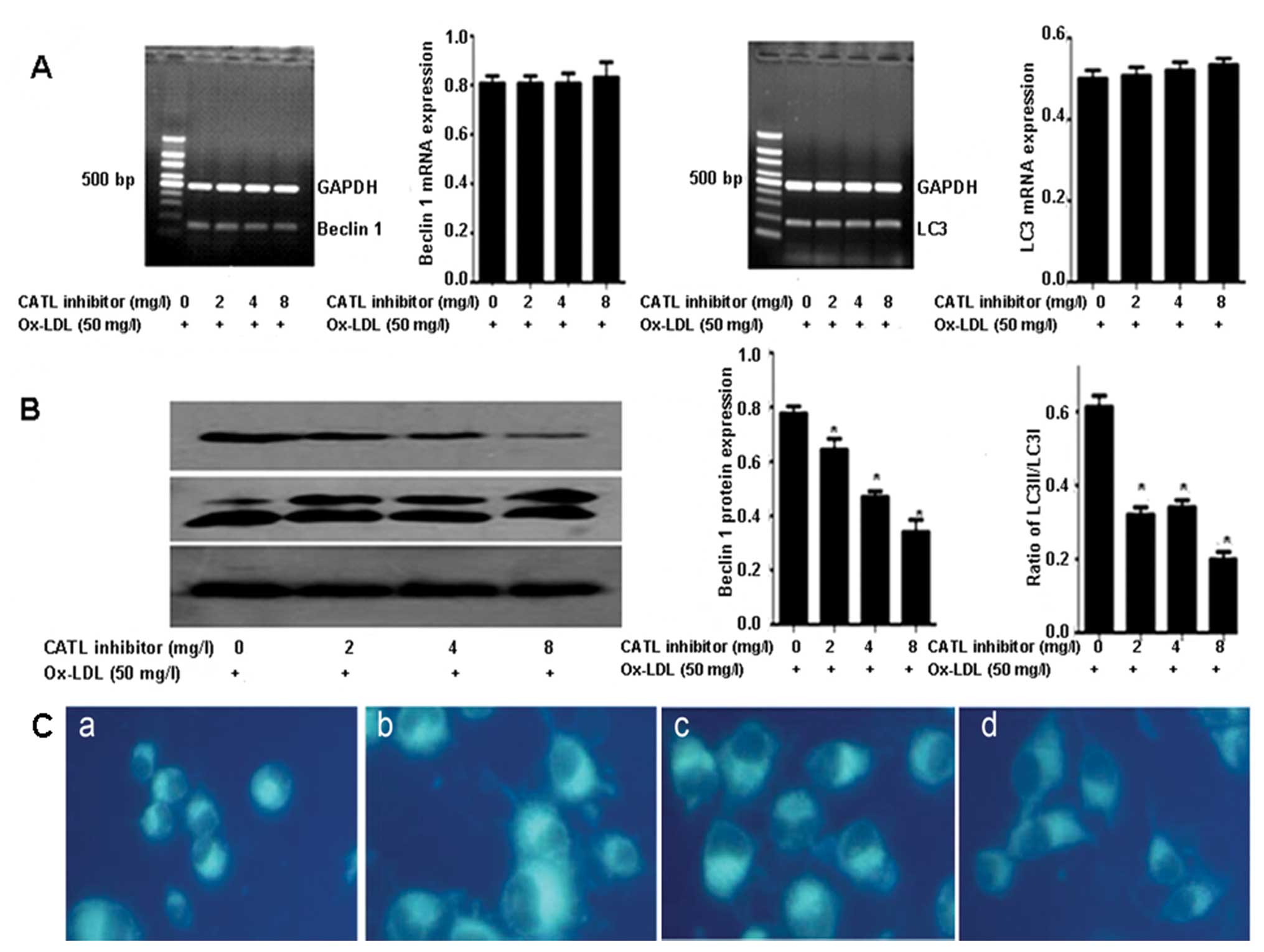
Pretreatment of ECs with a CATL inhibitor decreased
the protein levels of beclin 1 and the ratio of LC3II/LC3I
(Fig. 3B). However, no obviously
change in beclin 1 and LC3 mRNA expression was observed (Fig. 3A). The MDC-stained vesicles were
attenuated in response to CATL inhibitor treatment (Fig. 3C).
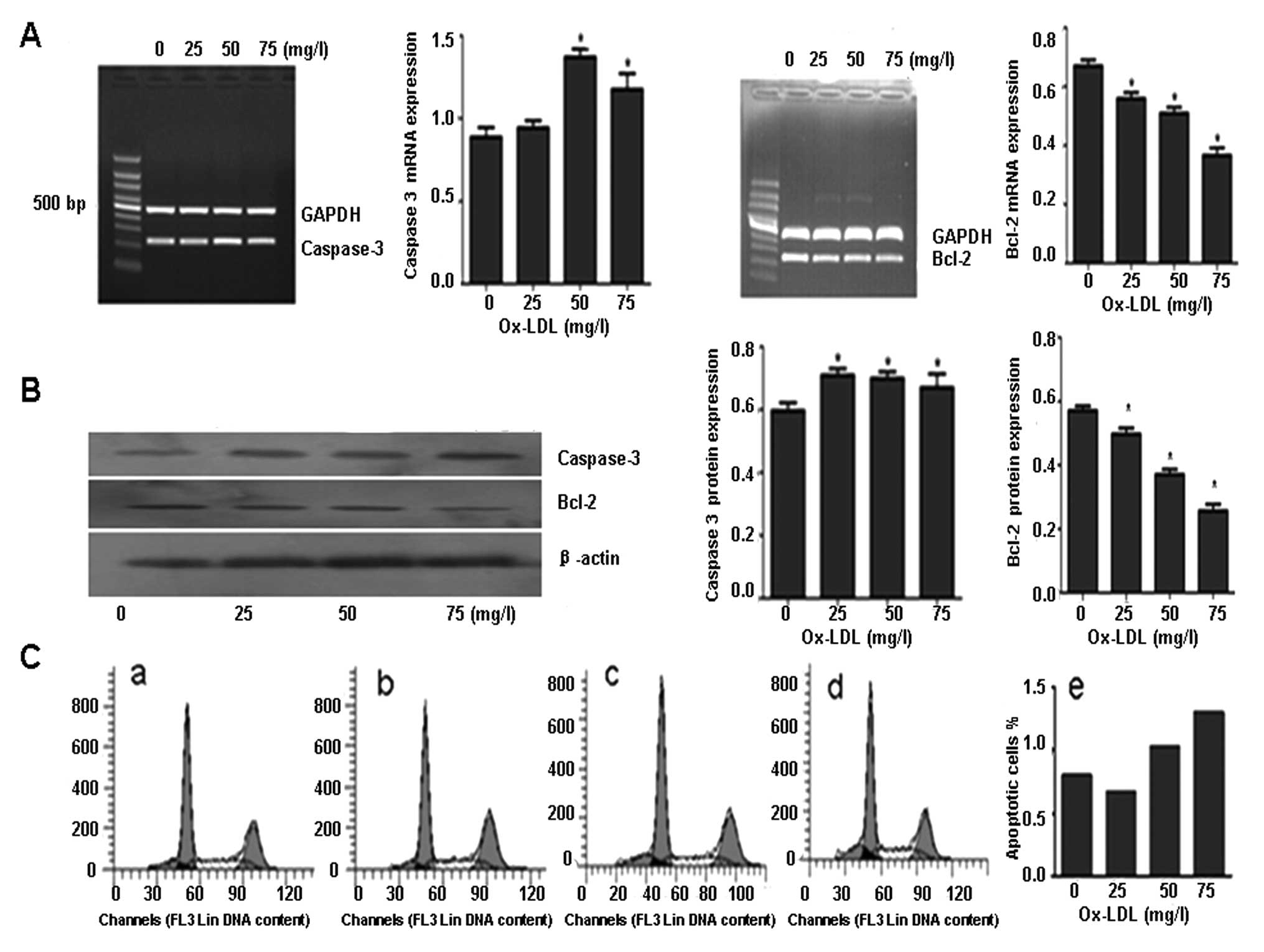
CATL inhibitor increases ox-LDL-induced
apoptosis
ECs treated with ox-LDL exhibited increased
caspase-3 expression and a reduction in Bcl-2 expression in a
dose-dependent manner (Fig. 4A and
B). Flow cytometry demonstrated that the percentage of
apoptotic ECs was increased following ox-LDL treatment (Fig. 4C).
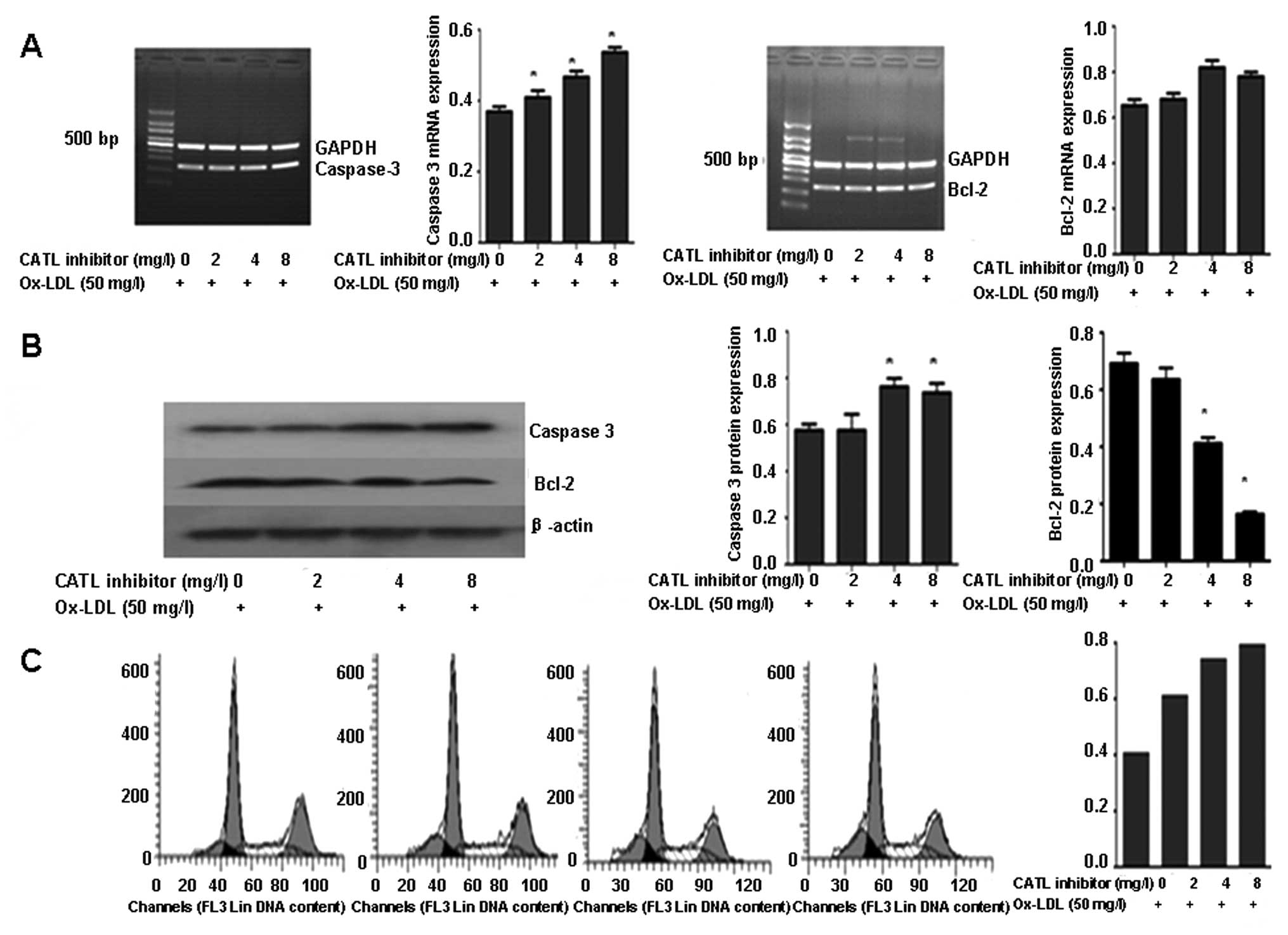
After pretreatment with a CATL inhibitor, the mRNA
and protein expression of caspase-3 was upregulated in response to
the CATL inhibitor in a concentration-dependent manner (Fig. 5A and B). The expression of the
Bcl-2 protein was further decreased (Fig. 5B). However, no significant change
in mRNA levels of caspase-3 and Bcl-2 was observed (Fig. 5A). The apoptotic ratio of the ECs
showed a concentration-dependent increase in response to the CATL
inhibitor (Fig. 5C).
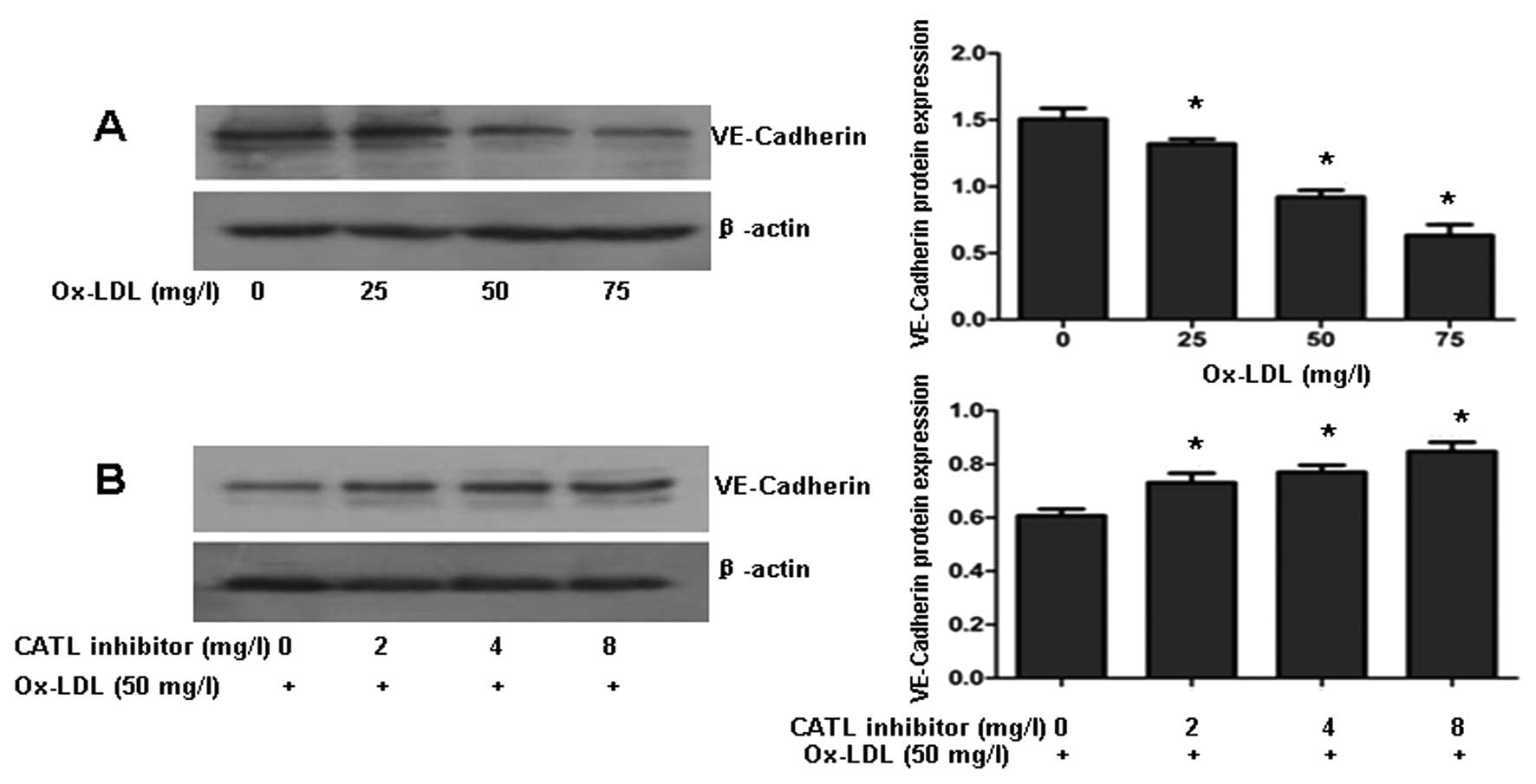
CATL inhibitor attenuates the
ox-LDL-induced decrease in VE-cadherin
VE-cadherin is one of the most important molecules
in the maintenance of endothelial integrity via its role in
adherens junctions. Western blotting revealed that ox-LDL decreased
VE-cadherin protein levels in a dose-dependent manner (Fig. 6A). When ECs were preincubated with
a CATL inhibitor for 24 h, followed by exposure to ox-LDL (50
mg/l), downexpression of VE-cadherin protein was reversed by the
CATL inhibitor (Fig. 6B).
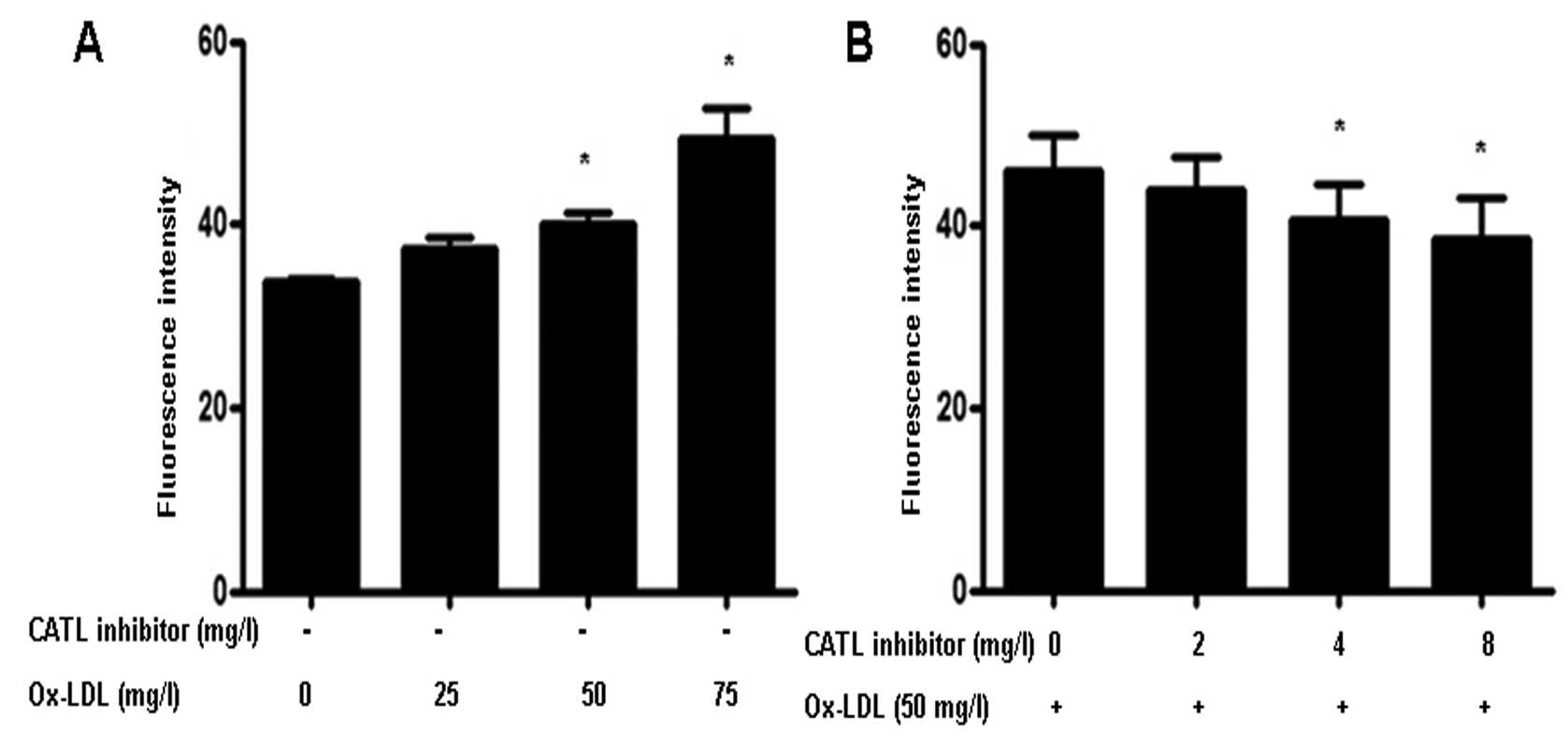
Effect of the CATL inhibitor on
endothelial monolayer permeability
Treatment of ox-LDL significantly increased
endothelial cell permeability (Fig.
7A). However, when ECs were pretreated with a CATL inhibitor,
the ox-LDL-induced increase in permeability was attenuated
(Fig. 7B).
Discussion
CATL has previously been reported to be involved in
the pregression of advanced atherosclerotic lesions. The present
study investigated the effect of CATL on ox-LDL-induced early
atherosclerotic events and its potential mechanisms. The results
revealed that ox-LDL increased CATL protein expression and
activation, inducing EC autophagy and apoptosis and increasing EC
monolayer permeability. When ECs were pretreated with a CATL
inhibitor, ox-LDL-induced autophagy was partly inhibited, while
apoptosis was further increased. Concomitantly, the ox-LDL-induced
decrease in VE-cadherin expression and increased EC monolayer
permeability was attenuated by the CATL inhibitor.
It is important to note that the death of ECs by
apoptosis is observed in the early stages of atherosclerosis. The
permeability of cultured bovine aortic endothelial cell monolayers
was found to be highly correlated with their rate of apoptosis and
that inhibiting apoptosis lowers the permeability of monolayers to
LDL (6). Several cathepsin family
members have been implicated in apoptosis. In human cancer cells,
cathepsin S siRNA induces autophagy and subsequent apoptosis
(7). Cathepsin B induces
mitochondrial release of cytochrome c and activates
caspase-3, signifying the onset of apoptotic cell death (8). In the present study, we found that
pretreatment with a CATL inhibitor resulted in increased apoptotic
cell death induced by ox-LDL, which means that the activation of
CATL may inhibit the apoptosis of ECs.
In addition to apoptosis, the death of vascular
cells by autophagy was also observed in this study. Autophagy is a
catabolic pathway for the bulk turnover of long-lived proteins and
organelles via lysosomal degradation. Basal autophagy is a survival
mechanism safeguarding vascular cells against oxidative injury,
metabolic stress and inflammation (9,10);
it is protective against EC injury and was observed in
athero-sclerotic plaques (11–15). Degradation of autophagolysosomal
content is impaired in CATL(−/−) mice (16). In this study, we examined the mRNA
and protein levels of beclin 1 (Bcl-2 interacting protein) and
microtubule-associated protein 1 light chain 3 (LC3), which have
previously been shown to promote autophagy. In ECs, the control
cells exhibited few autophagic features. After treatment with
ox-LDL, autophagy responses increased significantly. However,
autophagy was significantly inhibited in the presence of the CATL
inhibitor, indicating that upregulation of CATL may promote EC
autophagy.
Previous observations indicate that autophagy and
apoptosis are often induced by the same stimuli; they share similar
effectors and regulators, and are subjected to complex crosstalk
mechanisms. Recent studies have shown that beclin 1 and PI3K are
substrates of caspases-3, -7 and -8. Cleavage fragments lose the
autophagy-inducing capacity, while enhancing apoptosis by promoting
the release of proapoptotic factors from the mitochondria. It is
worth noting that the C-terminal fragments, Beclin-1-C, localized
predominantly at the mitochondria sensitized the cells to apoptosis
(17). Thus, the possible
mechanism of the CATL inhibitor-mediated increase in EC apoptosis
may be explained, at least in part, by increased caspase-3 protein
levels, which in turn create cleavaged beclin 1 consequently
promoting EC apoptosis. Hence, we speculated that increased
autophagic activity via CATL may overcome cell-death stimulation
and decrease apoptotic cell death, which represents an adaptive
process to benefit EC survival against ox-LDL damage.
The intercellular junctions of endothelial cells
also have an important barrier function that regulates
permeability. VE-cadherin is one of the main components of the
endothelial cell-cell junction, which determines the strength of
cell-cell adhesion and dictates monolayer properties (18). In our study, ox-LDL decreased the
VE-cadherin protein levels in a dose-dependent manner. However,
upon pretreatment with the CATL inhibitor, VE-cadherin protein
contents were markedly increased. In particular, overexpression of
CATL may contribute to the degradation of VE-cadherin induced by
ox-LDL. Consistent with the importance of VE-cadherin to
endothelial integrity, we found that pretreatment with the CATL
inhibitor decreased the permeability induced by ox-LDL.
Taken collectively, we propose that the role of CATL
in the alteration of ox-LDL-induced EC monolayer permeability is
due to two factors. First, the inhibition of autophagy and
induction of apoptosis by the CATL inhibitor suggest that the
upregulation of the expression of CATL may provide the benefit of
reducing endothelial cell monolayer permeability. Second,
upregulation of the expression of CATL may involve the degradation
of VE-cadherin, and this may be attributed to an increase in EC
barrier permeability induced by ox-LDL. As a result, the
proatherogenic effect of CATL may be neutralized by induction of EC
autophagy.
Acknowledgements
The present research is supported by
the National Natural Science Foundation of China (30800449), the
Science and Technology Innovative Research Team in Higher
Educational Institutions of Hunan Province and the Visiting Scholar
Foundation of Key Laboratory of Biorheological Science and
Technology (Chongqing University), the Ministry of Education.
References
|
1.
|
Koch S and Nusrat A: Dynamic regulation of
epithelial cell fate and barrier function by intercellular
junctions. Ann NY Acad Sci. 1165:220–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Tarbell JM: Mass transport in arteries and
the localization of atherosclerosis. Annu Rev Biomed Eng. 5:79–118.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Jormsjo S, Wuttge DM, Sirsjo A, Whatling
C, Hamsten A, Stemme S and Eriksson P: Differential expression of
cysteine and aspartic proteases during progression of
atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol.
161:939–945. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Rebbaa A, Chu F, Sudha T, Gallati C, Dier
U, Dyskin E, Yalcin M, Bianchini C, Shaker O and Mousa SA: The
anti-angiogenic activity of NSITC, a specific cathepsin L
inhibitor. Anticancer Res. 29:4473–4481. 2009.PubMed/NCBI
|
|
5.
|
Mahmood DF, Jguirim-Souissi I, Khadija
el-H, Blondeau N, Diderot V, Amrani S, Slimane MN, Syrovets T,
Simmet T and Rouis M: Peroxisome proliferator-activated receptor
gamma induces apoptosis and inhibits autophagy of human
monocyte-derived macrophages via induction of cathepsin L:
potential role in atherosclerosis. J Biol Chem. 286:28858–28866.
2011. View Article : Google Scholar
|
|
6.
|
Cancel LM and Tarbell JM: The role of
apoptosis in LDL transport through cultured endothelial cell
monolayers. Atherosclerosis. 208:335–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chen KL, Chang WS, Cheung CH, Lin CC,
Huang CC, Yang YN, Kuo CP, Kuo CC, Chang YH, Liu KJ, Wu CM and
Chang JY: Targeting cathepsin S induces tumor cell autophagy via
the EGFR-ERK signaling pathway. Cancer Lett. 317:89–98. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bhoopathi P, Chetty C, Gujrati M, Dinh DH,
Rao JS and Lakka S: Cathepsin B facilitates autophagy-mediated
apoptosis in SPARC overexpressed primitive neuroectodermal tumor
cells. Cell Death Differ. 17:1529–1539. 2010. View Article : Google Scholar
|
|
9.
|
Ouimet M, Franklin V, Mak E, Liao X, Tabas
I and Marcel YL: Autophagy regulates cholesterol efflux from
macrophage foam cells via lysosomal acid lipase. Cell Metab.
13:655–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T,
Xiao GD, Yang YP and Liu CF: The autophagy-lysosome pathway: a
novel mechanism involved in the processing of oxidized LDL in human
vascular endothelial cells. Biochem Biophys Res Commun.
394:377–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Liao X, Sluimer JC, Wang Y, Subramanian M,
Brown K, Pattison JS, Robbins J, Martinez J and Tabas I: Macrophage
autophagy plays a protective role in advanced atherosclerosis. Cell
Metab. 15:545–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Mei S, Gu H, Ward A, Yang X, Guo H, He K,
Liu Z and Cao W: p38 mitogen-activated protein kinase (MAPK)
promotes cholesterol ester accumulation in macrophages through
inhibition of macroautophagy. J Biol Chem. 287:11761–11768. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Zhaorigetu S, Yang Z, Toma I, McCaffrey TA
and Hu CA: Apolipoprotein L6, induced in atherosclerotic lesions,
promotes apoptosis and blocks Beclin 1-dependent autophagy in
atherosclerotic cells. J Biol Chem. 286:27389–27398. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Xie Y, You SJ, Zhang YL, Han Q, Cao YJ, Xu
XS, Yang YP, Li J and Liu CF: Protective role of autophagy in
AGE-induced early injury of human vascular endothelial cells. Mol
Med Rep. 4:459–464. 2011.PubMed/NCBI
|
|
15.
|
Martinet W and De Meyer GR: Autophagy in
atherosclerosis. Curr Atheroscler Rep. 10:216–223. 2008. View Article : Google Scholar
|
|
16.
|
Dennemärker J, Lohmüller T, Müller S,
Aguilar SV, Tobin DJ, Peters C and Reinheckel T: Impaired turnover
of autophagolysosomes in cathepsin L deficiency. Biol Chem.
391:913–922. 2010.PubMed/NCBI
|
|
17.
|
Wirawan E, Vande Walle L, Kersse K,
Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R,
Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S
and Vandenabeele P: Caspase-mediated cleavage of Beclin-1
inactivates Beclin-1-induced autophagy and enhances apoptosis by
promoting the release of proapoptotic factors from mitochondria.
Cell Death Dis. 1:e182010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sakamoto N, Segawa K, Kanzaki M, Ohashi T
and Sato M: Role of p120-catenin in the morphological changes of
endothelial cells exposed to fluid shear stress. Biochem Biophys
Res Commun. 398:426–432. 2010. View Article : Google Scholar : PubMed/NCBI
|